
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light photooxidation of nitrate: the dawn of a nocturnal radical†
T.
Hering
a,
T.
Slanina
a,
A.
Hancock
b,
U.
Wille
*b and
B.
König
*a
aInstitut für Organische Chemie, Universität Regensburg, Universitätsstrasse 31, D-93053 Regensburg, Germany. E-mail: burkhard.koenig@ur.de
bSchool of Chemistry and BIO21 Molecular Science and Biotechnology Institute, The University of Melbourne, 30 Flemington Road, Parkville, VIC 3010, Australia. E-mail: uwille@unimelb.edu.au
First published on 10th March 2015
Abstract
Highly oxidizing nitrate radicals (NO3˙) are easily accessed from readily available nitrate salts by visible light photoredox catalysis using a purely organic dye as the catalyst and oxygen as the terminal oxidant. The interaction of the excited catalyst and nitrate anions was studied by spectroscopic methods to elucidate the mechanism, and the method was applied to the NO3˙ induced oxidation of alkynes and alcohols.
The nitrate radical (NO3˙) is the most important nocturnal free radical oxidant in the troposphere and thus accounts for the majority of the oxidative reactions at night-time.1 In the atmosphere NO3˙ oxidizes a broad scope of volatile organic species including alkenes,2,3 alcohols,4,5 terpenes,1 esters,6 and sulfides.1 It is a highly reactive and chemically versatile O-centered radical7 with an oxidation potential of +2.00 V (vs. SCE in MeCN).8 Apart from electron transfer (ET),9,10 NO3˙ also reacts by addition to π systems1,11 and by hydrogen atom abstraction (HAT).8,12,13 Overall, the reactivity of NO3˙ with organic molecules can be seen in between that of hydroxyl radicals (OH˙) and sulfate radical anions (SO4˙−).14
Despite its high chemical versatility, it is surprising that only limited synthetic applications of NO3˙ are available so far. Shono reported the addition of electrochemically generated NO3˙ to alkenes.11 The reaction of NO3˙ with cyclic alkynes and alkynones was employed to obtain cis-fused bicyclic ketones in self-terminating oxidative radical cyclizations.15,16 This concept was later extended to alkyne ethers yielding tetrasubstituted tetrahydrofurans.17,18 One reason for the limited use of NO3˙ as a reagent in organic transformations is its rather difficult accessibility. Common methods for NO3˙ generation on preparative scale in solution are the reaction of nitrogen dioxide and ozone,1,19 electrooxidation of nitrate anions11 or the photolysis of (NH4)2Ce(NO3)6 (CAN) with UV light (λ = 350 nm)14,20 However, the use of toxic gases, high electrode potentials,8 or UV irradiation are so far limiting the applications and lead to undesired side reactions.
We were pleased to observe that, upon excitation of the organic photocatalyst 9-mesityl-10-methylacridinium perchlorate (1) with blue light, oxidation of nitrate anions to NO3˙, readily occurs (Scheme 1), thus providing a convenient access to NO3˙ on a preparative scale. 9-Mesityl-10-methylacridinium perchlorate (1) was chosen, because it is known to have a strong oxidizing capacity in the excited state.21,22 To the best of our knowledge, this is the first visible light mediated generation of nitrate radicals.
