
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel cobalt oxide hollow nanosponges as advanced electrocatalysts for the oxygen evolution reaction†
Chengzhou
Zhu
a,
Dan
Wen
a,
Susanne
Leubner
a,
Martin
Oschatz
b,
Wei
Liu
a,
Matthias
Holzschuh
a,
Frank
Simon
c,
Stefan
Kaskel
b and
Alexander
Eychmüller
*a
aPhysical Chemistry, TU Dresden, Bergstrasse 66b, 01062 Dresden, Germany. E-mail: alexander.eychmueller@chemie.tu-dresden.de; Fax: +49 351 463/37164; Tel: +49 351 463/39843
bInorganic Chemistry, TU Dresden, Bergstrasse 66, 01062 Dresden, Germany
cLeibniz Institute of Polymer Research Dresden, Hohe Straße 6, 01069 Dresden, Germany
First published on 30th March 2015
Abstract
A class of novel nickel cobalt oxide hollow nanosponges were synthesized through a sodium borohydride reduction strategy. Due to their porous and hollow nanostructures, and synergetic effects between their components, the optimized nickel cobalt oxide nanosponges exhibited excellent catalytic activity towards oxygen evolution reaction.
The rational design of novel self-assembled architectures has received great consideration in recent years driven by their creative synergetic collective properties and widespread fields of application.1 Among them, three-dimensional (3D) porous nanostructures consisting of various functional nanomaterials have been a long sought-after class of materials due to their low relative density, high specific surface area, and gas permeability as well as size-enhanced functionalities in various applications.2 As one of the most important functional materials amongst transition metal oxides, nickel cobalt oxide has recently attracted considerable attention in applications like energy storage, catalysis, sensing, and field-emission, mainly because of its unique chemical and magnetic properties.3 Compared with NiO and Co3O4, binary nickel cobalt oxides are endowed with rich redox reactions and improved electrical conductivity, which are beneficial for a broad range of electrochemical applications, ranging from supercapacitors and fuel cells to water oxidation.4 The electrochemical efficiency of these materials strongly depends on their morphologies. Namely, the materials possessing high specific surface areas can offer abundant active sites for electrocatalysis, easy transport channels for electrolyte ions, and continuous passageways for charge carrier transport.5 Efforts to search for advanced nickel cobalt oxide nanomaterials, thus providing attractive opportunities to develop advanced electrochemical devices, lead to the accurate design of a variety of 3D porous nanostructures.
Meanwhile, highly active and cost-effective electrocatalysts for the oxygen evolution reaction (OER) are essential components of renewable energy technologies, such as solar fuel synthesis and providing a hydrogen source for powering fuel cells.6 Currently, precious metal oxides (e.g. RuO2 and IrO2) are regarded as the most robust and efficient catalysts for this oxidation reaction.7 However, the high cost and scarcity severely limit their widespread application in the OER. In this context, intensive efforts have been devoted to the development of low-cost alternatives with high activity.8 Among various OER catalysts, nickel cobalt oxides have received enormous attention due to their low cost and large number of active centers.9 To date, a number of approaches to obtain functional nickel cobalt oxides have been reported, which can be commonly transformed from appropriate precursors such as metal hydroxides, carbonates, oxalate, and inorganic salts.4a,5,10 The various combinations of cations and tuning the composition within these materials provide more possibilities to change their chemical properties and thus optimize their electrocatalytic performance. Additionally, the synthesis of 3D porous nickel cobalt oxides is believed to enhance their OER performance because of the high surface area, profuse porosity as well as tunable compositions.11
Herein, we report on a series of nickel cobalt oxide hollow nanosponges (HNSs), successfully synthesized using a facile approach. 3D bimetallic nanosponges were first synthesized by a kinetically controlled sodium borohydride reduction process. After supercritical drying, 3D nickel cobalt oxide HNSs can be obtained via annealing at high temperatures (Scheme 1). Taking advantage of the remarkable features of the novel materials, such as their porous and hollow nanostructures as well as the synergetic effect between the components, the resulting nickel cobalt oxide HNSs abundant in the spinel structure exhibit greatly enhanced OER activity.
