
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh/Cu-catalyzed multiple C–H, C–C, and C–N bond cleavage: facile synthesis of pyrido[2,1-a]indoles from 1-(pyridin-2-yl)-1H-indoles and γ-substituted propargyl alcohols†
Ting
Li
,
Zhen
Wang
 ,
Mingliang
Zhang
,
Hui-Jun
Zhang
and
Ting-Bin
Wen
*
,
Mingliang
Zhang
,
Hui-Jun
Zhang
and
Ting-Bin
Wen
*
Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, Fujian, P. R. China. E-mail: chwtb@xmu.edu.cn; Tel: +86 592-218-6085
First published on 10th March 2015
Abstract
An unusual Rh/Cu-catalyzed synthesis of pyrido[2,1-a]indoles starting from 1-(pyridin-2-yl)-1H-indoles and γ-substituted propargyl alcohols was presented. The multi-step cascade transformations formally involve the cleavage of two C–H, three C–C, and one C–N bonds with concomitant construction of two C–H, four C–C, and one C–N bonds with excellent chemoselectivity in one-pot reaction.
The selective cleavage and construction of chemical bonds has always been a central topic in modern synthetic organic chemistry.1 In recent decades, transition metal-catalyzed direct cleavage of inert chemical bonds such as C–H, C–C and C–N bonds, and thus associated transformations have garnered increasing attention by the synthetic community. In particular, significant developments have been acquired in the area of C–H activation.2 However, reports of C–C activation are less prominent in the literature probably owing to the thermodynamic stability of these bonds and the kinetic factors arising from the greater steric hindrance as well as the competing C–H activation.3,4 Therefore, examples for C–C bond activation are still mostly limited to strained C–C bonds, decarboxylation reactions, or performed with the help of strong chelation assistants.3,4 Similarly, due to the high C–N bond dissociation energy, catalytic transformations involving selective cleavage of C–N bonds have been scarce so far.5,6 Not surprisingly, the simultaneous cleavage of both C–C and C–N bonds under one set of conditions would be even more challenging, which is limited to a few reports.7