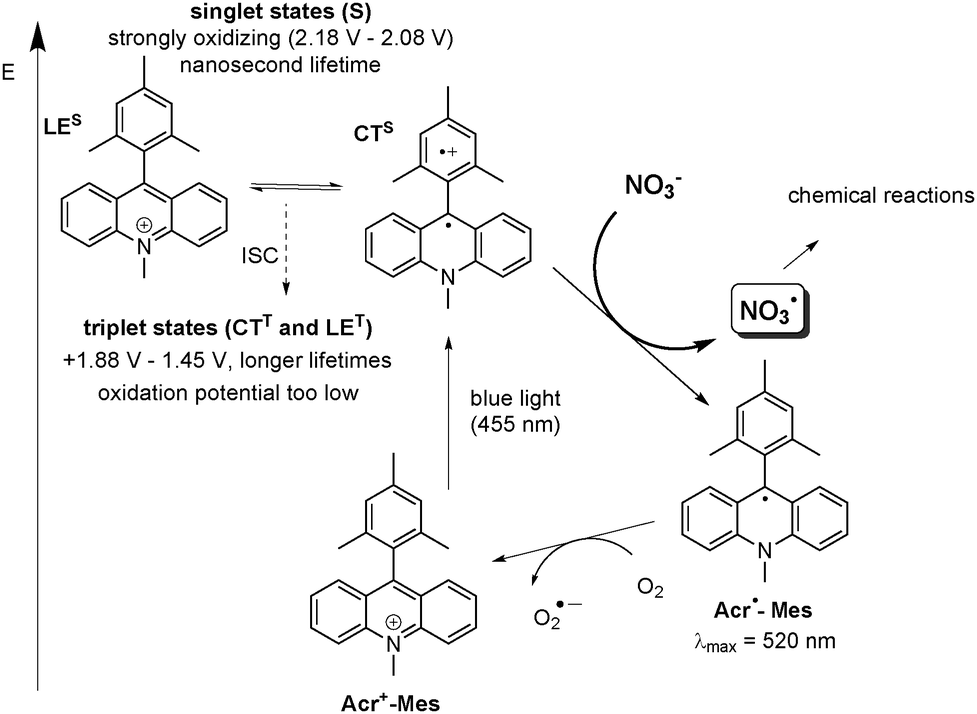 | ||
| Scheme 1 Proposed mechanism of visible light mediated generation of NO3˙ via photocatalytic oxidation by Acr+-Mes (1). The electron transfer from NO3− occurs from the short-lived singlet state (LES or CTS) with sufficient oxidative capacity to generate the reduced catalyst Acr˙-Mes and NO3˙, the longer lived transient triplet species (CTT or LET) is not reactive towards NO3−. The reduced catalyst Acr˙-Mes is regenerated by oxygen. (All oxidation potentials are given vs. SCE in MeCN or PhCN).23,25,26 | ||
In order to elucidate the mechanism of the NO3˙ formation, we monitored the generation of reduced catalyst Acr˙-Mes in the presence of LiNO3 upon continuous irradiation of a 5 μM solution of Acr+-Mes (1) in MeCN with 455 nm light under anaerobic conditions. The differential absorption spectrum shows the appearance of Acr˙-Mes with a maximum at 520 nm21,23 after irradiation for 120 s and 240 s. (see ESI,† Fig. S6) This observation suggests a direct oxidation of NO3− by the excited catalyst thus demonstrating that NO3− can act as an electron donor to the excited catalyst. The reduced catalyst Acr˙-Mes is stable under argon, however, the signal vanishes completely after aeration of the reaction mixture due to reoxidation of Acr˙-Mes to the ground state catalyst Acr+-Mes by oxygen (see Scheme 1).24 The negative signal at λ < 460 nm in the differential absorption spectrum is caused mainly by the decrease of the ground state absorption of Acr+-Mes as a result of Acr˙-Mes formation and partial photobleaching of Acr+-Mes.‡ The long-lived triplet state with a microsecond lifetime is generally discussed as the reactive state in most oxidative reactions.25,26 The exact nature of this state is controversial and could be both a CTT state with an oxidation potential of +1.88 V vs. SCE, as reported by Fukuzumi25 or a locally excited triplet state, LET, with an oxidation potential of +1.45 V vs. SCE as reported by Verhoeven,26 However, neither would have the oxidative capacity to oxidize NO3−. Recent detailed mechanistic investigations by the group of Nicewicz revealed that for substrates with oxidation potentials exceeding +1.88 V (vs. SCE), a reaction should occur out of the short-lived excited singlet state (mainly CTS), which has an estimated oxidation potential of 2.08 V (Scheme 1).23 Since both singlet states are fluorescent (ϕF ∼ 8%), whereas the triplet states do not emit,23 we performed fluorescence quenching experiments to explore the nature of the reactive state involved in NO3− oxidation. A clear quenching of the fluorescence by LiNO3 confirms that oxidation of NO3− occurs from the singlet excited state of 1 (see ESI,† Fig. S6). Moreover, laser flash photolysis experiments confirmed that no interaction of the long lived triplet state and NO3− can be observed (Fig. S8 in the ESI†). Based on these findings, we suggest that the reaction proceeds via a singlet excited state as depicted in Scheme 1.