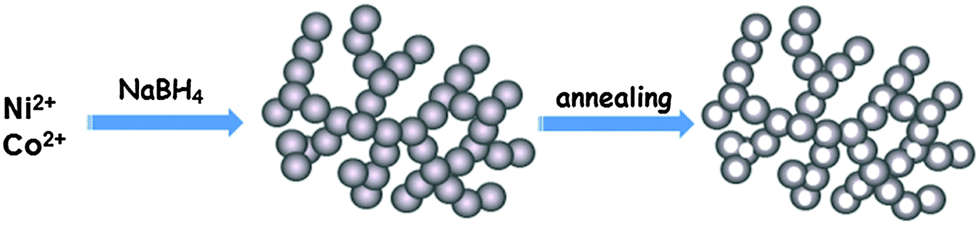 | ||
| Scheme 1 A schematic illustration of the synthesis of Ni–Co oxide HNSs. | ||
With regard to the synthesis of bimetallic Co–Ni nanosponges, 6 mL of the metal precursors containing 4.0 mL of CoCl2 (0.1 M) solution and 2.0 mL of NiCl2 (0.1 M) solution was quickly injected into 30 mL of an aqueous solution of 0.1 M NaBH4 with stirring. More detailed experimental steps are presented in the ESI.† Bimetallic Ni–Co2 (the numerical subscripts denote the molar ratio of the metal precursors) nanosponges are formed within 5 min (Fig. S1, ESI†). Metal nanoparticles are formed instead of porous nanostructures when the concentration of precursors and reducing agent is low.12 After supercritical drying, oxide (Ni–Co2–O) HNSs are obtained via annealing at 600 °C.
Scanning electron microscopy (SEM) was first employed to investigate the morphology and structure of the as-prepared nickel cobalt oxide HNSs. Fig. 1A and B reveal that Ni–Co2–O HNSs are composed of porous and interconnected networks. Furthermore, this sponge-like feature was confirmed by transmission electron microscopy (TEM) imaging. As can be seen from Fig. 1C and D, both bimetallic Ni–Co2 (Fig. S2, ESI†) and their derived oxide nanosponges are not nanoparticle-based aggregates but are fused architectures, similar to metal aerogels reported previously.13 The obtained Ni–Co2–O HNSs consist of irregular building blocks with an enlarged diameter in comparison with the pristine Ni–Co2 nanosponges. The sponge-like features can be well inherited after annealing treatment. A close inspection of the resultant Ni–Co2–O shows the presence of hollow structures in the product, which is ascribed to the Kirkendall effect, a vacancy flux and subsequent void formation resulting from diffusivity differences at inorganic interfaces.14 The highly porous structure and the large void space within the Ni–Co2–O HNSs result in an ultralow density of around 0.08 g cm−3. High-resolution TEM (HRTEM) was carried out to illustrate the detailed features. As shown in Fig. 1E, no obvious interstitial space was observed among these fused nanostructures, which is different from the aggregated type originating from well-dispersed nanoparticles.15 The lattice fringes can be readily indexed to the (111) crystal plane of the NiCo2O4 phase. In addition, the corresponding selected area electron diffraction (SAED) pattern (see inset in Fig. 1E) indicates the polycrystalline nature of these HNSs, and the diffraction rings can be readily indexed to the (220), (311), (400), and (440) planes of the NiCo2O4 phase. The energy-dispersive X-ray spectroscopy (EDX) (Fig. 1F) results show the presence of Co and Ni within Ni–Co2–O and the sample has an overall Co/Ni molar ratio of ∼2.1, which is in accordance with the feeding molar ratio. The effect of the composition on the morphology of the final product was also studied. As expected, the as-prepared Ni2–Co–O shows the same porous features as well as hollow nanostructures (Fig. S3, ESI†). With regard to the monometallic system, Ni nanosponges and the thereof derived oxide HNSs were also synthesized (Fig. S3, ESI†). In contrast, only Co and the thereof derived Co3O4 nanoparticles (Fig. S4, ESI†) precipitate rather than forming 3D nanosponges using our kinetically controlled reduction method. Moreover, no hollow structures were found within Co3O4 after annealing. This synthesis process is obviously different from that of other Co nanoassemblies.16 Additionally, the Co nuclei formed are not inclined to assemble spontaneously. The detailed mechanism is still to be unravelled. Obviously, the introduction of Ni species plays a significant role in the synthesis of 3D bimetallic Ni–Co and their oxide HNSs.