On the other hand, transition-metal mediated direct C–H alkynylation for the introduction of the alkynyl functionality has received much attention recently.8 Although terminal alkynes,9 alkynylhalides10 as well as benziodoxolone-based hypervalent iodine reagents11 have been used as alkynyl coupling partners to achieve direct C–H alkynylation of intrinsically different arenes, the substrate scope remains limited. It is still necessary to develop highly efficient and general alkynylation methods for broadly defined arenes via a C–H activation pathway. In this context, it is worth noting that tert-propargyl alcohols have been involved in Sonogashira-type reactions as masked terminal alkynes.12 Moreover, Miura discovered that [Rh(OH)(COD)]2 could catalyze regio- and stereo-selective homo-coupling of γ-arylated tert-propargyl alcohols via β-carbon elimination with liberation of ketone, in which an alkynyl rhodium generated in situ was proposed as the key intermediate.13 Furthermore, given the great success and advantages of rhodium catalysis in the construction of C–X bonds (X = C, N, O, etc.) based on C–H activation in recent years,14,15 we were prompted to design the Rh-catalyzed direct C–H alkynylation of arenes utilizing γ-substituted tert-propargyl alcohols as the alkynyl coupling partner. Following this idea, we have thus examined the reactions of 1-(pyridin-2-yl)-1H-indoles with tert-propargyl alcohols under the Rh/Cu catalyst system, which serendipitously led to the formation of pyrido[2,1-a]indoles instead of the expected alkynylation products (Scheme 1). Herein, we communicate this unusual cascade reaction, whereby at least six bonds, including two C–H, three C–C and one C–N bonds, were cleaved and seven bonds (two C–H, four C–C and one C–N bonds) were constructed in one pot. The reaction reported here is unprecedented especially when the efficiency is considered.16
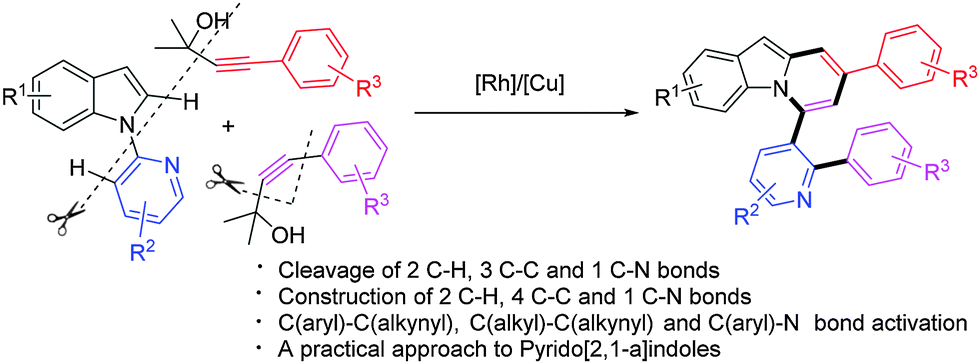 | ||
| Scheme 1 Rh/Cu-catalyzed facile synthesis of pyrido[2,1-a]indoles via multiple C–H, C–C, and C–N bond cleavage. | ||
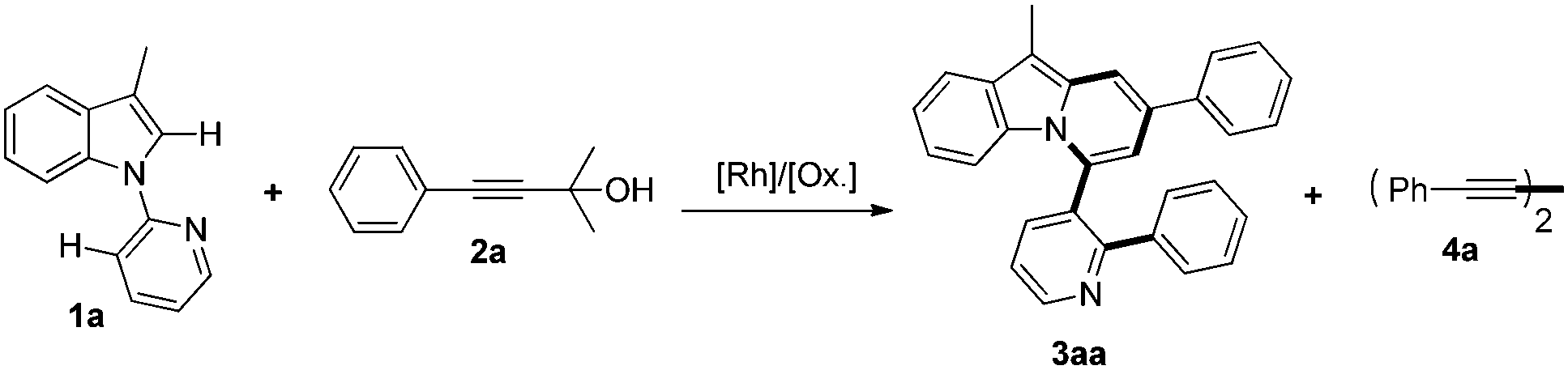
