
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepurposing de novo designed entities reveals phosphodiesterase 3B and cathepsin L modulators†
Tiago
Rodrigues
a,
Yen-Chu
Lin
a,
Markus
Hartenfeller
ab,
Steffen
Renner
b,
Yi Fan
Lim
a and
Gisbert
Schneider
*a
aSwiss Federal Institute of Technology (ETH), Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 4, 8093 Zürich, Switzerland. E-mail: gisbert.schneider@pharma.ethz.ch
bNovartis Pharma AG, Postfach, 4002 Basel, Switzerland
First published on 1st April 2015
Abstract
Using computational bioactivity prediction models we identified phosphodiesterase 3B (PDE3B) and cathepsin L as macromolecular targets of de novo designed compounds. By disclosing the most potent cathepsin L activator known to date, small molecule repurposing by target panel prediction represents a feasible route towards innovative leads for chemical biology and molecular medicine.
Computer-assisted de novo molecular design is a maturing technology for generating new chemical entities (NCEs) in chemical biology and molecular medicine.1–5 At the same time, the computational prediction of macromolecular target engagement by small molecules may offer a platform for expeditious profiling of drug-like molecules.6–9 Here we combine both concepts and demonstrate that predictive quantitative bioaffinity models8,10 are suited for the identification of the macromolecular targets of de novo designed drug-like molecules. We report tool compounds for gauging the effects of phosphodiesterase 3B (PDE3B) inhibition on lipolysis, and disclose a pioneering and best-in-class cathepsin L activator.
The prediction of target profiles for NCEs is of current interest in drug discovery.11,12 Although it is widely accepted that promiscuity is ubiquitous across drug-like chemical space,13 only few robust computational tools are presently available for the in silico qualitative projection of polypharmacology.6,9,14–17 Importantly, only one of these tools (SPiDER) was constructed for explicit use with de novo designed molecules.6 We have recently reported a machine-learning approach (Gaussian process regression, GP) for quantitative structure–activity relationship (SAR) modeling for a large panel of human drug targets, including a confidence estimation.8,10
We here present the application of this quantitative approach to de novo designed NCEs that had failed to engage the originally intended on-targets. As part of our de novo design efforts we used two of our ligand-based software tools (DOGS18,19 and its extension libDOGS)20 to generate libraries of potential matrix metalloproteinase (MMP) and human immunodeficiency virus-1 (HIV-1) protease inhibitors. Structurally diverse non-zinc chelating small molecules21–24 served as template structures for prototyping innovative MMP inhibitors (ESI†). We selected compounds 1–4 for synthesis (Fig. 1) and biochemical profiling against MMP-1, -3, -8, -12 and -13, but no significant inhibition was observable at a concentration of 100 μM. Similarly, compounds 5–6 (Fig. 1), which had been designed as mimetics of amprenavir and obtained through Mitsunobu chemistry, were profiled against HIV-1 protease with a negative outcome. Although bioactivity against MMPs and HIV-1 protease was not obtained as intended, the low structural similarity of 1–6 to the design templates (Tanimoto similarity < 0.50; Table 1) corroborates the scaffold hopping capabilities of the design algorithm and designates these compounds as potentially valuable chemical agents.
 | ||
| Fig. 1 De novo designed entities 1–6 and their retrosyntheses. | ||
| Cpd | De novo designed entities | Templates | ||||
|---|---|---|---|---|---|---|
| Structure | EC50/μM | LEa | Functional activity | Nearest neighbors | T c | |
a Ligand efficiency (LE) = −1.4 × log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) EC50/HA; HA = non-hydrogen atoms.
b Tanimoto coefficient (Tc) calculated with Morgan substructure fingerprints (radius = 2, nbits = 2048, pathlength = 1–7).
c Percentage of inhibition at a ligand concentration of 50 μM. EC50/HA; HA = non-hydrogen atoms.
b Tanimoto coefficient (Tc) calculated with Morgan substructure fingerprints (radius = 2, nbits = 2048, pathlength = 1–7).
