
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis(triphenylphosphine)silver(I) perrhenate, a cyclic dimer†
F.
Deiser
a,
F.
Kraus
*ab and
H.
Schmidbaur
*a
aDepartment Chemie, Technische Universität München, Lichtenbergstrasse 4, 85747 Garching b. M., Germany. E-mail: h.schmidbaur@lrz.tum.de; florian.kraus@chemie.uni-marburg.de
bFachbereich Chemie, Philipps Universität Marburg, Hans-Meerwein-Strasse 4, 35032 Marburg, Germany
First published on 10th March 2015
Abstract
The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and 1:2 complexes of silver perrhenate and triphenylphosphine, [(Ph3P)4Ag]+ ReO4− and [(Ph3P)2AgReO4]2, have been prepared and their structures determined in the solid state by X-ray diffraction. The former is composed of independent ions, while in the latter the ions are aggregated into cyclic dimers. The silver centers are tetracoordinated including contact with two bridging perrhenate anions, setting this structure apart from that of its gold analogue [(Ph3P)2Au]+ ReO4− where the gold centers are strictly two-coordinate.
:4 and 1:2 complexes of silver perrhenate and triphenylphosphine, [(Ph3P)4Ag]+ ReO4− and [(Ph3P)2AgReO4]2, have been prepared and their structures determined in the solid state by X-ray diffraction. The former is composed of independent ions, while in the latter the ions are aggregated into cyclic dimers. The silver centers are tetracoordinated including contact with two bridging perrhenate anions, setting this structure apart from that of its gold analogue [(Ph3P)2Au]+ ReO4− where the gold centers are strictly two-coordinate.
The perrhenate anion ReO4− is a common component of many salt-like compounds1–4 where it is associated with a large variety of monoatomic or more complex onium cations M+[ReO4]−.5–16 Owing to the very similar structure and charge characteristics of the anions, there are many analogies not only with the congeners, permanganate MnO4− or pertechnetate TcO4−, but also with perchlorate ClO4−, perbromate BrO4− and periodate IO4−. To mention an example, the cesium salts Cs[ReO4] and Cs[BrO4] are isotypical.17 However, in an earlier report it has been noticed that the structural analogy does not lead to a set of comparable physical properties, and this is particularly true for silver(I) salts: a striking difference has been noticed between the very high solubility of AgClO4 in both water and benzene on the one hand, and the very low solubility of AgReO4 in the same pair of solvents on the other.18 This observation suggests a much stronger donor–acceptor interaction of the ReO4− anion with the silver(I) centers, which is not overcome upon solvation with water or the formation of arene complexes, respectively.19
However, compounds in which the perrhenate anion is coordinatively attached to acceptor atoms, or forms covalent bonds with a substituent, are generally rare and in most cases of limited stability.1,2 This is particularly true for the highly unstable esters of perrhenic acid ROReO3, showing an analogy with the explosive esters of permanganic, pertechnetic and perchloric acids.18 An exceptional stability was found only for the silyl esters R3SiOReO3 which can be readily prepared and are stable at ambient temperature.20 The crystal structure of Me3SiOReO3 has been determined and has been shown to have a single discrete Si–O–Re linkage.21 Detailed 185,187Re NQR measurements have confirmed that in this ester the ReO4 tetrahedron is indeed strongly distorted.22 Similar overall characteristics were found for organogermanium, -tin and -indium perrhenates.23–27
The combination of perrhenate with the heavy coinage metals, Ag and Au, is poorly presented in the literature. Contrary to silver(I) perrhenate AgReO4,5,28 gold(I) perrhenate AuReO4 is still an unknown compound. In a report published in 2013 on complexes with triphenylphosphine as the ligand it has been shown that in the 1:2 complex, [(Ph3P)2Au]+ ReO4−, the gold center has no acceptor affinity for the anion and remains strictly linearly two-coordinate.29 This result has recently been confirmed for a series of other gold(I) complexes with tertiary phosphines, where again the ReO4− counterion is not part of the coordination sphere of the metal atoms.30 In subsequent investigations attempts have been made to synthesize the corresponding silver(I) complexes, where perrhenate complexation and hence activation may arise because relativistic effects, which strongly favour two-coordination at gold(I), are strongly reduced at silver(I) centres.31,32
Upon the reaction of a mixture of AgReO4 with PPh3 in the molar ratio of 1:2 in dry acetonitrile at reflux temperature under nitrogen and with protection against incandescent light a crystalline product could be isolated in 92% yield. The pale rose colour of the primary product vanished upon careful crystal growth in acetonitrile. The expected 1:2 composition of the product was confirmed by an elemental analysis (ESI†). Solutions in acetonitrile-d3 showed a sharp single resonance in the 31P NMR spectrum with δ 11.25 ppm at 25 °C. No couplings 1J(107/109Ag–31P) were observed even upon lowering the temperature to the solubility limit, probably owing to the rapid ligand exchange, very common for silver(I) complexes of tertiary phosphines.33,34 The spectrum also suggests that the eight-membered ring found in the crystal structure is flexible or subject to partial dissociation in solution, which is rapid on the NMR time scale.
