
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple coordination complex-derived three-dimensional mesoporous graphene as an efficient bifunctional oxygen electrocatalyst†
Kyung Joo
Lee
a,
Young Jin
Sa
a,
Hu Young
Jeong
b,
Christopher W.
Bielawski
ac,
Sang Hoon
Joo
ad and
Hoi Ri
Moon
*a
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 689-798, Republic of Korea. E-mail: hoirimoon@unist.ac.kr
bUNIST Central Research Facilities (UCRF), Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 689-798, Republic of Korea
cCenter for Multidimensional Carbon Materials, Institute for Basic Science (IBS), Ulsan 689-798, Republic of Korea
dSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Ulsan 689-798, Republic of Korea
First published on 9th March 2015
Abstract
3D mesoporous graphene (mesoG) was synthesized from [Ni2(EDTA)] (EDTA = ethylenediaminetetraacetate). The material is comprised of interconnected 4 nm-sized hollow carbon shells composed of 3–4 layers of graphene and exhibits high bifunctional electrocatalytic activity as well as high durability for use in oxygen evolution and reduction reactions.
Due to their numerous potential applications in gas adsorption,1 energy storage,2 and catalysis,3 porous carbon materials have attracted tremendous interest. In particular, given the high porosity of carbon-based graphitic frameworks, such materials are expected to exhibit excellent electrochemical and mechanical properties4,5 as well as high thermal conductivity.6 Of the several forms of graphitic porous carbons, 3D porous graphene-like materials are attractive because they are expected to display high conductivities and large surface areas. While ideal graphene is composed of a single layer of sp2 hybridized carbon atoms with a high theoretical surface area (2630 m2 g−1), its strong stacking affinity via the formation of π–π interactions can hamper the full utilization of its intrinsic properties in practical applications.4,7 To integrate the functionality of graphene with structural features that prevent undesired stacking, efforts have been directed toward the synthesis of 3D porous graphene-like networks. For instance, Chen et al. reported the synthesis of graphene foams via a template-directed chemical vapor deposition (CVD) that utilized nickel foam both as a template and as a catalyst.8 Similarly, 3D porous graphene networks have been prepared by CVD in the presence of templates such as 3D assembled colloidal silica9 and porous MgO10 along with additional catalysts to facilitate graphitization. However, such template-assisted CVD methods are commonly unsuitable for large-scale syntheses and often require templates. An alternative approach is the assembly of materials which are related graphene, such as graphene oxide (GO) and reduced graphene oxide (rGO), followed by activation.11 Despite the potential advantage of mass production, such a method might yield lower electrochemical performances caused by damage of the GO and rGO surfaces. Another method is the hybridization of graphene with organic or inorganic materials that effectively function as ‘spacers’ between the graphene layers.12,13 However, this approach is susceptible to the restacking of 2D graphene layers due to low pillar densities as compared to the overall surface area.13