c Percentage of inhibition at a ligand concentration of 50 μM.
|
||||||
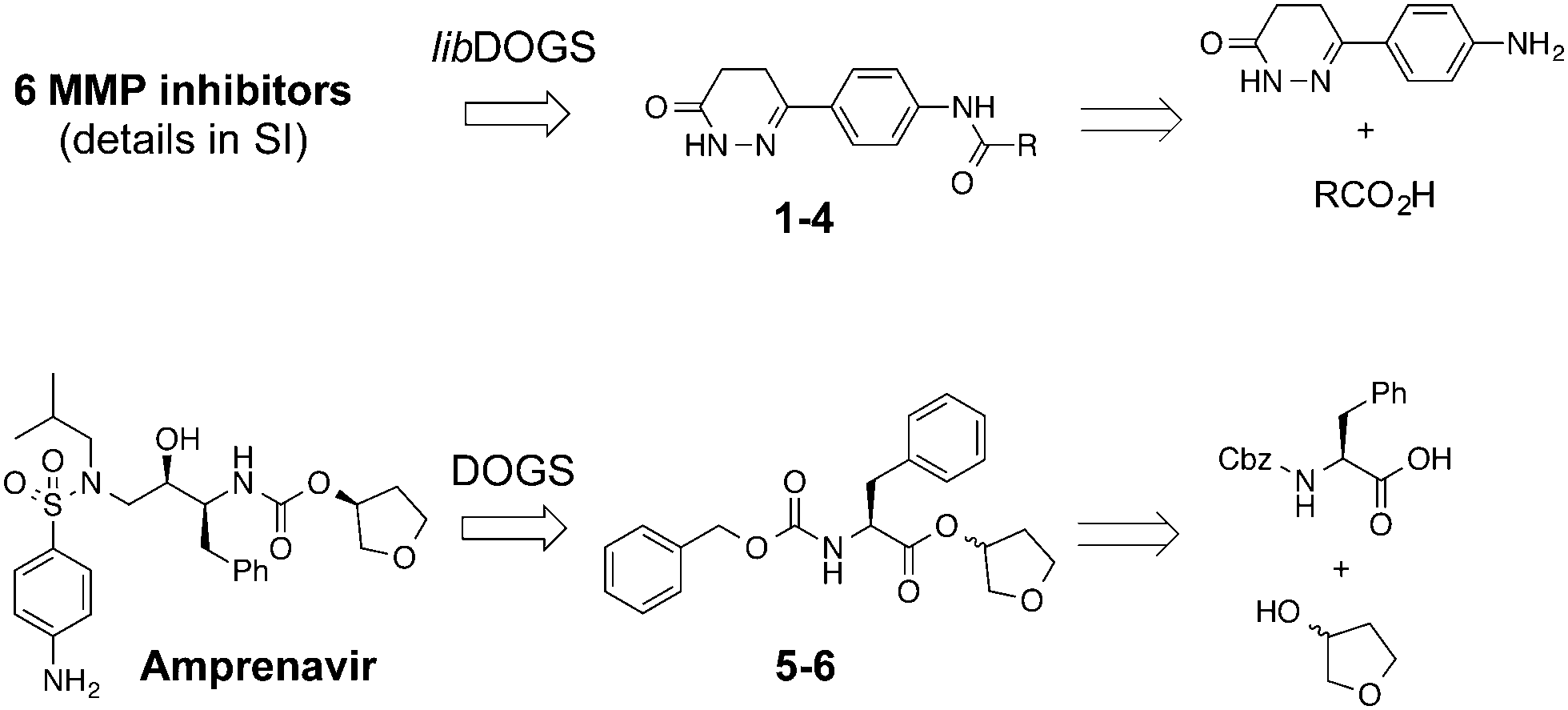
| 1 |
|
38 ± 1%c | <0.22 | PDE3B inhibitor |
|
0.17 |
| 2 |
|
47 ± 10%c | ∼0.27 | PDE3B inhibitor |
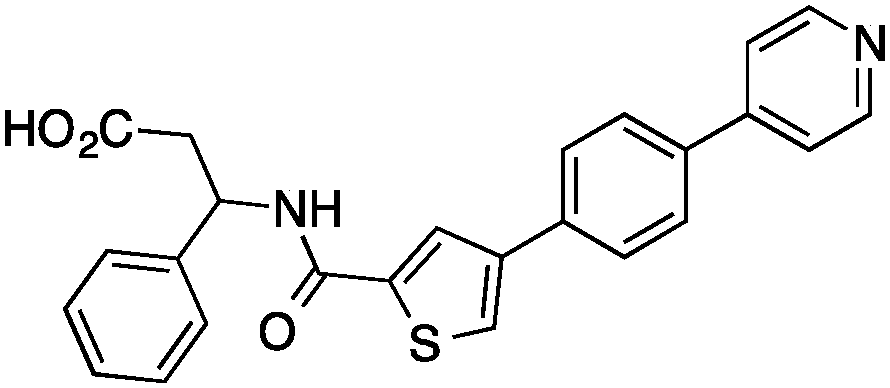
|
0.19 |
| 3 |
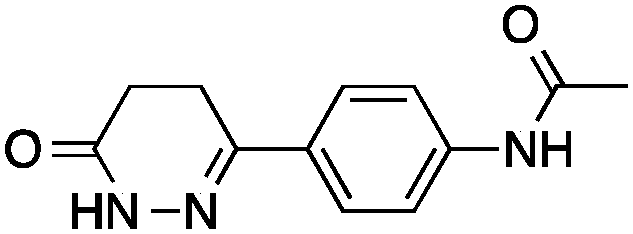
|
11 ± 1 | 0.41 | PDE3B inhibitor |

|
0.15 |
| 4 |
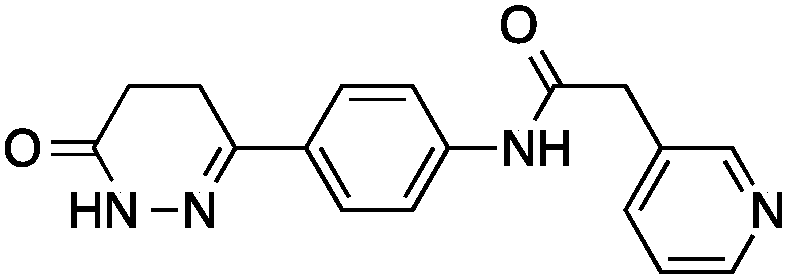
|
6 ± 1 | 0.32 | PDE3B inhibitor |
|
0.18 |
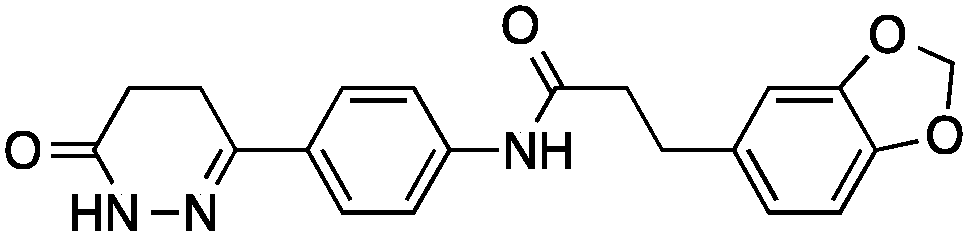
| 5 |
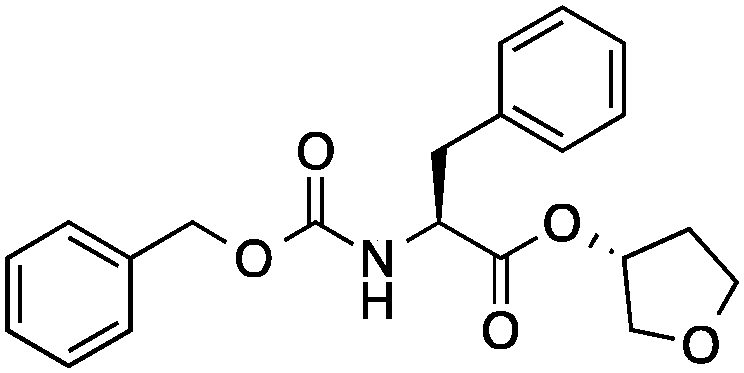
|
38 ± 6 | 0.24 | Cathepsin L inhibitor |
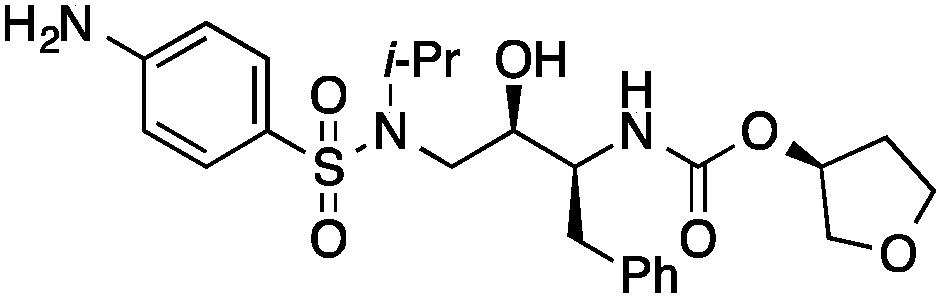
|
0.45 |
| 6 |
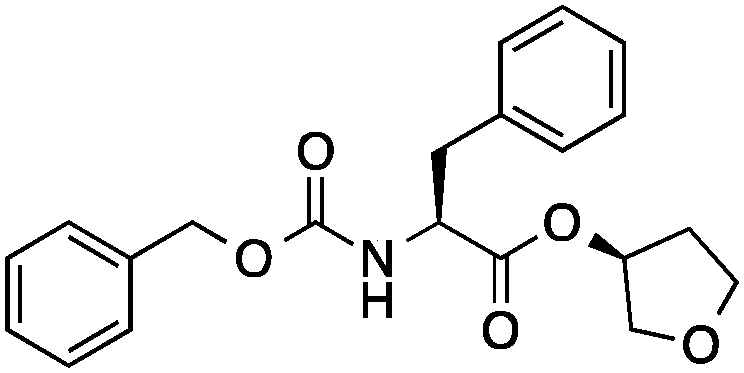
|
6 ± 2 | 0.27 | Cathepsin L activator |
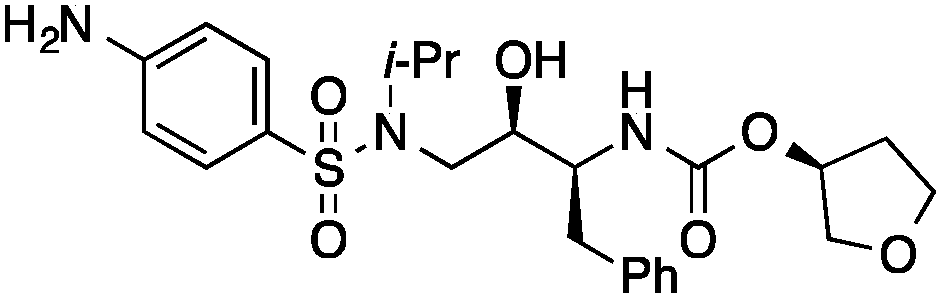
|
0.45 |
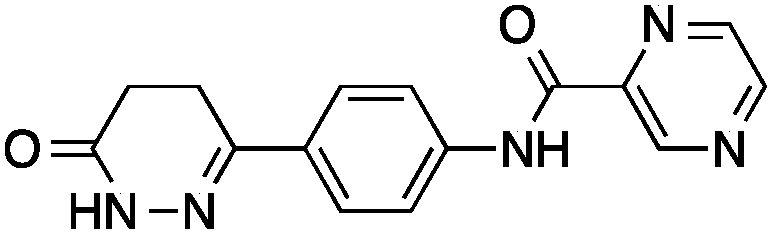
Having drug-like compounds 1–6 in hand we hypothesized that their macromolecular targets could be discovered with our GP models constructed from annotated ChEMBL data.25 The models predict pAffinity values using CATS226 pharmacophores and Morgan substructure fingerprints. To ensure non-trivial predictions the software calculates the Mahalanobis distance (MD) of the predicted values to predictions made for a collection of randomly selected molecules.10 We nominated potential drug targets based on pAffinity >6 and/or MD >4 values (ESI†). For 1–4 our prediction suggested PDE3B, which is involved in the metabolic regulation of lipolysis, and its inhibitors may find applicability for treating obesity.27 In