We initiated our study by choosing 1-(pyridin-2-yl)-1H-indole 1a and 2-methyl-4-phenyl-3-butyn-2-ol 2a as model substrates. Surprisingly, when [Cp*RhCl2]2 and Cu(OAc)2·H2O were employed as the catalytic system, the unexpected product 3aa, which contains a pyrido[2,1-a]indole skeleton, was isolated in 24% yield (Table 1, entry 1). The definite structures of 3 came from the X-ray crystallographic studies of a series of pyrido[2,1-a]indole products 3af, 3ag (vide infra, see ESI†), 3la (Table 3), and 3jga (Scheme 3). To the best of our knowledge, efficient strategies for the construction of pyrido[2,1-a]indoles, a recurring structural motif found in many pharmaceuticals and functional materials, have rarely been reported.17,18 After screening of a variety of rhodium precatalysts, [RhCl(COD)]2 was found to be the best choice and 3aa was obtained in 78% isolated yield (entries 2–5). Investigation of solvent effects indicated that toluene was the most suitable solvent (entries 5–8). Control experiments revealed that no desired product 3aa was detected in the absence of either [RhCl(COD)]2 or Cu(OAc)2·H2O (entries 9 and 10). Notably, the reaction was suddenly shut down in the presence of an extra basic additive, K2CO3 (entry 11). The use of other common oxidants such as AgOAc or Ag2CO3 did not provide 3aa at all (entries 12 and 13). 125 °C was found to be the optimal temperature (entries 14 and 15). We have also examined the possibility of employing a simple terminal alkyne instead of 2a for this transformation, which gave 3aa in only 21% yield accompanied by a large amount of Glaser coupling product 4a in 71% yield (entry 16). This result clearly showed that Glaser coupling was inhibited effectively in this reaction by using tert-propargyl alcohol as the alkynylating reagent instead of phenylacetylene.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [M] | Oxidant | Solvent | Temp. (°C) | Yield (%) |
| 3aa | |||||
| a Reaction conditions: 1a (0.20 mmol), 2a (0.80 mmol, 4 equiv.), [M] (1.5 mol%), oxidant (2.2 equiv.) and solvent (2 mL) under air for about 6 h. b Yield of isolated products based on 1a. c Yield of isolated products based on 2a. d 2.0 equiv. of K2CO3 was added. e Phenylacetylene instead of 2a. | |||||
| 1 | [Cp*RhCI2]2 | Cu(OAc)2·H2O | PhMe | 125 | 24/(4a 64c) |
| 2 | [Cp*Rh(CH3CN)3][SbF6]2 | Cu(OAc)2·H2O | PhMe | 125 | 32 |
| 3 | Cp*Rh(OAc)2 | Cu(OAc)2·H2O | PhMe | 125 | 40 |
| 4 | [RhOH(COD)]2 | Cu(OAc)2·H2O | PhMe | 125 | 57/(4a 54) |