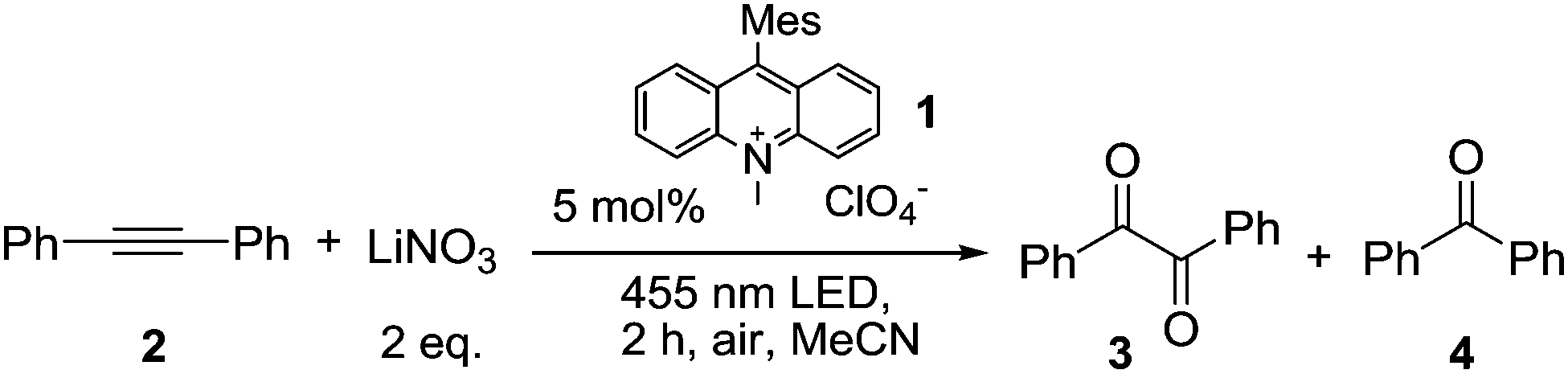
Having demonstrated the pathway for photocatalytic NO3˙ generation, we selected the well-studied reaction of NO3˙ with diphenylacetylene (2) yielding benzil (3) and benzophenone (4) to explore the synthetic application of this new method and to compare it with the previously reported methods. The results are compiled in Table 1. Under photocatalytic conditions using 5 mol% of Acr+-Mes (1), 0.25 mmol of alkyne 2 and 2 eq. of LiNO3, diketone 3 and ketone 4 were obtained after 2 h of irradiation with blue light (λ = 455 nm) with yields comparable to previous methods.27 When oxygen was replaced by ammonium persulfate as the electron acceptor in a degassed system, the yield and product ratio were not changed significantly (entry 5). This shows that potential interfering reactions by singlet oxygen could be excluded. In the absence of light or catalyst no reaction occurred (entries 7 and 9). However, small amounts of diketone 3 were formed in the direct reaction of 2 with the excited catalyst in the absence of nitrate ions (entry 8).