Herein, we report that the direct thermal conversion of a NiII coordination complex followed by etching can yield 3D mesoporous graphene (mesoG). The method is cost effective, may be performed on the gram scale, and can proceed without the aid of additional templates, catalysts or CVD techniques (Scheme 1). During the thermal conversion of the NiII complex under an inert atmosphere, the NiII ionic centers are transformed in situ into 4 nm-sized Ni NPs (Fig. 1a), which act as catalysts that facilitate the graphitization of amorphous carbon from the organic ligands while also serving as a template for the creation of mesopores. The resulting 3D mesoG material is composed of a 3D interconnected structure of hollow carbon shells comprised of 3–4 graphene layers. In addition, the material features small, uniform mesopores (∼4 nm in diameter), a large surface area, and a high C/O ratio. The 3D mesoG also exhibits high activity and durability as a bifunctional electrocatalyst for oxygen reduction and evolution reactions (ORR and OER, respectively), revealing its promise as a non-precious metal electrocatalyst for use in energy conversion and storage devices.14
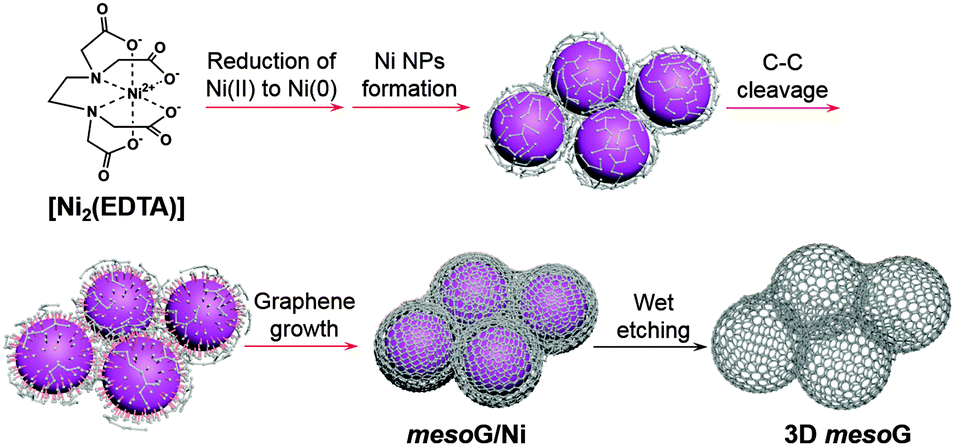 | ||
| Scheme 1 Schematic representation of the thermal conversion of [Ni2(EDTA)] into three-dimensional mesoporous graphene, 3D mesoG. The processes indicated by the red arrows occurred during the one-step heat treatment of [Ni2(EDTA)]. | ||
 | ||
| Fig. 1 TEM image of (a) mesoG/Ni during the thermal conversion at 400 °C, and (b) mesoG/Ni after heat treatment at 1000 °C, and (c–e) 3D mesoG at various magnifications. | ||
The NiII coordination complex used herein can be readily prepared via the coordination of NiII ions with ethylenediaminetetraacetic acid (H4EDTA) in the presence of N,N,N-triethylamine (TEA), which serves to deprotonate the carboxylic acid. In the corresponding product, [Ni2(EDTA)],15 which is a blue powder, the NiII center is chelated by an EDTA ligand to form six coordination bonds with four oxygen atoms of the carboxylates and two nitrogen atoms of the tertiary amines.
With [Ni2(EDTA)] in hand, 1.15 g of the complex was heated to 1000 °C at a ramp rate of 10 °C min−1 under a nitrogen flow of 500 mL min−1. After maintaining the temperature of the reaction at 1000 °C for 1 h, the solid was cooled to room temperature, which resulted in the formation of 0.32 g of a black powder. Elemental composition analysis of the resulting powder revealed that during the thermolysis of [Ni2(EDTA)], most of the H, N, and O were removed, while the Ni and 30% of the C contents remained (mesoG/Ni, Table S1 and Fig. S2, ESI†). Transmission electron microscopy (TEM) (Fig. 1b) revealed that after the thermal treatment, the black powder was composed of 3D carbon materials and ∼50 nm-sized Ni NPs (mesoG/Ni). The Ni NPs could be removed by a wet etching process to obtain a relatively metal-free carbon material (∼0.1 g) where approximately 1 wt% of the Ni remained in the 3D mesoG after etching, as determined by thermogravimetric analysis (TGA) under oxygen (Table S1 and Fig. S3, ESI†). Interestingly, the 3D mesoG also contained 1.7 wt% of nitrogen, presumably from the EDTA ligands, and thus can be considered an N-doped carbon. Fig. 1c and d show that the obtained carbon materials consisted of small hollow carbon shells in addition to large shells. A closer observation (Fig. 1e) via atomic resolution TEM (AR-TEM) revealed that these small hollow carbon shells were composed of 3–4 layers of graphene with an inner diameter of ∼4 nm, and were interconnected to produce the 3D nanostructured mesoG. We note that even after etching the Ni NPs from hollow carbon, the 3D mesoG structure did not collapse. Furthermore, elemental composition analysis of the 3D mesoG (Table S1, ESI†) revealed that etching with a strong acid had a negligible effect on the chemical state of the 3D mesoG. Scanning electron microscopy (SEM) analysis of the 3D mesoG revealed the external microscopic morphology of the integrated graphene shells (see Fig. S4, ESI† for an image of this material). Notably, the synthetic method can be scaled and at least tens of grams of the [Ni2(EDTA)] precursor may be used.