| 5 | [RhCI(COD)]2 | Cu(OAc)2·H2O | PhMe | 125 | 78/(4a 41) |
| 6 | [RhCI(COD)]2 | Cu(OAc)2·H2O | Xylene | 125 | 67 |
| 7 | [RhCI(COD)]2 | Cu(OAc)2·H2O | DCE | 125 | 56 |
| 8 | [RhCI(COD)]2 | Cu(OAc)2·H2O | Dioxane | 125 | 20 |
| 9 | [RhCI(COD)]2 | NO | PhMe | 125 | 0 |
| 10 | NO | Cu(OAc)2·H2O | PhMe | 125 | 0/(4a 98) |
| 11 | [RhCI(COD)]2 | Cu(OAc)2·H2O | PhMe | 125 | Traced |
| 12 | [RhCI(COD)]2 | AgOAc | PhMe | 125 | 0 |
| 13 | [RhCI(COD)]2 | Ag2CO3 | PhMe | 125 | 0 |
| 14 | [RhCI(COD)]2 | Cu(OAc)2·H2O | PhMe | 110 | 0/(4a 84) |
| 15 | [RhCI(COD)]2 | Cu(OAc)2·H2O | PhMe | 140 | Trace |
| 16 | [RhCI(COD)]2 | Cu(OAc)2·H2O | PhMe | 125 | 21/(4a 71)e |
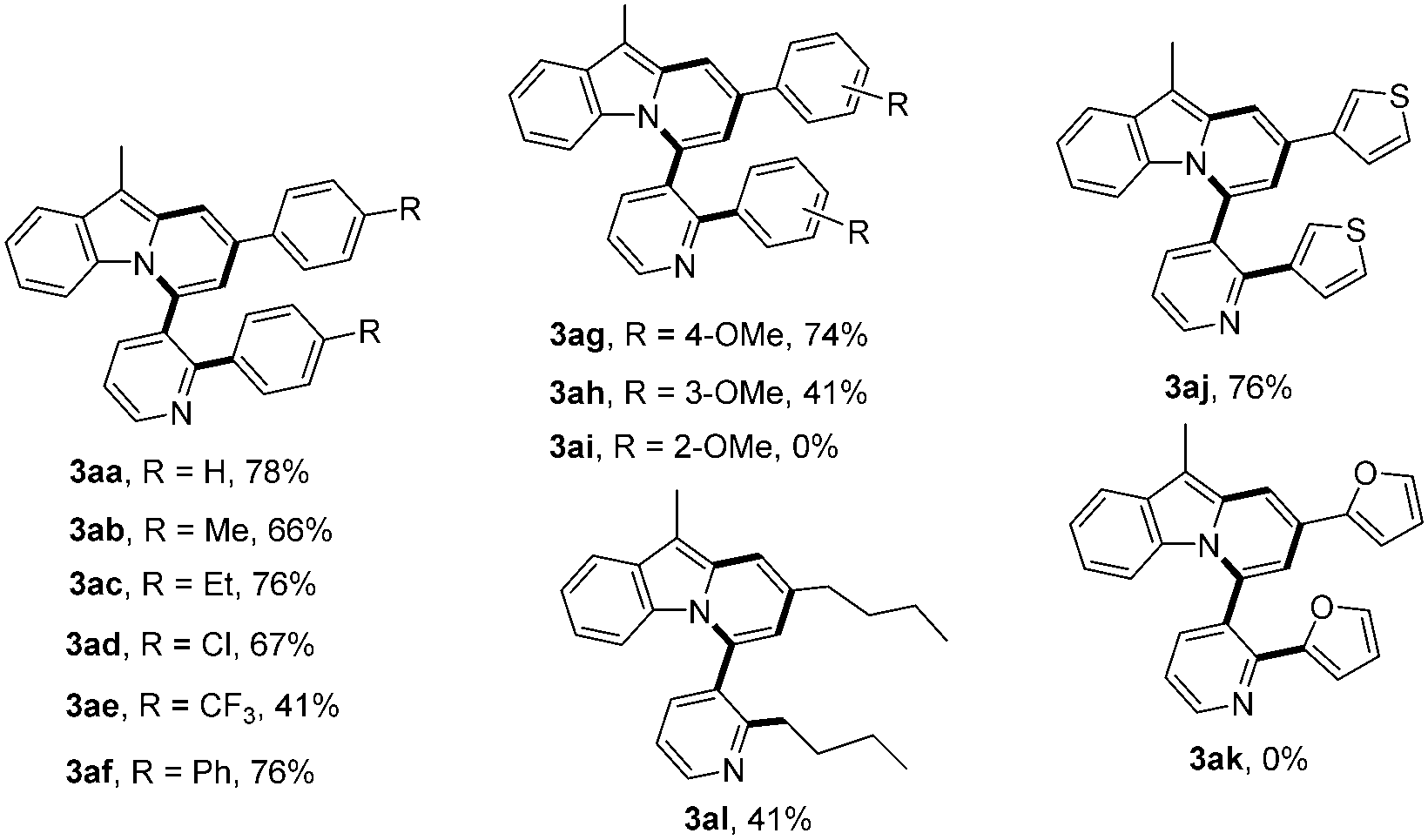
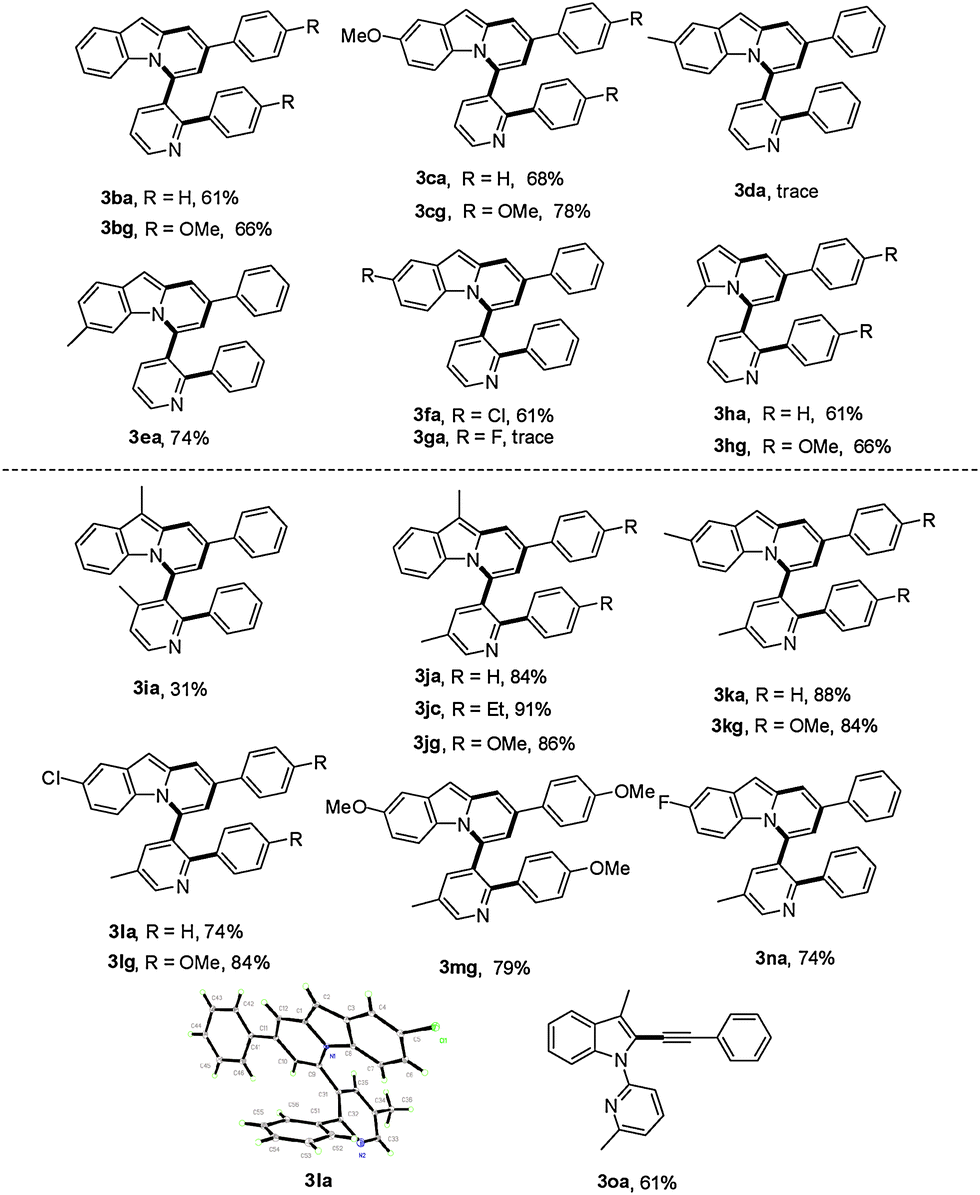
With these optimized reaction conditions in hand, the scope of this reaction was extensively studied. First, a series of γ-substituted tert-propargyl alcohols were investigated (Table 2). To our satisfaction, γ-arylated tert-propargyl alcohols with electron-donating or electron-withdrawing substituents, such as Me, Et, Cl, CF3, and Ph, at the para position on the phenyl ring were well tolerated to provide the corresponding products in moderate to good yields (3aa–3af, 41–78% yield). Substrates bearing methoxy groups at the meta and para positions on the phenyl ring proceeded well (3ag, 3ah), while that with a methoxy group at the ortho position on the phenyl ring failed to yield the desired product 3ai, possibly due to the steric hindrance. It is worth noting that the methodology could be further extended to thiophene containing substrate 2j to afford 3aj in 76% yield, while γ-furan tert-propargyl alcohol 2k did not work at all. Moreover, aliphatic 2-methyloct-3-yn-2-ol 2l was also investigated to produce 3al in 41% yield, which represents the first example of C(alkyl)–C(alkynyl) single bond cleavage.19
|
|
|---|
| a General reaction conditions: 1a (0.2 mmol), 2 (0.8 mmol), [RhCl(COD)]2 (1.5 mol%), Cu(OAc)2·H2O (2.2 equiv.), PhMe (2 mL), 125 °C, air, 6 h. Yield of isolated products based on 1a. |
|
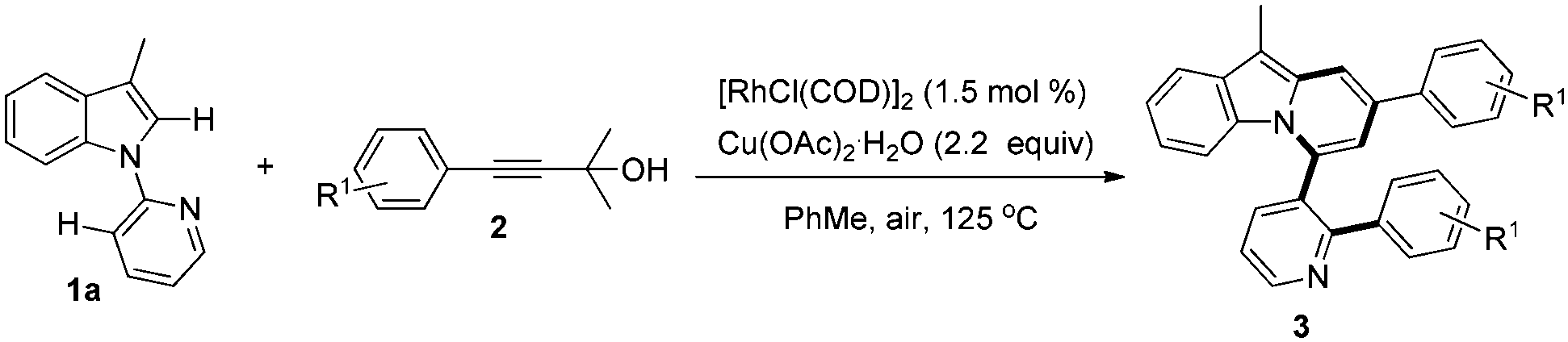
|
|
|---|