The IR spectrum of the solid (on ATR crystals) shows that the prominent ν3(Re–O) vibrational mode, which appears as a single band for the undistorted tetrahedral anionic ReO4 unit near 910 cm−1 (see below), is split into two bands at 896 and 925 cm−1 suggesting a significant distortion. In a single crystal X-ray diffraction analysis it has been shown that the perrhenate anions are indeed strongly coordinated to the silver atoms as shown in Fig. 1 (triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , for further crystallographic details see Table S1 in the ESI†).
, for further crystallographic details see Table S1 in the ESI†).
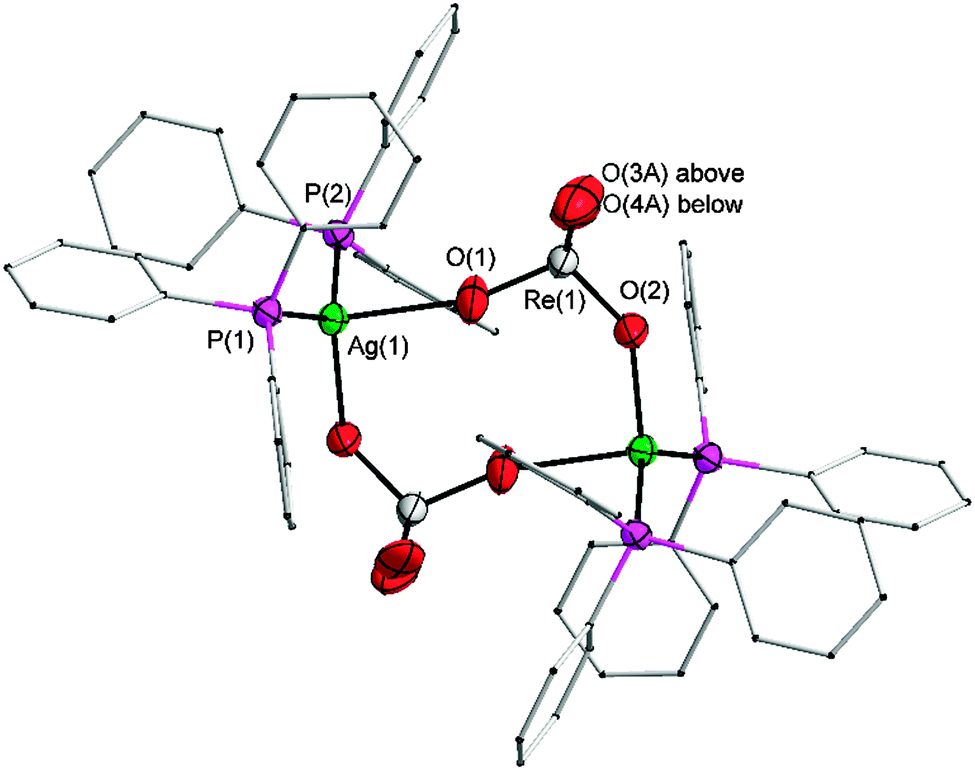 | ||
| Fig. 1 Molecular structure of [(Ph3P)2AgReO4]2. Thermal displacement parameters are shown at the 70% level at 123 K. H atoms are omitted and the C atoms are shown as a wire-frame for the sake of clarity. Only one orientation of the disordered O3 and O4 atoms is shown. | ||
Two formula units are associated into a cyclic dimer in which the ReO4− anions form triatomic bridges O–Re–O between the silver cations. The silver atoms are thus tetracoordinated with Ag–P and Ag–O bond lengths at 2.3997(8)/2.4227(7) and 2.366(2)/2.511(2) Å, respectively. The P–Ag–P angle of 133.38(3)° shows a strong deviation from the linear geometry observed in the gold analogue.29 The point group symmetry of the cyclic dimer is Ci, as it resides around a crystallographic inversion center. The Re–O distances for the Ag–O–Re bridges are observed for Re–O(1) 1.718(2) and Re–O(2) 1.705(2) Å. Due to the disorder of the terminal O atoms, a direct comparison of the distances to the terminal oxygen atoms (Re–O(3A/B) 1.60(2)/1.81(2) and Re–O(4A/B) 1.78(1)/1.65(2) Å) is not meaningful, but as expected the former Re–O distances are similar to, and the latter shorter than the reference distance in an undistorted ReO4− tetrahedron which is present in the gold analogue [1.710(7)–1.716(7) Å].29 However, it is interesting to note that in crystals of AgReO4 (Scheelite-type), where all four oxygen atoms of the anion are in close contact with the silver cations,5 the Re–O distances are as long as 1.732(2) Å, showing the strong influence of silver multiple-coordination on the structure of the anion. The strong donor properties of the perrhenate anion for silver(I) are also evident from a comparison with the corresponding tetrafluoroborate [(Ph3P)2Ag]+ BF4−. When this compound is prepared in acetonitrile, its 1:1 solvate [(Ph3P)2Ag(NCMe)]+ BF4− with a tricoordinate silver atom is formed.34 By contrast, perrhenate competes favourably with the solvent molecules and only perrhenate complexation is obtained.