full agreement with the prediction, 1–4 showed potent functional inhibition of PDE3B in a cell-based assay (Table 1, Fig. 2A). Publically available data further suggests that insertion of a methyl group in the dihydropyridazinone moiety increases activity.28,29 While compound 3 had previously been disclosed as a PDE3 inhibitor28 and pimobendan30 presents a related scaffold, compounds 1, 2 and 4 remained unreported.
 | ||
| Fig. 2 (A) EC50 curves of compounds 3 and 4 against phosphodiesterase 3B (PDE3B) in a cell-based assay; control: milrinone (IC50 = 0.67 μM, Hill slope = −0.8). (B) and (C) EC50 curves of compounds 5, NID-1 (B), and 6 (C) against cathepsin L; control: E6431 (IC50 = 8.6 nM, Hill slope = −1.3). | ||
1, 2 and 4 add new SAR information that can be used for future optimization of PDE3B inhibitors featuring the phenyldihydropyridazinone scaffold. These results suggest that our GP models provide a feasible and adequate solution for target identification of de novo designed small molecules.
Significantly, 3 and 4 did not show measurable colloidal aggregation at 50 μM, under dynamic light scattering, that could have resulted in unspecific binding to PDE3B and a false-positive readout. Moreover, screening of 4 against a PDE panel revealed a range of weak to potent inhibitory effects at 50 μM, in line with pAffinity (ESI†) and supporting specific binding to PDE3B. While ligand-efficient entities were identified, e.g.3 and 4, their similarity to compounds in the training data is apparent (ESI†). Still, low structural fingerprint similarity (Tc < 0.2) between these de novo designed entities and their respective nearest neighbors in ChEMBL suggests, in this case, that straightforward similarity searching would likely have failed in their retrieval. In fact, when using the SEA32 software for target prediction, PDE3B is not confidently predicted for 1, 2 and 4. On the other hand, the SPiDER6 model confidently predicts PDE3B for 1–4, which is in line with our GP affinity predictions.
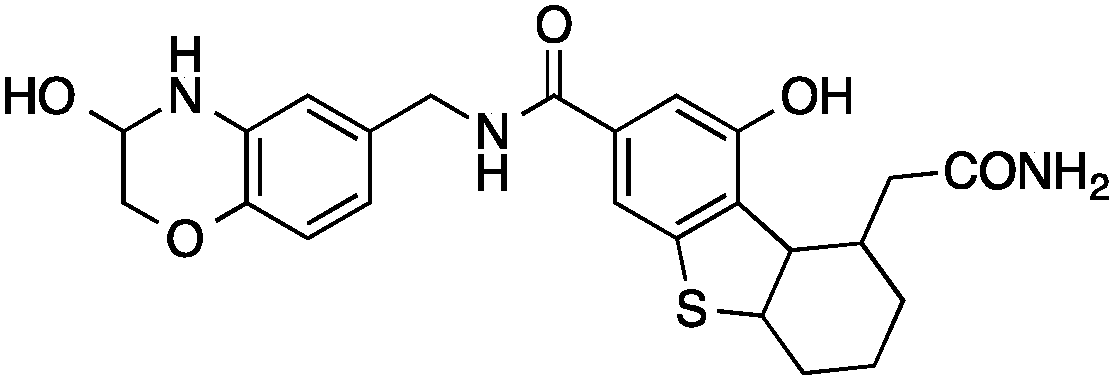
Then we set out to identify drug targets for 5 and 6, which had been designed as orthosteric site inhibitors of HIV-1 protease – a validated drug target for acquired immunodeficiency syndrome therapy.33 Poor modulation of HIV-1 protease by 5 and 6 at a concentration of 100 μM can be partly explained by the inexistence of a hydroxyl group capable of displacing a catalytic water molecule from the active site. Accordingly, our regression models did not suggest inhibition of HIV-1 protease, unlike SEA and SPiDER, but predicted cathepsin L, a lysosomal cysteine protease involved in several pathophysiological events including cancer,34–36 as a potential target of 5 and 6. Biochemical evaluation revealed moderate cathepsin L inhibition by (3′R)-5 (IC50 = 38 ± 6 μM; Table 1, Fig. 2B); in line with the predicted pAffinity value, and with similar potency to the nearest neighbor in ChEMBL (ESI†). Remarkably, the diastereomer (3′S)-6 presented the opposite functional