|
|
||
|---|---|---|
| Entry | Conditions | Yieldb3 + 4 (%) |
| a Reactions were carried out using diphenylacetylene (2, 0.25 mmol) and the respective amount of 9-mesityl-10-methylacridinium perchlorate (1) in 1 mL of MeCN unless otherwise noted with an irradiation time of 2 h. b Quantitative GC yields using acetophenone as internal standard. | ||
| 1 | 5 mol% 1, air | 50 (30 + 20) |
| 2 | 5 mol% 1, O2 | 55 (31 + 24) |
| 3 | NaNO3 | 41 (27 + 15) |
| 4 | 10 mol% 1 | 38 (24 + 14) |
| 5 | (NH4)2S2O8, N2 atmosphere | 46 (27 + 19) |
| 6 | DCM | 52 (32 + 20) |
| 7 | Without light | 0 |
| 8 | Without NO3− | 13 (3 only) |
| 9 | Without 1 | 0 |
According to computational studies, the mechanism for the NO3˙ induced oxidation of diphenylacetylene suggests formation of diketone 3 and benzophenone (4) through competing pathways in the initial vinyl radical adduct 5 (Scheme 2). While diketone 3 results from a 5-endo cyclization, followed by loss of NO˙, the key-step in the formation of benzophenone (4) is γ-fragmentation with elimination of NO2˙, and subsequent Wolff-rearrangement of the carbene intermediate 7 followed by oxidative decarboxylation.27
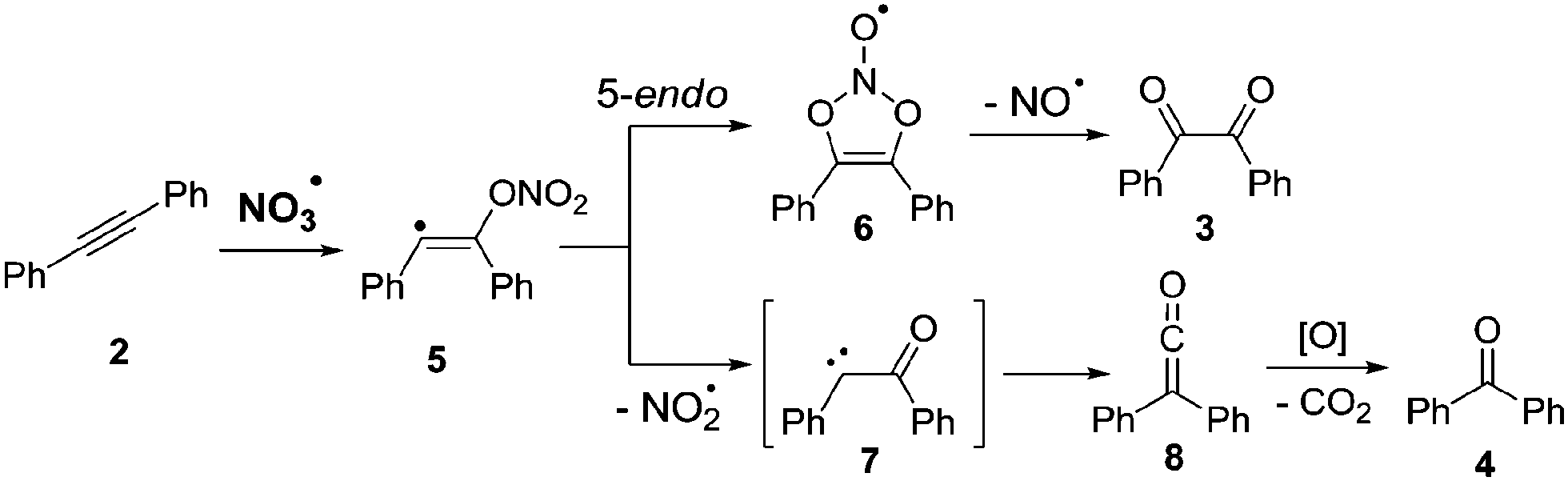 | ||
| Scheme 2 Proposed mechanism for the oxidation of aromatic alkynes by NO3˙.27 | ||
Next, we applied the photocatalytic NO3˙ formation to the synthesis of tetrasubstituted tetrahydrofurans, which proceeds via a self-terminating radical cascade that is initiated by NO3˙ addition to the triple bond in alkyne 9. The reaction was described previously using either anodic oxidation of lithium nitrate or CAN photolysis.17,18 The starting material 9 (Scheme 3) contains an aliphatic alkyne, which is more difficult to oxidize compared to 2 and thus decreases the background reaction that is caused by direct oxidation of 9 by the photocatalyst. The reaction of 9b with 2 eq. of LiNO3 and 5 mol% 1 gave the anticipated product 10b in a yield of 37% (67% based on conversion), with 45% of the starting material 9b being recovered. Methyl ether 9a gave lower yields and an incomplete conversion, which can be rationalized by a non-regioselective addition of NO3˙ to both ends of the alkyne,§ in accordance with previous reports. The low conversion (and resulting low product yield) is likely due to the fact that NO3˙ leads to degradation of catalyst 1. This effect could also be observed in UV/Vis measurements of the reaction mixture, which showed considerable photobleaching of the ground state during irradiation (see Fig. S7 in the ESI†). It is likely that the observed degradation proceeds via oxidation of the methyl groups on the mesityl moiety of the catalyst,8 which is a known degradation pathway that leads to loss of catalytic activity.28 The problem of low conversion could be partly overcome through slow addition of the catalyst via syringe pump.