To identify the origin of the formation of the hollow shell and the 3D graphene-like structure, variable temperature X-ray powder diffraction (VT-XRPD) experiments were performed during the treatment of [Ni2(EDTA)] (Fig. S5a, ESI†). When the temperature reached 400 °C, a broad signal near 2θ = 45° was detected. Upon further increase of the temperature, the peak at 45° became sharper and was accompanied by the formation of two newly evolved peaks at 52° and 76°, which were characteristic reflections of a cubic Ni0 structure (JCPDS file no. 04-0850). When the temperature was further increased to 1000 °C, a very small but clear peak at 26° was observed and a typical (002) reflection of graphitic carbon. Fig. S5b (ESI†) shows the size of Ni NPs at each temperature, estimated by applying the Debye–Scherrer equation to the (111) reflection. Below 600 °C, the sizes of the Ni NPs were less than 5 nm. However, above 700 °C, some of the small nanoparticles began to agglomerate to form larger Ni NPs (plotted as red and blue dots, respectively), as derived from the Debye–Scherrer equation with two deconvoluted peaks of the (111) reflection. Subsequently, when the temperature reached 900 °C, the small Ni NPs ceased to agglomerate and resulted in a single data point of ∼35 nm-sized Ni NPs. The number of expected peaks for deconvolution was determined from TEM images obtained at each temperature (Fig. S6, ESI†). Since the solids heated to less than 300 °C were unstable under the e-beam during TEM analysis, images were obtained for samples treated at 400, 500, 600, 700, 800, 900, and 1000 °C. Similar to the XRPD results, when the temperature was 400 °C, Ni NPs embedded in the carbon matrix were formed. As the temperature increased, the shapes of the Ni NPs became more regular and crystalline, and had diameters of less than 5 nm. Meanwhile, owing to the catalytic function of Ni NPs, the organic species from the EDTA began to decompose and subsequently formed sp2 carbons on the Ni NPs. A similar phenomenon has been observed with the growth of CNTs assisted by Ni catalysts.16 In the present system where the Ni NPs were embedded within carbon layers, the total directional contact of carbon with Ni NP surfaces results in spherical shells rather than tubular structures. Therefore, the size of the Ni NPs formed in situ dictates the inner diameter of the hollow carbon. In the TEM image of the solid treated at 700 °C (Fig. S6, ESI†), Ni NPs with diameters larger than 50 nm were observed in the presence of small Ni NPs with diameters of ca. 5 nm. We presume that this was due to the secondary agglomeration of the 4 nm Ni NPs that was initiated upon heating at the temperatures above ∼600 °C. The mobility of the Ni NPs can be explained by the melting-point depression in nanoscale materials.17 While the melting temperature of bulk Ni metal is 1453 °C, metal particles smaller than ∼100 nm can melt at significantly lower temperatures; indeed, the melting point of 4 nm Ni metal particles was calculated to be ∼700 °C, a temperature at which the Ni NPs began to agglomerate. Therefore, after heating at 1000 °C for 1 h, the 3D mesoG coexisted with large Ni NPs. Likewise, the surfaces of the large Ni NPs also act as catalysts to generate large carbon shells, as shown in Fig. S6 (ESI†).