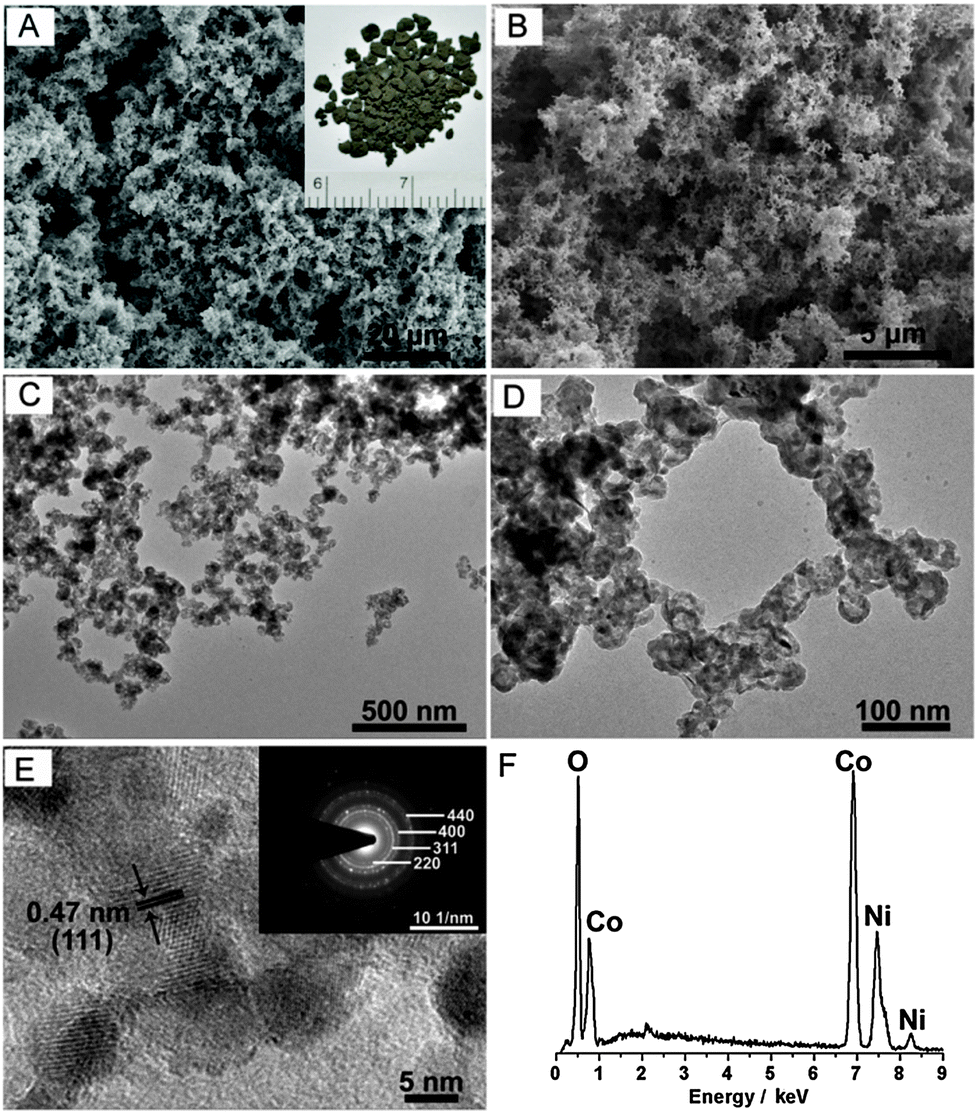 | ||
| Fig. 1 (A, B) SEM, (C, D) TEM, and (E) HRTEM images of the as-obtained Ni–Co2–O HNSs (inset: photograph of the as-prepared Ni–Co2–O HNSs). (E) The inset shows a SAED pattern. (F) EDX pattern of Ni–Co2–O HNSs. | ||
Fig. 2A shows the X-ray diffraction (XRD) patterns of the as-prepared nickel cobalt oxides. Broad reflections were observed in all patterns, which indicate the existence of nanocrystalline phases. The fringes in this figure are assigned to the NiCo2O4 spinel (JCPDF 73-1702) and NiO (JCPDF 73-1519) structures. For Ni–Co2–O, the formation of NiCo2O4 is predominant, which is well consistent with the SAED analysis mentioned above. Besides, a part of NiO can also be found within the product. The lattice constant value (0.8089 nm) determined from the crystal plane (311) of the Ni–Co2–O is considerably smaller than 0.8112 nm in JCPDS for NiCo2O4, which can be explained by the appearance of NiO and the partial decomposition of the spinel structure. Note that when the feeding molar ratio between Ni2+ and Co2+ increases to 2, the intensity of the diffraction peaks of NiO is enhanced, which indicates that the amount of NiO dominates within Ni2–Co–O.17 In the case of NiO HNSs, good crystallinity is obtained after heat treatment of Ni nanosponges. The bimetallic Ni–Co2 is not thermally stable, and it oxidizes upon calcination and transforms into the corresponding metal oxide. As confirmed by thermogravimetric analysis (TGA), the oxidation of Ni–Co2 occurred at about 150 °C and Ni–Co2 was completely transformed into Ni–Co2–O at around 600 °C (Fig. S5A, ESI†). With the increase in the annealing temperature, the product turned grey black gradually with the corresponding XRD patterns shown in Fig. S5B (ESI†). An amorphous structure is observed when the samples were annealed at 200 °C and a distinct spinel phase along with the NiO phase was formed at higher temperature (400 °C). With a further increase of the annealing temperature to 600 °C, Ni–Co2 was completely transformed into Ni–Co2–O and the peak intensities gradually increase whereas peak width decreases indicating the enlargement of the crystal domains with the increase of the particle sizes. In addition, XPS spectra show that the surface of the Ni–Co2–O materials has a composition of Co2+, Co3+, Ni2+ and Ni3+ (Fig. S6, ESI†). The Co/Ni atomic ratio at the surface of the materials is larger than that of the feeding molar ratio, indicating a minor enrichment in Co at the surface of the products (Table S1, ESI†).