| a General reaction conditions: 1 (0.2 mmol), 2 (0.8 mmol), [RhCl(COD)]2 (1.5 mol%), Cu(OAc)2·H2O (2.2 equiv.), PhMe (2 mL), 125 °C, air, 6 h. Yield of isolated products based on 1. |
|
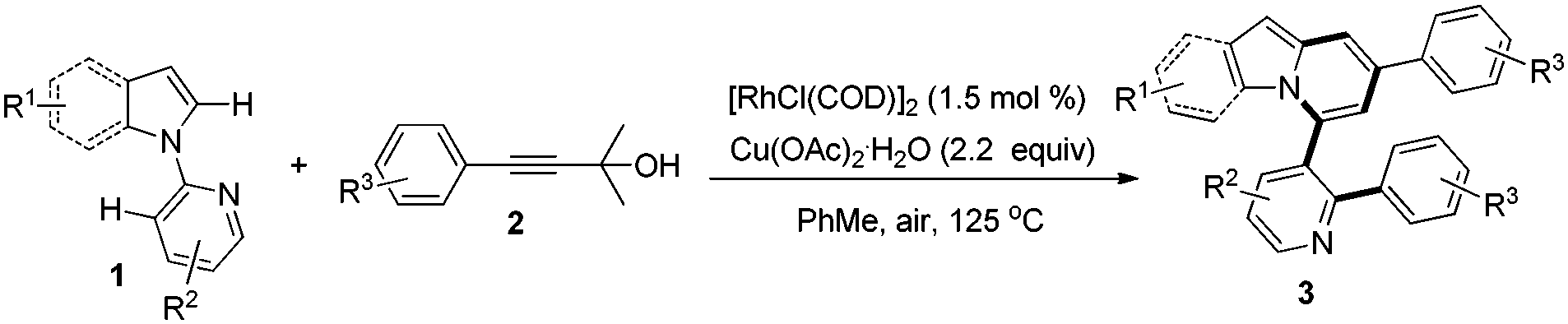
Subsequently, the scope of 1-(pyridin-2-yl)-1H-indoles was further investigated (Table 3). Gratifyingly, reactions of 1b with γ-arylated tert-propargyl achohols worked well to yield 3ba and 3bg in good yields. Other indoles with a substituent group such as OMe or Cl at the C-5 position on the indole ring were also compatible with this transformation, thus affording the corresponding products 3ca, 3cg, and 3fa, while substrates with Me or F at the C-5 position on the indole ring did not work (3da, 3ga). In contrast, when the Me group was at the C-6 position, the reaction proceeded smoothly to give 3ea in 74% yield. More importantly, when 2-(2-methyl-1H-pyrrol-1-yl)pyridine, 1h, was used as the substrate, indolizine derivatives 3ha and 3hg could be obtained in 61% and 66% yields respectively, which represent the rare examples of indolizine synthesis starting from pyrrole.18b,c
We next examined the influence of the substituent at the pyridyl directing groups for the reaction (Table 3). Firstly, when the C-3 position of the pyridyl group, which is subject to the consequent C–C bond formation, was blocked with a Me group, the reaction was completely suppressed to yield only 4a. In addition, installation of the Me substituent on the pyridyl C-4 position furnished product 3ia in only 31% yield. Further studies revealed that the 5-methylpyridin-2-yl directing group gave the best result, providing the target products in particularly high yields (3ja–3na, 74–91%), and even substrates with Me or F at the C-5 position on the indole ring were converted into the corresponding target products 3ka, 3kg, and 3na effectively. It should be noted that changing the directing group from pyridin-2-yl to 6-methylpyridin-2-yl significantly affected the reactivity and only the alkynylation product 3oa was isolated. The steric hindrance of the Me group at the C-6 position may account for this difference. Isolation of the alkynylation product 3oa indicated that the alkynylation step was possibly involved in the catalytic cycle for the formation of pyrido[2,1-a]indoles 3.