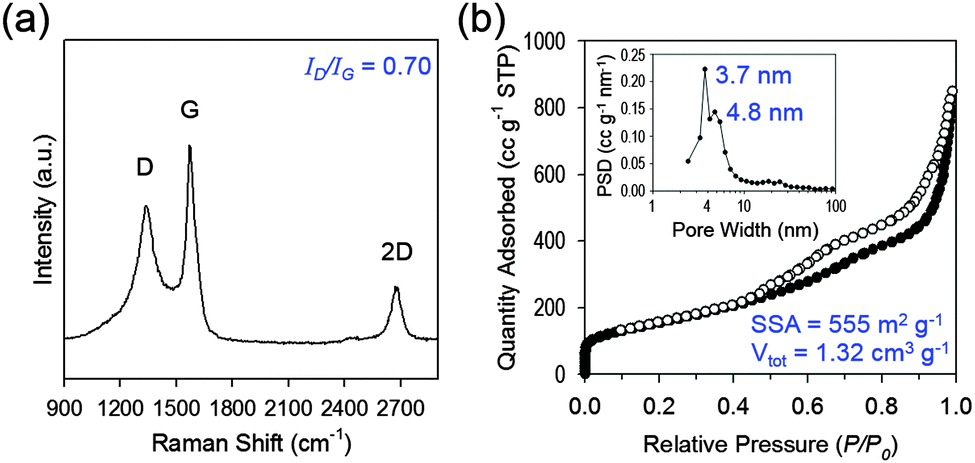 | ||
| Fig. 2 Structural and electronic characterization of 3D mesoG. (a) Raman spectrum. (b) N2 adsorption–desorption isotherm and the corresponding BJH pore size distribution curve (inset). | ||
To explore the structural and electronic properties of the 3D mesoG material, Raman spectroscopy was conducted (Fig. 2a). A broad D-band attributed to the disorder-induced mode and a relatively sharp G-band attributed to the E2g-mode from the sp2 carbon domain appeared at 1340 and 1575 cm−1, respectively, along with the 2D-band at 2676 cm−1. The G-peak indicated the presence of crystalline graphitic carbon in the 3D mesoG. The ratio between the intensity of the D- and G-bands (ID/IG), which is commonly used as an indicator for the degree of disorder in graphitic materials, was 0.70. This ID/IG ratio was comparable to or smaller than those measured for GO and rGO (ID/IG = ∼1)18 as well as CVD-grown multi-wall CNTs (ID/IG = 0.7–1.3),19 thus indicating that high quality 3D mesoG was formed (Table S2, ESI†). The broad D-peak might originate from the distorted nature of the hexagonal sp2 carbons because of significant lattice stress arising from high curvature of the 4 nm hollow graphene shell in 3D mesoG. As listed in Table S1 (ESI†), elemental analysis of 3D mesoG revealed a high C/O ratio of 55, which was higher than that of some forms of chemically or thermally reduced graphene oxide (rGO) (∼20).20 Regardless, such a high C/O atomic ratio implies the presence of limited oxygen-containing functionalities, which may lead to improved physical properties. In order to assess the porous structure of 3D mesoG, nitrogen adsorption–desorption measurements were conducted. As shown in Fig. 3b, 3D mesoG exhibited typical type IV isotherms with an H3 type hysteresis loop over a relative pressure range of 0.42 < P/P0 < 0.99, characteristic of a mesoporous material. The Barrett–Joyner–Halenda (BJH) pore size distribution curve (Fig. 2b, inset) indicated that the sizes of the mesopores were centered around 4 nm, which was in good agreement with the TEM analysis. The highly porous nature of 3D mesoG gave rise to a relatively large Brunauer–Emmett–Teller (BET) specific surface area (SSA) of 555 m2 g−1, which was comparable to the specific surface area of five-layered graphene calculated from the theoretical SSA of monolayer graphene (2630 m2 g−1). In addition, 3D mesoG exhibited a very high total pore volume (Vtot) of 1.32 cm3 g−1.