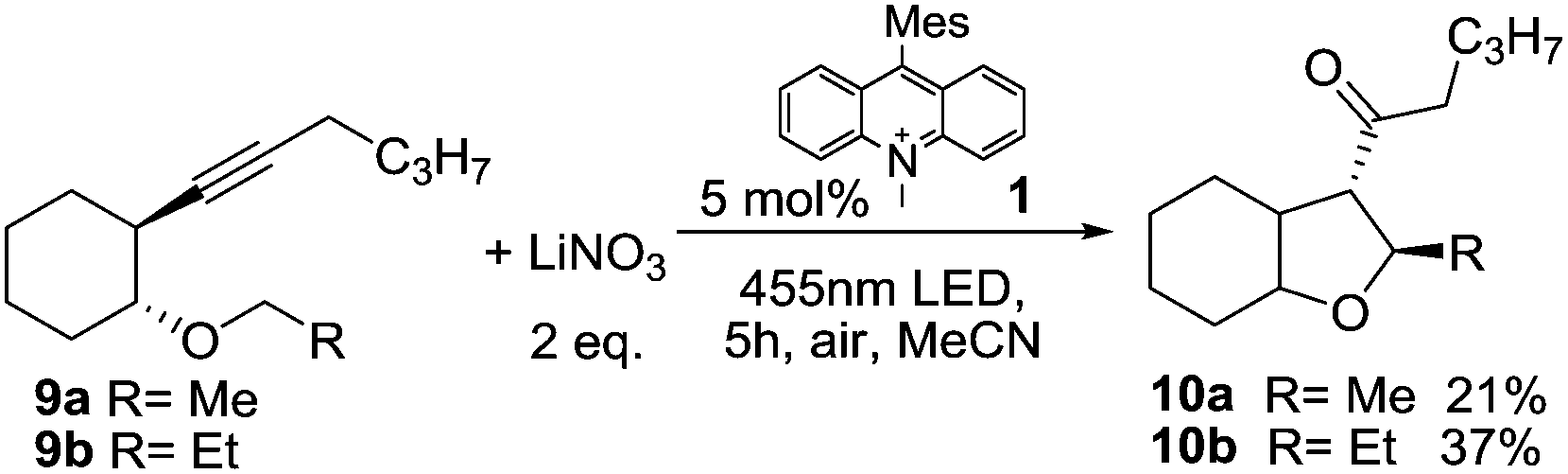 | ||
| Scheme 3 Self-terminating radical oxidative cyclization to tetrasubstituted tetrahydrofurans 10.17,18 | ||
Apart from addition to π systems, NO3˙ also reacts through hydrogen abstraction,8,12,13 which was explored in the catalytic oxidation of non-activated alcohols. In this reaction, NO3˙ acts as a redox mediator, which is regenerated during the catalytic cycle, according to the mechanism in Scheme 4. Initial HAT from the alcohol carbon atom by NO3˙29 leads to the regeneration of NO3− as nitric acid and formation of radical 12. The latter is subsequently oxidized by either NO3˙ or oxygen to give cationic intermediate 13, which deprotonates to yield ketone 14. The mechanism is similar to the indirect anodic oxidation of alcohols by nitrate.30 Donaldson and Styler reported the enhanced gas phase oxidation of propanol under UV irradiation using TiO2 co-embedded with KNO3. The finding was explained by formation of NO3˙ and its ability to abstract hydrogen atoms from the alcohol carbon atom.31
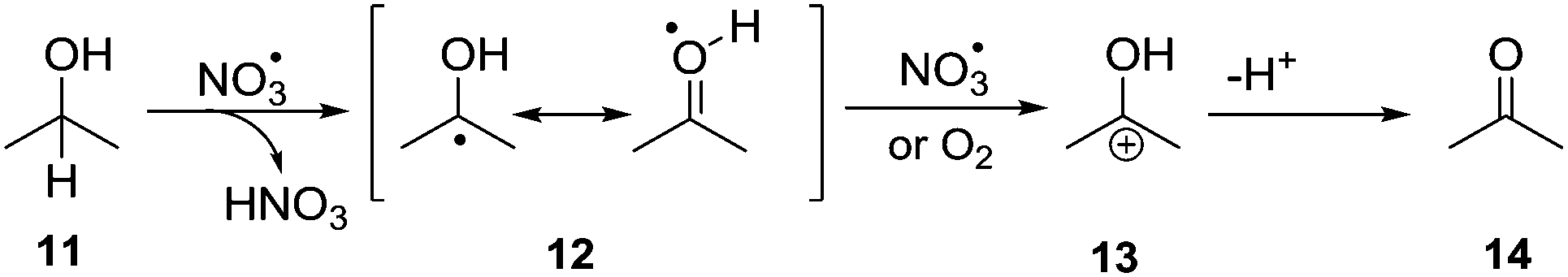 | ||
| Scheme 4 General mechanism of the nitrate mediated alcohol oxidation via initial hydrogen abstraction followed by oxidation and loss of a proton. | ||