 | ||
| Fig. 2 (A) XRD patterns of Ni–Co2–O, Ni2–Co–O and NiO HNSs. (B) Vertically offset (VO) N2 physisorption isotherms for the NiO (VO = 150 cm3 g−1), Ni2–Co–O (VO = 100 cm3 g−1), Ni–Co2–O (VO = 50 cm3 g−1) and Ni–Co2 nanosponges. | ||
Fig. 2B shows typical nitrogen adsorption–desorption isotherms of NiO, Ni2–Co–O, Ni–Co2–O and bimetallic Ni–Co2. It is clear that all the isotherms recorded are similar in shape and essentially of type II curve combined with a type IV contribution, which implies the typical pore characteristics of all samples. The amount of N2 adsorbed increases sharply at high relative pressures and does not reach a plateau, confirming the presence of macropores within these products. In addition, the slight hysteresis loop can be assigned to textural mesopores of fused nanostructures. The specific surface-area values determined using the Brunauer–Emmett–Teller (BET) model were found to be 43.6, 34.4, 24.8, and 32.4 m2 g−1 for NiO, Ni2–Co–O, Ni–Co2–O and bimetallic Ni–Co2, respectively. The introduction of Ni not only plays a key role in constructing these bimetallic Ni–Co nanosponges and their derived oxide HNSs but also affects the BET surface area greatly. In detail, the BET surface area increases with increasing Ni content within these oxides. Compared with the bimetallic Ni–Co2, both specific surface area and pore volume of the Ni–Co2–O HNSs decrease despite the formation of the hollow structures within this oxide. This can be explained by the growth and coalescence of the original nanostructures and the subsequent elimination of a certain porosity at higher temperature. Independent of the applied model, the corresponding pore-size evaluation shows the presence of a wide distribution of pore-sizes, further confirming the co-existence of micropores and mesopores in these materials (Fig. S7, ESI†). Compared with micropores, mesopores and macropores dominate the porosity of this class of nanomaterials (Table S2, ESI†). This range of pore size mainly originate from the void spaces within fused nanoparticles in the process of the assembly and hollow structures induced by the Kirkendall effect. The high surface area and abundant porous structures indicate that the inner edge and corner area are accessible from the outside, further facilitating mass diffusion/transport and furnishing a high electroactive surface area for catalytic applications.