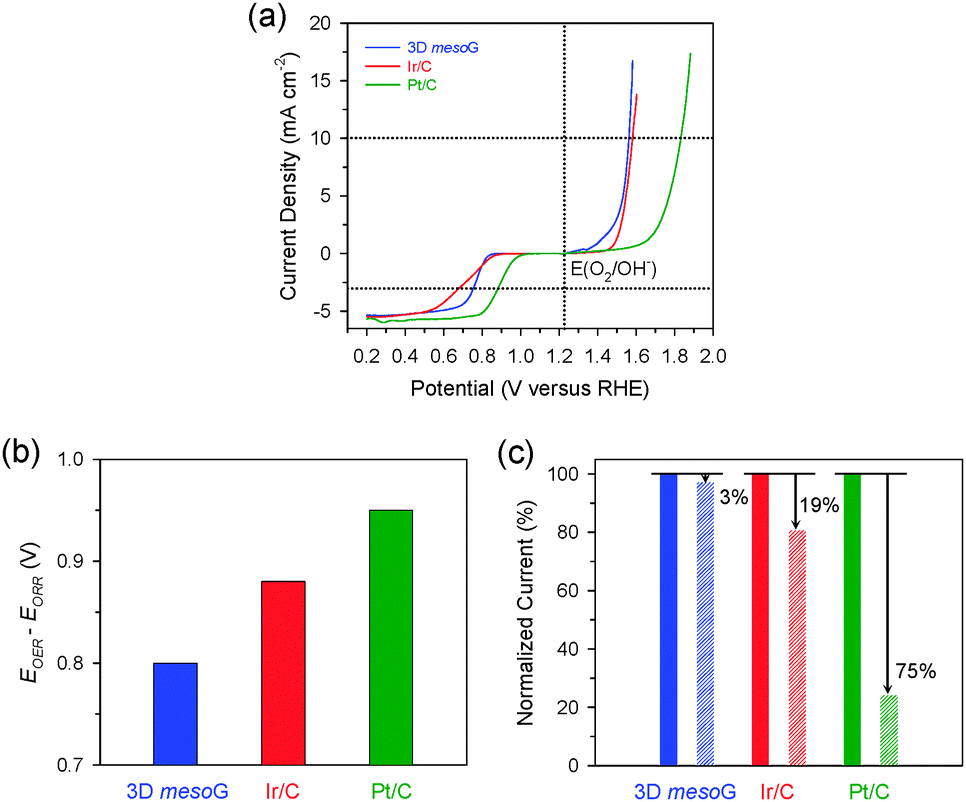 | ||
| Fig. 3 Electrocatalytic activity and durability of 3D mesoG in the OER and ORR. (a) Polarization curves showing ORR and OER activities of 3D mesoG, Ir/C, and Pt/C catalysts. (b) Bar graph showing the potential differences to drive 10 mA cm−2 for the OER and −3 mA cm−2 for the ORR. (c) The changes in relative current densities of 3D mesoG, Ir/C, and Pt/C catalysts for the OER before and after cycling tests. | ||
Overall, the characterization data were consistent with the material with large surface area from a readily-accessible Ni coordination complex. It is noteworthy that it was previously challenging to prepare 3D graphene with such small, monodisperse mesopores without the need for additional pore generation processes, such as the introduction and removal of various templates (e.g., Ni foams8 or silica beads9). Furthermore, the present method can proceed without the use of reductive gases (e.g., H2), which is typically required to form the carbons observed within the graphene layers. As such, this method represents a significant advancement in the preparation of 3D graphene materials in terms of simplicity of the synthetic process as well as the quality of the resulting materials.