The reaction was explored using tert-butyl cyclohexanol (11a) and the results are compiled in Scheme 5. To our delight, oxidation into the corresponding ketone 14a occurred upon irradiation with blue light in the presence of LiNO3 using 5 mol% of 1 in acetonitrile. No reaction was observed in the absence of nitrate, which clearly confirms the role of NO3˙ in this reaction. Stepwise reduction of the amount of LiNO3 from 2 eq. to 20 mol% did not affect the outcome, showing that NO3˙ can act as mediator in this reaction (Scheme 5). An acidification of the solution due to formation of nitric acid was observed, but no apparent influence on the reaction or the stability of the catalyst was found.¶
 | ||
| Scheme 5 Experimental conditions and results for the NO3˙ mediated oxidation of alcohols. | ||
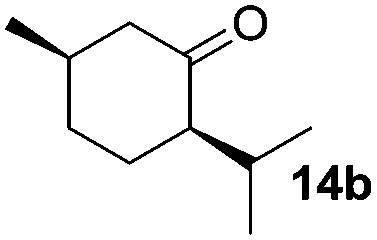
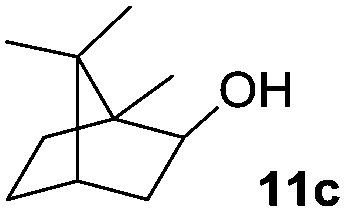
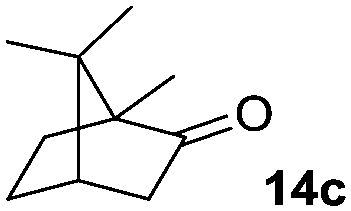
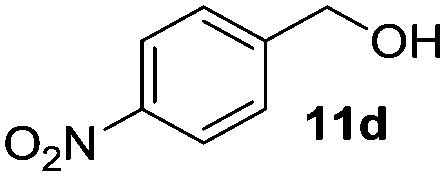
The scope of this method was explored towards other non-activated alcohols and electron deficient benzyl alcohols. All reactions were carried out by two sequential additions of 5 mol% of 1 in order to counteract the loss of catalytic activity caused by degradation of the catalyst. The reactions proceed with good selectivity (see Table 2, entries 1, 2, 4), but the conversion was incomplete and unreacted starting material was recovered. Aliphatic (entries 1–3) and benzylic alcohols (entries 4 and 6) were converted.