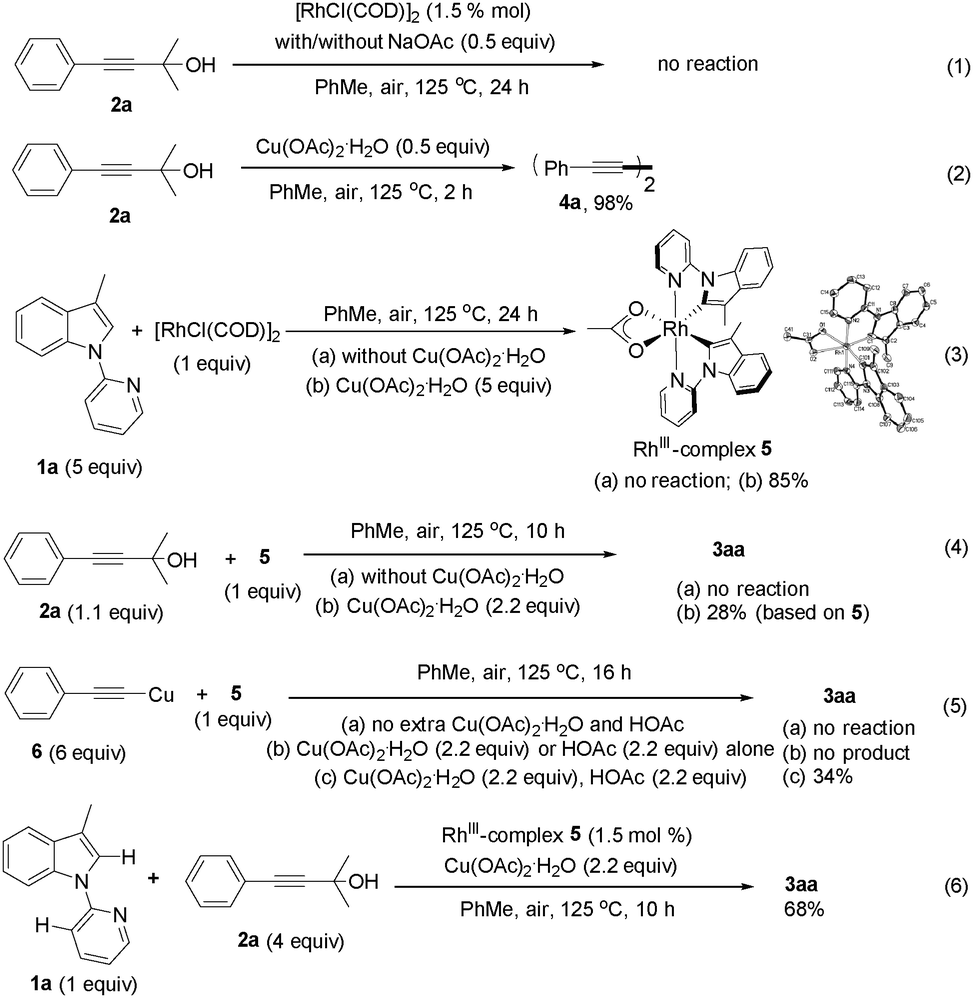
To get some mechanistic insights into this transformation, several experiments were carried out (Scheme 2). First, addition of TEMPO to the reaction mixture had a negligible effect on the reaction, which suggested that a free-radical pathway might be ruled out. Next, stoichiometric reaction of 2a with [RhCl(COD)]2 was performed and no reaction could be observed indeed, even if an excess of NaOAc was added (eqn (1)). Notably, it was found that the Glaser coupling product 4a was generated quantitatively when Cu(OAc)2·H2O was mixed with 2a at 125 °C (eqn (2)). These results indicated that it is Cu(OAc)2·H2O rather than [RhCl(COD)]2 that is responsible for the C–C bond cleavage of 2avia β-carbon elimination.13 Reaction of [RhCl(COD)]2 with 1a was also investigated and no appreciable reaction could be observed when the reaction was performed in the absence of Cu(OAc)2·H2O. However, the biscyclometalated Rh(III) complex 5 was obtained in 85% yield when the reaction was performed in the presence of Cu(OAc)2·H2O (eqn (3)), which clearly indicated that the Csp2–H bond cleavage of substrate 1a was enforced by the RhIII species. The structure of 5 was confirmed by single-crystal X-ray diffraction. Furthermore, the reactivity of complex 5 was tested. Again, no reaction took place when complex 5 was mixed with 2a at 125 °C (eqn (4)). In sharp contrast, the reaction of 5 and 2a in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 in the presence of Cu(OAc)2·H2O did proceed to yield the target product 3aa (eqn (4)). However, further efforts to isolate intermediates from the reaction mixtures by adjusting the ratio of 5 and 2a failed. Reaction of 5 with copper phenylacetylide 6 was also investigated to give product 3aa, albeit in 34% yield. Of note, the addition of extra Cu(OAc)2·H2O as an oxidant and HOAc was essential to promote the reaction. No desired product could be observed in the absence of Cu(OAc)2·H2O or HOAc (eqn (5)). These observations implied that the acetylide copper species generated from the reaction of 2a with Cu(OAc)2·H2O could undergo transmetalation of the alkynyl group to rhodium to afford acetylide rhodium, which then participated in the subsequent transformations. The catalytic activity of complex 5 under standard conditions was also tested to provide 3aa in 68% yield, which indicated that complex 5 could be a possible intermediate in the catalytic cycle (eqn (6)).
:1.1 in the presence of Cu(OAc)2·H2O did proceed to yield the target product 3aa (eqn (4)). However, further efforts to isolate intermediates from the reaction mixtures by adjusting the ratio of 5 and 2a failed. Reaction of 5 with copper phenylacetylide 6 was also investigated to give product 3aa, albeit in 34% yield. Of note, the addition of extra Cu(OAc)2·H2O as an oxidant and HOAc was essential to promote the reaction. No desired product could be observed in the absence of Cu(OAc)2·H2O or HOAc (eqn (5)). These observations implied that the acetylide copper species generated from the reaction of 2a with Cu(OAc)2·H2O could undergo transmetalation of the alkynyl group to rhodium to afford acetylide rhodium, which then participated in the subsequent transformations. The catalytic activity of complex 5 under standard conditions was also tested to provide 3aa in 68% yield, which indicated that complex 5 could be a possible intermediate in the catalytic cycle (eqn (6)).