To gain insight into the effect of Ni on the OER process, the electrochemical properties were first investigated in 0.1 M KOH, and the corresponding cyclic voltammograms (CVs) are given in Fig. 3A. For the pure Co3O4 and NiO electrodes, the CVs exhibit two pronounced peaks in the anodic process at 1.183 and 1.406 V, which mainly result from redox reactions related to Co2+/Co3+ and Ni2+/Ni3+, respectively.3a The CV of Ni–Co2–O shows only one broad anodic peak at around 1.249 V and a cathodic peak at around 1.145 V. Due to the very close exchange potentials of Co3+/Co4+ and Ni2+/Ni3+, this broad anodic peak can be correlated to the M2+/M3+ and M3+/M4+ (M = Co and Ni).18 The anodic peak of Ni2–Co–O shifts positively in comparison with Ni–Co2–O. We note that the redox peak shifts negatively with the increase in the Co/Ni molar ratio due to the synergetic effect from the Co addition, which was reported to increase the activity and reversibility of Ni2+/Ni3+.18b Clearly, Ni–Co2–O possesses the highest current density amongst these samples, indicating the largest active surface area and the highest amount of stored charge. Among them, the high charge densities corresponding to the transitions of M3+/M4+ (M = Co and Ni) are particularly favourable for enhancing the OER performance.19 As shown in Fig. 3B, the current density increases with an increase in the scan rate for Ni–Co2–O HNSs. The inset shows the dependence of the current density on the square root of the scan rate, demonstrating that the diffusion of OH− is the rate controlling step in the whole process.
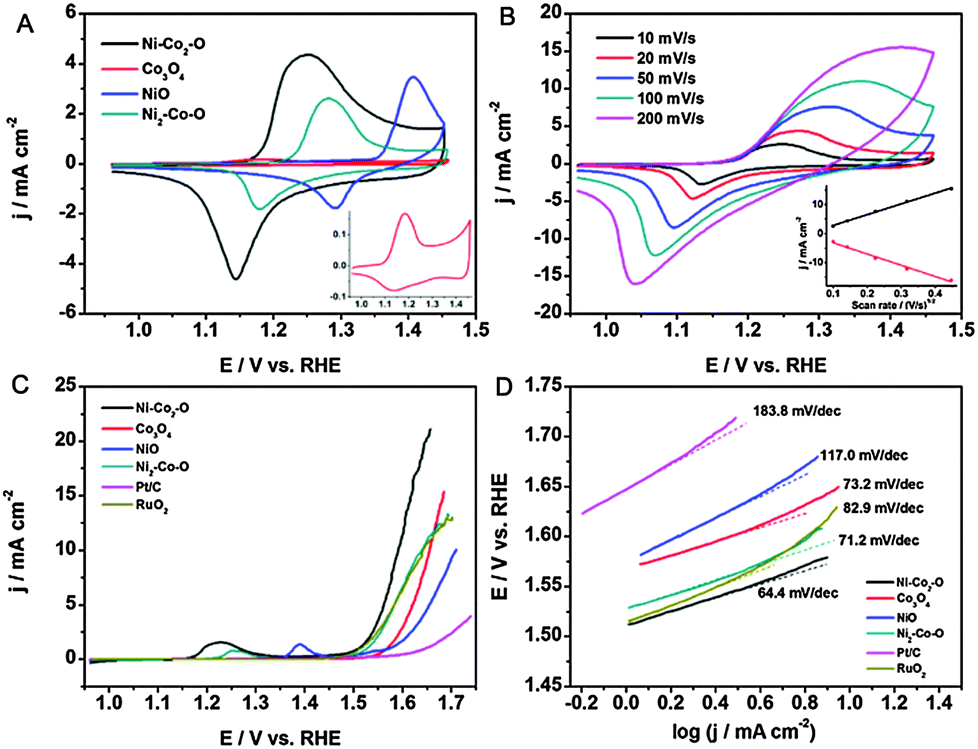 | ||
| Fig. 3 (A) CVs of pure NiO, Ni2–Co–O, Ni–Co2–O hollow nanosponges, and Co3O4 nanoparticles at a scan rate of 20 mV s−1 (inset: magnifies the CVs of pure Co3O4 nanoparticles). (B) CVs of Ni–Co2–O hollow nanosponges at various scan rates ranging from 10 to 200 mV s−1 (inset: the plots of anodic and cathodic peak current density against the square root of the scan rate). (C) LSV curves for OER on modified GCE comprising Ni–Co2–O NiO, Ni2–Co–O hollow nanosponges, Co3O4 nanoparticles, and commercial Pt/C in 0.1 M O2-saturated KOH, respectively. Catalyst loading: ∼0.2 mg cm−2. Sweep rate: 5 mV s−1. (D) Tafel plot derived from (C). | ||
Subsequently, the OER catalytic performance of the Ni–Co2–O HNSs and their corresponding reference materials was investigated by linear sweep voltammetry (LSV) in 0.1 M O2-saturated KOH solution (Fig. 3C). Compared with the other materials, Ni–Co2–O exhibits the highest catalytic activity towards the OER with the onset potential being as low as 1.501 V (see Table S3, ESI†). The current density of 10 mA cm−2, which is a metric relevant to solar fuel synthesis, can be achieved at a small overpotential (η) of 0.362 V with our constructed catalyst, being advantageous over e.g. the Mn3O4–CoSe2 nanocomposites8c and the nanoporous carbon–cobalt-oxide hybrids20 and comparable to the performance of Co3O4/graphene