Experiments with a molar ratio of 1:4 of the reactants in a mixture of dry acetonitrile and benzene [2:5 v/v] at 25 °C under nitrogen and protection against light led to the formation of the complex [(Ph3P)4Ag]+ ReO4− in 89% yield. A compound of this composition has been prepared in a previous report, but no structural data are available.18 Crystals grown from the reaction mixture were colourless and stable to air, moisture and light. Their composition was confirmed by elemental analysis (ESI). Solutions in acetonitrile-d3 at 25 °C showed a singlet resonance at δ 10.8 ppm in the 31P NMR spectrum, again without 107/109Ag–31P couplings. The crystals (trigonal, space group R3) are related to those of known compounds containing the [(Ph3P)4Ag]+ cation associated with other tetrahedral anions like ClO4− or BF4− (space groups R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , heavily disordered tetrahedral anions due to site symmetry).35,36 It is possible that for these cases the space group type R3 would be a better choice as well. For crystallographic details, information on the choice of the space group, and crystal structure solution and refinement, see the ESI.†
, heavily disordered tetrahedral anions due to site symmetry).35,36 It is possible that for these cases the space group type R3 would be a better choice as well. For crystallographic details, information on the choice of the space group, and crystal structure solution and refinement, see the ESI.†
The bond lengths and angles of the cations are within the range of data collected in earlier studies. The tetrahedron of the anion is largely undistorted as also seen in the IR spectrum of the crystals, where only a single sharp band is observed for the ν3(ReO) mode at 906.8 cm−1. Attempts to prepare crystals of compounds with the 1:1 and 1:3 stoichiometry were unsuccessful.
In summary, in the present work structural details of silver-perrhenate coordination have been provided for a molecular system. The results help us to explain the unusual silver-perrhenate affinity suggested by a number of earlier qualitative observations. This affinity causes distinct differences in the structural chemistry of silver(I) and gold(I) compounds. The radii of Ag+ and Au+ are known to be virtually the same for both the two- and four-coordinate states owing to strong relativistic contraction of the frontier orbitals of the latter.37,38 The differences in the Ag/Au ← O–ReO3 acceptor strengths are the result of the distinct preference for two-coordination with high s-character in the two dative bonds at gold(I), which is less pronounced for silver(I). Along the same lines, the higher donor strengths of the perrhenate anion as compared to e.g. the perchlorate anion (the former is coordinated to Ag, while the latter is not) are related to strong relativistic effects on rhenium which strongly modify the donor properties of the ReO4− anion through electronegativity enhancement and orbital rehybridization affecting the nature of the Re–O bonds.39,40
Notes and references
- G. Bandoli, A. Domella, M. Porchias, F. Refosco and F. Tisato, Coord. Chem. Rev., 2001, 214, 43 CrossRef CAS.
- M. C. Chakravorti, Coord. Chem. Rev., 1990, 106, 205 CrossRef CAS.
- C. Rouschias, Chem. Rev., 1974, 74, 531 CrossRef.
- R. D. Peacock, The Chemistry of Technetium and Rhenium, Elsevier, Amsterdam, 1966 Search PubMed.