| Entry | Alcohol | Product | Yield productb (%) | Recovered starting materialb (%) |
|---|---|---|---|---|
| a Reactions carried out using 0.25 mmol of the alcohol 11, 1 eq. of LiNO3 and 10 mol% of 1 (two subsequent additions of 5 mol%) in 1 mL of MeCN with an irradiation time of 6 h. b Isolated yields, in brackets yield based on conversion. c Background reaction without LiNO3 is 9%. d Decomposition of substrate 11e. | ||||
| 1 |
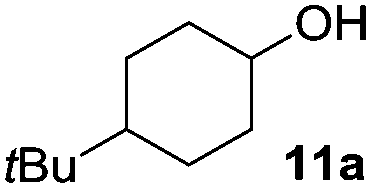
|
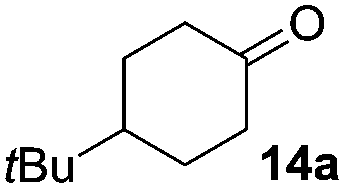
|
45 (79) | 44 |
| 2 |
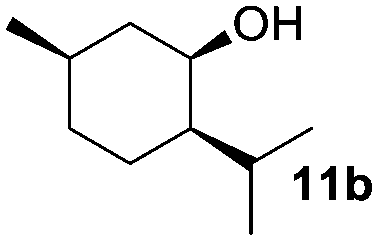
|
|
42 (95) | 56 |
| 3 |
|
|
40 (40) | — |
| 4c |
|
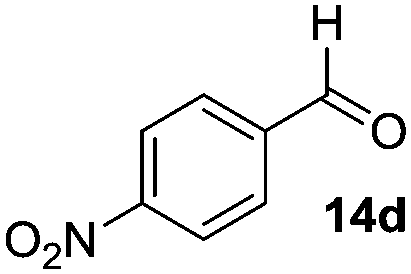
|
55 (100) | 45 |
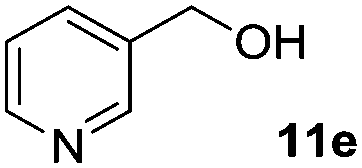
| 5d |
|
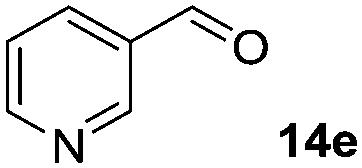
|
—d | —d |
| 6 |
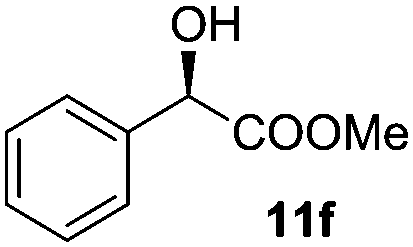
|
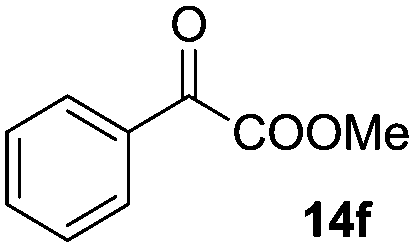
|
16 (20) | 17 |
In the oxidation of isomenthol (11b) (entry 2) the configuration of the stereocenter remained unchanged, while the basic substrate 11e gave no product, which is most likely due to an acid–base reaction of pyridine with nitric acid that is generated during this reaction|| by the H-abstraction by NO3˙ or a possible direct oxidation of the nitrogen of pyridine by the photocatalyst or NO3˙ (entry 5).32
In conclusion, we described a new and simple access to highly reactive nitrate radicals using visible light photocatalysis with an organic dye as the photoredox catalyst. This method avoids the use of toxic compounds, or high electrochemical potentials and is, to the best of our knowledge, the first method yielding NO3˙ in a catalytic process using visible light. We verified the formation of nitrate radicals by observation of the reduced catalyst Acr˙-Mes and showed that the mechanism is proceeding via the singlet excited state of the catalyst. By investigating the addition to aromatic alkynes, a previously well studied model reaction of NO3˙, we showed that the photocatalytic procedure is as efficient as the previously employed methods.
Financial support by the Deutsche Forschungsgemeinschaft (DFG), the GRK 1626 and the Australian Research Council is acknowledged. TH thanks the Fonds der Deutschen Chemischen Industrie for a fellowship.
Notes and references
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. Le Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, Atmos. Environ., Part A, 1991, 25, 1–203 CrossRef.
- M. P. Pérez-Casany, I. Nebot-Gil, J. Sánchez-Marín, F. Tomás-Vert, E. Martínez-Ataz, B. Cabañas-Galán and A. Aranda-Rubio, J. Org. Chem., 1998, 63, 6978–6983 CrossRef PubMed.
- H. Gong, A. Matsunaga and P. J. Ziemann, J. Phys. Chem. A, 2005, 109, 4312–4324 CrossRef CAS PubMed.