 | ||
| Scheme 2 Mechanistic studies. | ||
 | ||
| Scheme 3 Cross reactions. | ||
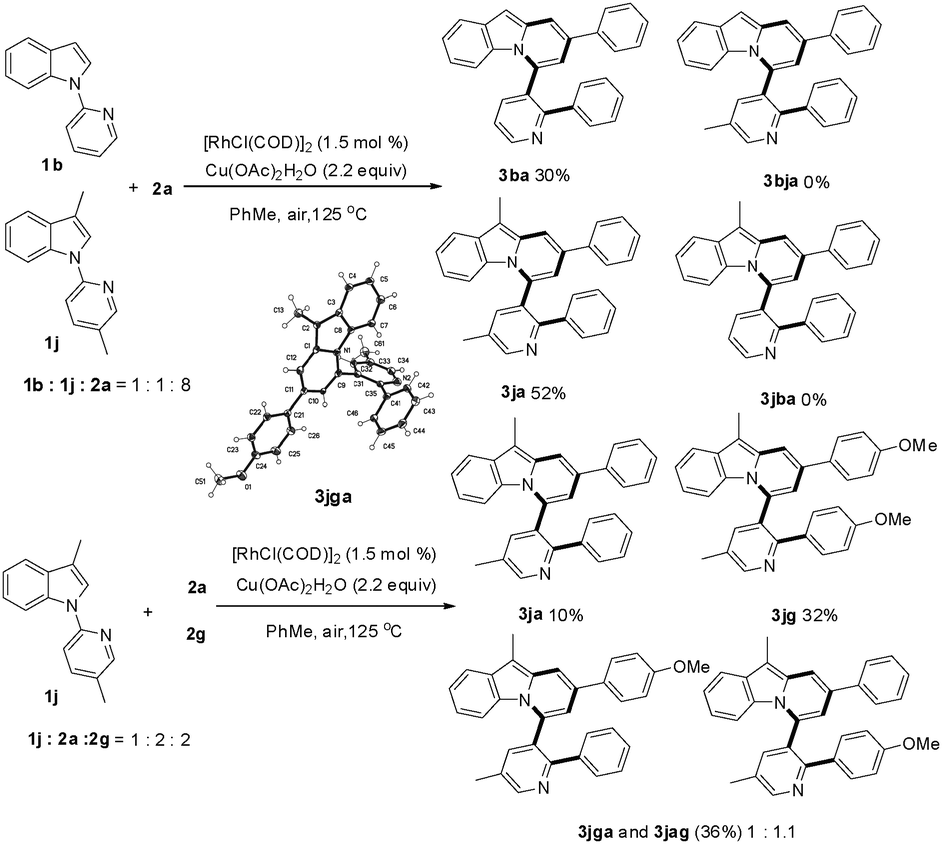
The possibility of cross reactions between two different indoles or propargyl alcohols was also investigated (Scheme 3). The reaction of indole substrates 1b and 1j with 2a afforded only the corresponding 3ba and 3ja, while the crossover products 3bja and 3jba were not detected at all, clearly indicating that the C–N bond cleavage and the concomitant transformations proceeded in an intramolecular manner. In contrast, reaction of 1j with a mixture of 2a and 2g led to the isolation of products 3ja and 3jg as well as the expected crossover products 3jga and 3jag with the latter two as a mixture.
Given the results of the above control experiments, it is reasonable to believe that the reaction is presumably initiated by the cyclometallation of 1-(pyridin-2-yl)-1H-indoles with RhIII species. Transmetalation of the alkynyl group from the acetylide copper species generated in situ to Rh followed by a reductive elimination yields the alkynylation product (see ESI†), which then proceeds to undergo a series of transformations to afford the target product finally. The mechanistic details for the further transformation of the alkynylation product to give the pyrido[2,1-a]indole skeleton are unclear yet.
In conclusion, we have discovered a novel Rh/Cu catalyzed cascade reaction involving multiple C–H, C–C, and C–N bond cleavage and formation, giving rise to a practical approach for the construction of pyrido[2,1-a]indoles. Remarkably, the highly selective cleavage of several types of extremely inert chemical bonds such as C(aryl)–C(alkynyl), C(alkyl)–C(alkynyl), and C(aryl)–N bonds was realized in this transformation. Further studies aimed at revealing the detailed mechanism of this complicated reaction and applications of this protocol are currently being investigated in our laboratory.
We thank the National Basic Research Program of China (973 Program 2012CB821600), the National Natural Science Foundation of China (21072161), and Program for Changjiang Scholars and Innovative Research Team (PCSIRT) in the University.
Notes and references
- M. B. Smith and J. March, March's Advanced Organic Chemisty: Reactions Mechanisms and Structure, Wiley, Hoboken, NJ, 2007 Search PubMed.
- Selected reviews: (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) C. I. Herrerías, X. Yao, Z. Li and C.-J. Li, Chem. Rev., 2007, 107, 2456 CrossRef PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (e) J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed; (f) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (g) J. Wencel-Delord, T. Drge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (h) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (i) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- For selected reviews, see: (a) R. H. Crabtree, Nature, 2000, 408, 415 CrossRef CAS PubMed; (b) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC; (c) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed; (d) Y. Horino, Angew. Chem., Int. Ed., 2007, 46, 2144 CrossRef CAS PubMed; (e) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC; (f) C. Winter and N. Krause, Angew. Chem., Int. Ed., 2009, 48, 2460 CrossRef CAS PubMed; (g) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS PubMed.