catalysts21 at a similar loading (Table S4, ESI†). In addition, the Ni–Co2–O electrode shows the smallest Tafel slope value (64.4 mV dec−1) amongst all the samples investigated, corresponding to the most favourable OER kinetics (Fig. 3D). However, after 500 cycles, the anodic current decreased to some extent, largely because the physically attached catalyst on the electrode lost contact with the electrode over repeated catalytic cycles due to the production of oxygen (Fig. S8, ESI†). The remarkable enhancement in the OER performance of the porous Ni–Co2–O can be attributed to its high specific surface area and profuse porosity, which provide effective diffusion channels for the electrolyte ions leading to an improved electrochemical performance. More importantly, the favorable composition factor plays a dominating role in enhancing the electrocatalytic activity towards the OER. Of the three different oxides, Ni–Co2–O HNSs abound in the spinel structure, which provides more active sites, enhance the conductivity, accelerate the adsorption of H2O, and efficiently detach gas bubbles and are thus beneficial for surface electrocatalytic conversions.
In summary, we have demonstrated a kinetically controlled strategy for the synthesis of a class of novel 3D nickel cobalt oxide HNSs. Significantly, taking advantage of the remarkable features of the novel materials, such as their porous and hollow nanostructures, and synergetic effects between their components, the optimized Ni–Co2–O HNSs with spinel structure exhibit excellent catalytic activity, which makes them ideal candidates for the next generation OER catalysts. This class of nickel cobalt oxides can offer very attractive prospects and could be extended to electrochemical applications in various fields, such as supercapacitors, fuel cells, batteries and electrochemical sensors.
This work was supported by the Alexander von Humboldt Foundation and by the Cluster of Excellence cfAED. Work on metal aerogel based electrocatalysts is also funded by the EU through the ERC Advanced Grant AEROCAT. We thank Susanne Goldberg for the SEM measurements and Christine Damm (IFW Dresden e.V.) for assistance in performing TEM imaging.
Notes and references
- (a) T. Wang, J. Zhuang, J. Lynch, O. Chen, Z. Wang, X. Wang, D. LaMontagne, H. Wu, Z. Wang and Y. C. Cao, Science, 2012, 338, 358–363 CrossRef CAS PubMed; (b) H.-W. Liang, Q.-F. Guan, L.-F. Chen, Z. Zhu, W.-J. Zhang and S.-H. Yu, Angew. Chem., Int. Ed., 2012, 51, 5101–5105 CrossRef CAS PubMed.
- (a) B. C. Tappan, S. A. Steiner and E. P. Luther, Angew. Chem., Int. Ed., 2010, 49, 4544–4565 CrossRef CAS PubMed; (b) L. Lu and A. Eychmüller, Acc. Chem. Res., 2008, 41, 244–253 CrossRef CAS PubMed.
- (a) X.-C. Dong, H. Xu, X.-W. Wang, Y.-X. Huang, M. B. Chan-Park, H. Zhang, L.-H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed; (b) T.-Y. Wei, C.-H. Chen, H.-C. Chien, S.-Y. Lu and C.-C. Hu, Adv. Mater., 2010, 22, 347–351 CrossRef CAS PubMed.