To utilize the structural properties exhibited by 3D mesoG, we investigated its bifunctional electrocatalytic activity in ORR and OER, which are relevant for renewable energy technologies.14,21 The activities of Ir/C (20 wt% Ir, Premetek) and Pt/C (20 wt% Pt, Johnson-Matthey) were also evaluated as benchmarks. The polarization curves for the ORR and OER were measured in 0.1 M KOH at 1600 rpm using the rotating disk electrode (RDE) technique (Fig. 3a). In the OER, the 3D mesoG material showed a high activity that was comparable to that of noble-metal-based Ir/C and was significantly higher than that of Pt/C. The potential to drive 10 mA cm−2 for 3D mesoG was 1.56 V (vs. reversible hydrogen electrode (RHE), overpotential (η) = 330 mV), which was slightly better than that of Ir/C (1.58 V, η = 350 mV) and significantly better than that of Pt/C (1.83 V, η = 600 mV). In the ORR, 3D mesoG also showed high activity, which approached that of Pt/C and exceeded that of the Ir/C catalyst. The potentials required to reach −3 mA cm−2 during the ORR were 0.76 V, 0.88 V, and 0.70 V for 3D mesoG, Pt/C, and Ir/C, respectively. To quantitatively compare the bifunctional activity (oxygen electrode activity) of the catalysts for the OER and ORR, the potential difference to drive 10 mA cm−2 and −3 mA cm−2 (or the sum of overpotentials at those current densities) was calculated (Fig. 3b and Table S3, ESI†).22 Remarkably, the 3D mesoG material exhibited the highest oxygen electrode activity among the catalysts compared: indeed, the potential difference of the 3D mesoG (0.80 V) was lower than that of the noble metal-based Ir/C (0.88 V) and Pt/C (0.95 V) catalysts. It should also be noted that the comparison of the oxygen electrode activities of the reported catalysts indicated that the 3D mesoG may be ideally suited for use as a carbon-based bifunctional catalyst for the ORR and OER (Table S4, ESI†) and a promising catalyst for use in regenerative fuel cells as well as rechargeable metal–air batteries. The high bifunctional activity of 3D mesoG may originate from its very large surface area, with numerous defect sites due to the high curvature of its structure. In addition, the presence of nitrogen and nickel species in the carbon framework also contributed to the high catalytic activity.23
Finally, we assessed the structural stability of the 3D mesoG material via durability tests during the OER. Specifically, the 3D mesoG material as well as Ir/C and Pt/C catalysts were cycled from 1.20 to 1.70 V 100 times (Fig. S7, ESI†). A comparison of the current densities at 1.70 V before and after the cycling tests revealed that 3D mesoG underwent only a 3% decay in the current density, whereas the OER activities of Ir/C and Pt/C catalysts declined by 19% and 75%, respectively (Fig. 3c). Carbon-supported metal nanoparticles are subject to various deactivation processes, such as carbon oxidation, metal dissolution, and Ostwald ripening, which are more pronounced at high oxidative OER potentials (>1.4 V). The superior stability of the 3D mesoG material can be attributed to the highly graphitic carbon framework, which appears in the aforementioned experiments to be more resistant to oxidation than the amorphous carbon in Ir/C and Pt/C.
In conclusion, we describe a straightforward and cost-effective method to synthesize 3D mesoporous graphene frameworks from a Ni(II) molecular coordination compound. The resulting 3D mesoG was composed of 3D interconnected hollow N-doped graphene shells consisting of 3–4 layers with small, uniform mesopores of ca. 4 nm. The 3D mesoG material exhibited high bifunctional electrocatalytic activity and durability in the ORR and OER, which in our case was found to be superior to that of noble metal catalysts such as Pt/C and Ir/C. Similar to recent studies regarding the conversion of metal–organic frameworks,24 we believe that the method described herein can be extended to a variety of porous graphene frameworks, including heteroatom-doped graphene, and utilized as a high-performance material for use in energy conversion and storage applications. Furthermore, we envisage that the conversion of small molecular metal complexes into nanostructured materials can be exploited for the preparation of novel functional materials.
This work was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea (NRF-2013R1A1A3010846; NRF-2014M1A8A1049255; NRF-2013R1A1A2012960) as well as the IBS (IBS-R019-D1 to CWB).
Notes and references
- H. Y. Yang, Z. J. Han, S. F. Yu, K. L. Pey, K. Ostrikov and R. Karnik, Nat. Commun., 2013, 4, 2220 Search PubMed.