- For selected recent examples, see: (a) C.-H. Jun, H. Lee and S.-G. Lim, J. Am. Chem. Soc., 2001, 123, 751 CrossRef CAS; (b) A. Sattler and G. Parkin, Nature, 2010, 463, 523 CrossRef CAS PubMed; (c) C. Qin, P. Feng, Y. Ou, T. Shen, T. Wang and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 7850 CrossRef CAS PubMed; (d) M. Saydou and A. M. Echavarren, Angew. Chem., Int. Ed., 2013, 52, 13468 CrossRef PubMed; (e) H. M. Ko and G. Dong, Nat. Chem., 2014, 6, 739 CrossRef CAS PubMed; (f) L. Souillart, E. Parker and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 3001 CrossRef CAS PubMed; (g) X. Huang, X. Li, M. Zou, S. Song, C. Tang, Y. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS PubMed.
- For reviews of C–N bond cleavage, see: (a) K. Khumtaveeporn and H. Alper, Acc. Chem. Res., 1995, 28, 414 CrossRef CAS; (b) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed; (c) N. J. Turner, Chem. Rev., 2011, 111, 4073 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Koreeda, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 7238 CrossRef CAS PubMed; (b) W. Geng, W.-X. Zhang, W. Hao and Z. Xi, J. Am. Chem. Soc., 2012, 134, 20230 CrossRef CAS PubMed; (c) B.-J. Li, H.-Y. Wang, Q.-L. Zhu and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948 CrossRef CAS PubMed; (d) M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587 CrossRef CAS PubMed.
- For some recent reports on both C–C and C–N bond cleavage, see: (a) C. Li, W.-T. Zhang and X.-S. Wang, J. Org. Chem., 2014, 79, 5847 CrossRef CAS PubMed; (b) P. Feng, X. Sun, Y. Su, X. Li, L.-H. Zhang, X. Shi and N. Jiao, Org. Lett., 2014, 16, 3388 CrossRef CAS PubMed.
- (a) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS; (b) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS PubMed; (c) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 RSC; (d) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC.
- Selected examples, see: (a) Y. Wei, H. Zhao, J. Kan, W. Su and M. Hong, J. Am. Chem. Soc., 2010, 132, 2522 CrossRef CAS PubMed; (b) N. Matsuyama, M. Kitahara, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2358 CrossRef CAS PubMed; (c) S. H. Kim, J. Yoon and S. Chang, Org. Lett., 2011, 13, 1474 CrossRef CAS PubMed; (d) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed.
- Selected examples, see: (a) K. Kobayashi, M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8528 CrossRef CAS PubMed; (b) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742 CrossRef CAS PubMed; (c) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS PubMed; (d) J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387 CrossRef CAS PubMed.
- (a) J. P. Brand, L. Charpentier and J. Waster, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (b) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 2722 CrossRef CAS PubMed; (c) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780 CrossRef CAS PubMed; (d) K. D. Collins, F. Lied and F. Glorius, Chem. Commun., 2014, 50, 4459 RSC.
- (a) T. Nishimura, H. Ariki, Y. Maeda and S. Uemura, Org. Lett., 2003, 5, 2997 CrossRef CAS PubMed; (b) T. Nishimura, H. Ariki, Y. Maeda and S. Uemura, Org. Lett., 2003, 5, 2997 CrossRef CAS PubMed.
- A. Funayama, T. Satoh and M. Miura, J. Am. Chem. Soc., 2005, 127, 15354 CrossRef CAS PubMed.
- See for selected reviews: (a) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- Rh-catalyzed C–H alkynylation, see ref. 11b–d.
- (a) B. M. Trost, Science, 1991, 254, 1471 CAS; (b) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed.
- (a) F. De Simone, J. Gertsch and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 5767 CrossRef CAS PubMed; (b) M. Mizutani, F. Inagaki, T. Nakanishi, C. Yangagihara, I. Tamai and C. Mukai, Org. Lett., 2011, 13, 1796 CrossRef CAS PubMed.
- (a) A. Ohsawa, T. Kawaguchi and H. Igeta, J. Org. Chem., 1982, 47, 3497 CrossRef CAS; (b) H. Zhu, J. Stöckigt, Y. Yu and H. Zou, Org. Lett., 2011, 13, 2792 CrossRef CAS PubMed; (c) D. C. Rogness, N. A. Markina, J. P. Waldo and R. C. Larock, J. Org. Chem., 2012, 77, 2743 CrossRef CAS PubMed; (d) S. Samala, P. Pallavi, R. Kumar, R. K. Arigela, G. Singh, R. S. Ampapathi, A. Priya, S. Datta, A. Patra and B. Kundu, Chem. – Eur. J., 2014, 20, 14344 CrossRef CAS PubMed.
- Rare examples for C(aryl)–C(alkynyl) bond cleavage, see: (a) M. Gaydou and A. M. Echavarren, Angew. Chem., Int. Ed., 2013, 52, 13468 CrossRef CAS PubMed; (b) X.-R. Song, Y.-P. Han, Y.-F. Qiu, Z.-H. Qiu, X.-Y. Liu, P.-F. Xu and Y.-M. Liang, Chem. – Eur. J., 2014, 20, 12046 CrossRef CAS PubMed; (c) ref. 4c ; (d) C. Müller, C. N. Iverson, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 9718 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details. CCDC 1032402–1032406. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc01412c |
| This journal is © The Royal Society of Chemistry 2015 |