- D. Yu. Naumov, A. V. Virovets, S. V. Korenev and A. I. Gubanov, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, IUC99000097 Search PubMed.
- A. Atzesdorfer and K.-J. Range, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 1417 CAS.
- P. Rögner and K.-J. Range, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 233 Search PubMed.
- P. Rögner and K.-J. Range, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 685 Search PubMed.
- R. J. C. Brown, B. M. Powell and S. N. Stuart, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 214 CrossRef.
- K.-J. Range, P. Rögner, A. M. Heyns and L. C. Prinsloo, Z. Naturforsch., B: J. Chem. Sci., 1992, 47, 1513 CAS.
- T. Betz and R. Hoppe, Z. Anorg. Allg. Chem., 1983, 500, 23 CrossRef CAS.
- G. J. Kruger and E. C. Reynhardt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 259 CrossRef.
- B. Krebs and K.-D. Hasse, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1334 CrossRef.
- C. J. L. Locke and G. Turner, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 1764 CrossRef.
- H. Beyer, A. Müller and B. Krebs, Z. Phys. Chem., 1967, 234, 432 Search PubMed.
- J. Beintema, Z. Kristallogr., 1937, 97, 300 CAS.
- P. Rögner, U. Schießl and K.-J. Range, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 235 Search PubMed.
- A. A. Woolf, J. Less-Common Met., 1978, 61, 151 CrossRef CAS.
- H. Schmidbaur, Angew. Chem., 1985, 97, 893 CrossRef CAS and ref. therein.
- M. Schmidt and H. Schmidbaur, Chem. Ber., 1959, 92, 2667 CrossRef CAS.
- G. M. Sheldrick and W. S. Sheldrick, J. Chem. Soc. A, 1969, 2160 RSC.
- H. Schmidbaur, D. Koth and P. Burkert, Chem. Ber., 1974, 107, 2697 CrossRef CAS.
- M. Schmidt and I. Ruidisch, Angew. Chem., 1961, 73, 408 Search PubMed.
- H. Schmidbaur and D. Koth, Chem.-Ztg., 1976, 100, 290 CAS.
- W. A. Herrmann, J. G. Kuchler, J. K. Felixberger, E. Herdtweck and W. Wagner, Angew. Chem., Int. Ed. Engl., 1988, 27, 394 CrossRef.
- E. Herdtweck, P. Kiprof, W. A. Herrmann, J. G. Kuchler and I. A. Degnan, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 937 CAS.
- H. Schmidbaur and D. Koth, Naturwissenschaften, 1976, 63, 482 CrossRef CAS.
- D. Yu. Naumov, A. V. Virovets, S. V. Korenev and A. I. Gubanov, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 8 Search PubMed.
- S. A. Baer, A. Pöthig, S. M. Bawaked, H. Schmidbaur and F. Kraus, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 1173 CAS.
- A. B. Browne, N. Deligonul, B. L. Anderson, A. L. Rheingold and T. G. Gray, Chem. – Eur. J., 2014, 20, 17552 CrossRef CAS PubMed.
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151 CrossRef CAS PubMed.
- P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef.
- E. L. Muetterties and C. W. Allegranti, J. Am. Chem. Soc., 1972, 94, 6386 CrossRef CAS.
- R. E. Bachman and D. F. Andretta, Inorg. Chem., 1998, 37, 5657 CrossRef CAS PubMed.
- L. M. Engelhardt, C. Pakawatchal, A. H. White and P. C. Healy, J. Chem. Soc., Dalton Trans., 1985, 125 RSC.
- X. Huang, Q.-M. Qiu, X. Wang, Q.-H. Jin and C.-L. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m706 CAS.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006 CrossRef CAS.
- U. M. Tripathi, A. Bauer and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1997, 2865 RSC.
- C. J. Massena, A. M. S. Riel, G. F. Neuhaus, D. A. Decato and O. B. Berryman, Chem. Commun., 2015, 51, 1417 RSC.
- E. A. Katayev, N. V. Boev, V. N. Krustalev, Y. A. Ustynyuk, I. G. Tananaev and J. L. Sessler, J. Org. Chem., 2007, 72, 2886 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of the described compounds, ATR-IR spectra, NMR and elemental analysis data, selected crystallographic data, details on structure solution and refinement, and additional figures. CCDC 1052824 and 1052825. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc01446h |
| This journal is © The Royal Society of Chemistry 2015 |