- (a) X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed; (b) C. Merlet, B. Rotenberg, P. A. Madden, P.-L. Taberna, P. Simon, Y. Gogotsi and M. Salanner, Nat. Mater., 2012, 11, 306 CrossRef CAS PubMed; (c) Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- (a) C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275 CrossRef CAS PubMed; (b) D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782 CrossRef CAS PubMed; (c) S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS PubMed.
- J.-C. Yoon, J.-S. Lee, S.-I. Kim, K.-H. Kim and J.-H. Jang, Sci. Rep., 2013, 3, 1788 Search PubMed.
- C. Cui, W. Qian, Y. Yu, C. Kong, B. Yu, L. Xiang and F. Wei, J. Am. Chem. Soc., 2014, 136, 2256 CrossRef CAS PubMed.
- (a) Z.-S. Wu, Y. Sun, Y.-Z. Tan, S. Yang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 19532 CrossRef CAS PubMed; (b) L. Zhang, F. Zhang, X. Yang, G. Long, Y. Wu, T. Zhang, K. Leng, Y. Huang, Y. Ma, A. Yu and Y. Chen, Sci. Rep., 2013, 3, 1408 Search PubMed; (c) C. Wu, X. Huang, G. Wang, L. Lv, G. Chen, G. Li and P. Jiang, Adv. Funct. Mater., 2013, 23, 506 CrossRef CAS.
- (a) K. H. Kim, M. H. Yang, K. M. Cho, Y.-S. Jun, S. B. Lee and H.-T. Jung, Sci. Rep., 2013, 3, 3251 Search PubMed; (b) Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723 CrossRef CAS PubMed; (c) J. W. Burress, S. Gadipelli, J. Ford, J. M. Simmons, W. Zhou and T. Yildirim, Angew. Chem., Int. Ed., 2010, 49, 8902 CrossRef CAS PubMed.
- T. K. Kim, J. Y. Cheon, K. Yoo, J. W. Kim, S. Hyun, H. S. Shin, S. H. Joo and H. R. Moon, J. Mater. Chem. A, 2013, 1, 8432 CAS.
- (a) S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331 RSC; (b) P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- E. Coronado, M. Drillon, A. Fuertes, D. Beltran, A. Mosset and J. Galy, J. Am. Chem. Soc., 1986, 108, 900 CrossRef CAS.
- M. Kumar, Carbon Nanotubes – Synthesis, Characterization, Applications, in Carbon Nanotube Synthesis and Growth Mechanism [Online], ed. S. Yellampalli, InTech, 2011, ch. 8, http://www.intechopen.com/books/carbon-nanotubes-synthesis-characterization-applications/carbon-nanotube-synthesis-and-growth-mechanism, accessed February 5, 2015 Search PubMed.
- M. Anna, G. N. Albert and I. K. Esko, J. Phys.: Condens. Matter, 2013, 15, S3011 Search PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- (a) M. Sveningsson, R.-E. Morjan, O. A. Nerushev, Y. Sato, J. Bäckström, E. E. B. Campbell and F. Rohmund, Appl. Phys. A: Mater. Sci. Process., 2001, 73, 409 CrossRef CAS; (b) K. Kwok and W. K. S. Chiu, Carbon, 2005, 43, 437 CrossRef CAS PubMed; (c) W. Li, H. Zhang, C. Wang, Y. Zhang, L. Xu, K. Zhu and S. Xie, Appl. Phys. Lett., 1997, 70, 2684 CrossRef CAS PubMed.
- C. K. Chun and M. Pumera, Chem. Soc. Rev., 2014, 43, 291 RSC.
- R. Cao, J.-S. Lee, M. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816 CrossRef CAS.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- (a) T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940 CrossRef CAS PubMed; (b) J. H. Lee, Y. J. Sa, T. K. Kim, H. R. Moon and S. H. Joo, J. Mater. Chem. A, 2014, 2, 10435 RSC; (c) K. J. Lee, T.-H. Kim, T. K. Kim, J. H. Lee, H.-K. Song and H. R. Moon, J. Mater. Chem. A, 2014, 2, 14393 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc01123j |
| This journal is © The Royal Society of Chemistry 2015 |