- (a) G. Zhang and X. W. Lou, Adv. Mater., 2013, 25, 976–979 CrossRef CAS PubMed; (b) G. Zhang, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2013, 26, 2408–2412 CrossRef PubMed; (c) X. Zou, J. Su, R. Silva, A. Goswami, B. R. Sathe and T. Asefa, Chem. Commun., 2013, 49, 7522–7524 RSC.
- H. B. Wu, H. Pang and X. W. Lou, Energy Environ. Sci., 2013, 6, 3619–3626 CAS.
- (a) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed; (b) D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- (a) V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Angew. Chem., Int. Ed., 2010, 49, 4813–4815 CrossRef CAS PubMed; (b) B. Johnson, F. Girgsdies, G. Weinberg, D. Rosenthal, A. Knop-Gericke, R. Schlögl, T. Reier and P. Strasser, J. Phys. Chem. C, 2013, 117, 25443–25450 CrossRef CAS.
- (a) A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed; (b) M.-R. Gao, Y.-F. Xu, J. Jiang, Y.-R. Zheng and S.-H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed; (c) Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- (a) Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926–1929 CrossRef CAS PubMed; (b) S. Chen and S.-Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed; (c) B. Cui, H. Lin, J.-B. Li, X. Li, J. Yang and J. Tao, Adv. Funct. Mater., 2008, 18, 1440–1447 CrossRef CAS.
- (a) Q. Lu, Y. Chen, W. Li, J. G. Chen, J. Q. Xiao and F. Jiao, J. Mater. Chem. A, 2013, 1, 2331–2336 RSC; (b) J. Xiao and S. Yang, RSC Adv., 2011, 1, 588–595 RSC.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- C. Zhu, S. Guo and S. Dong, Chem. – Eur. J., 2013, 19, 1104–1111 CrossRef CAS PubMed.
- (a) N. C. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed; (b) A.-K. Herrmann, P. Formanek, L. Borchardt, M. Klose, L. Giebeler, J. Eckert, S. Kaskel, N. Gaponik and A. Eychmüller, Chem. Mater., 2013, 26, 1074–1083 CrossRef.
- (a) J. G. Railsback, A. C. Johnston-Peck, J. Wang and J. B. Tracy, ACS Nano, 2010, 4, 1913–1920 CrossRef CAS PubMed; (b) W. Ni, H. B. Wu, B. Wang, R. Xu and X. W. Lou, Small, 2012, 8, 3432–3437 CrossRef CAS PubMed.
- J. Li, H.-E. Fu, L.-J. Wu, A.-X. Zheng, G.-N. Chen and H.-H. Yang, Anal. Chem., 2012, 84, 5309–5315 CrossRef CAS PubMed.
- (a) Y. Zhai, J. Zhai and S. Dong, Chem. Commun., 2010, 46, 1500–1502 RSC; (b) M.-J. Hu, Y. Lu, S. Zhang, S.-R. Guo, B. Lin, M. Zhang and S.-H. Yu, J. Am. Chem. Soc., 2008, 130, 11606–11607 CrossRef CAS PubMed.
- H. Wang, Q. Gao and L. Jiang, Small, 2011, 7, 2454–2459 CAS.
- (a) B. Lu, D. Cao, P. Wang, G. Wang and Y. Gao, Int. J. Hydrogen Energy, 2011, 36, 72–78 CrossRef CAS PubMed; (b) G. Wu, N. Li, D.-R. Zhou, K. Mitsuo and B.-Q. Xu, J. Solid State Chem., 2004, 177, 3682–3692 CrossRef CAS PubMed.
- (a) B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed; (b) P. Rasiyah, A. Tseung and D. Hibbert, J. Electrochem. Soc., 1982, 129, 1724–1727 CrossRef CAS PubMed.
- W. Chaikittisilp, N. L. Torad, C. Li, M. Imura, N. Suzuki, S. Ishihara, K. Ariga and Y. Yamauchi, Chem. – Eur. J., 2014, 20, 4217–4221 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional characterization results. See DOI: 10.1039/c5cc01558h |
| This journal is © The Royal Society of Chemistry 2015 |