- J. C. Harrison and J. R. Wells, Int. J. Chem. Kinet., 2012, 44, 778–788 CrossRef CAS.
- D. Rousse and C. George, Phys. Chem. Chem. Phys., 2004, 6, 3408–3414 RSC.
- S. Langer, E. Ljungstrom and I. Wangberg, J. Chem. Soc., Faraday Trans., 1993, 89, 425–431 RSC.
- O. Ito, S. Akiho and M. Iino, J. Phys. Chem., 1989, 93, 4079–4083 CrossRef CAS.
- E. Baciocchi, T. D. Giacco, S. M. Murgia and G. V. Sebastiani, J. Chem. Soc., Chem. Commun., 1987, 1246–1248 RSC.
- H. Suzuki and T. Mori, J. Chem. Soc., Perkin Trans. 2, 1996, 677–683 RSC.
- E. Baciocchi, I. Del Giacco, C. Rol and G. V. Sebastiani, Tetrahedron Lett., 1985, 26, 541–544 CrossRef CAS.
- T. Shono, M. Chuankamnerdkarn, H. Maekawa, M. Ishifune and S. Kashimura, Synthesis, 1994, 895–897 CrossRef CAS PubMed.
- A. A. Fokin, S. A. Peleshanko, P. A. Gunchenko, D. V. Gusev and P. R. Schreiner, Eur. J. Org. Chem., 2000, 3357–3362 CrossRef CAS.
- M. Mella, M. Freccero, T. Soldi, E. Fasani and A. Albini, J. Org. Chem., 1996, 61, 1413–1422 CrossRef CAS.
- U. Wille, Chem. – Eur. J., 2002, 8, 340–347 CrossRef CAS.
- U. Wille, J. Am. Chem. Soc., 2001, 124, 14–15 CrossRef PubMed.
- U. Wille, Chem. Rev., 2012, 113, 813–853 CrossRef PubMed.
- U. Wille and L. Lietzau, Tetrahedron, 1999, 55, 11465–11474 CrossRef CAS.
- U. Wille and L. Lietzau, Tetrahedron, 1999, 55, 10119–10134 CrossRef CAS.
- L. F. Gamon, J. M. White and U. Wille, Org. Biomol. Chem., 2014, 12, 8280–8287 CAS.
- D. C. E. Sigmund and U. Wille, Chem. Commun., 2008, 2121–2123 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
- N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima and S. Fukuzumi, Res. Chem. Intermed., 2013, 39, 205–220 CrossRef CAS.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- A. C. Benniston, A. Harriman, P. Li, J. P. Rostron, H. J. van Ramesdonk, M. M. Groeneveld, H. Zhang and J. W. Verhoeven, J. Am. Chem. Soc., 2005, 127, 16054–16064 CrossRef CAS PubMed.
- U. Wille and J. Andropof, Aust. J. Chem., 2007, 60, 420–428 CrossRef CAS.
- A. C. Benniston, K. J. Elliott, R. W. Harrington and W. Clegg, Eur. J. Org. Chem., 2009, 253–258 CrossRef CAS.
- S. Langer and E. Ljungstrom, J. Chem. Soc., Faraday Trans., 1995, 91, 405–410 RSC.
- D. Kyriacou, Modern Electroorganic Chemistry, Springer-Verlag, Berlin, Heidelberg, 1994 Search PubMed.
- S. A. Styler and D. J. Donaldson, Environ. Sci. Technol., 2011, 45, 10004–10012 CrossRef CAS PubMed.
- A. Thellend, P. Battioni, W. Sanderson and D. Mansuy, Synthesis, 1997, 1387–1388 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc01580d |
| ‡ After aeration the ground state absorption of Acr+-Mes cannot be fully recovered (see ESI†). |
| § For the mechanism of this reaction see ESI.† |
| ¶ The addition of different bases (LiNO3, LiOAc, pyridine, lutidine) did not influence the outcome of the reaction or the stability of the catalyst. |
| || Based on the assumption that both the initial hydrogen abstraction and the oxidation of 12 are done by nitrate radicals. |
| This journal is © The Royal Society of Chemistry 2015 |