effect (Fig. 2C), potently activating cathepsin L (EC50 = 6 ± 2 μM, Table 1). We ruled out artifact readouts by re-screening 6 against different cathepsin L batches, and by measuring auto-fluorescence as well as colloidal aggregation up to 100 μM. We then compared 6 to NID-1 (Fig. 2B), which has also been reported as a cathepsin L activator.37 Surprisingly, we were unable to reproduce activation of cathepsin L at approximately 16 μM as reported by Varma et al.37 Our data rather suggest concentration-dependent enzyme inhibition by NID-1 (IC50 = 43 ± 4 μM, Fig. 2B) and possible “frequent hitter”34 pan-assay interference according to PAINS38,39 and REOS40 filters. Conversely, our tool compound 6 does not feature potentially problematic moieties according to PAINS and REOS, further supporting its appropriateness as a chemical probe. Nevertheless, in vitro cytotoxicity against MCF7 cells corroborates the previously identified cell death promotion by NID-1 (ESI†). In the same assay system, both 5 and 6 showed negligible cytotoxicity.
Urea had been reported as an activator of cathepsin L.41 In a concentration of 4 M and using Z-Phe–Arg–4NNapOMe as substrate, it was employed to develop a selective colorimetric assay for cathepsin L activity monitoring in crude homogenates. Our results suggest that compound 6 does not simply act as a cathepsin L substrate because the screening assay was devoid of urea. While binding to an allosteric site may be expected and responsible for the observed activation, recognition by the orthostetic site, with Phe sitting in the P2 site, cannot be irrevocably ruled out. In fact, allosteric modulation of the related cathepsin K has been reported recently.42
Cathepsin L-like proteases (cathepsin L, B, S and V) share high amino acid sequence identity in the active site and substrate specificities.43–45 In a preliminary focused screening campaign we further probed selectivity by 5 and 6 for cathepsin L. Using the same assay technology, 5 and 6 were confirmed inactive against cathepsin S up to a concentration of 100 μM (ESI†), advocating specific cathepsin L binding and engagement. This result is noteworthy as it may provide introductory evidence of a positive allosteric effect of 6 on cathepsin L, considering the similar substrate specificities between the protease subtypes. Taking into account the paramount role of cathepsin L for presenting peptides on MHC class II molecules,46,47 protein processing in the heart,48 and as a putative tumor suppressor in the K14-HPV16 mouse model of epidermal cancer,49 the tool compound unveiled here may find multiple areas of application.
While the exact molecular mechanism of recognition of 6 by cathepsin L and its biological implications are currently unclear, our re-purposing approach delivered a pioneering tool compound to leverage studies on the role of cathepsin L activation in (patho)physiological events.
We identified PDE3B inhibitors and the best-in-class cathepsin L activator as a tool compound for mechanistic elucidation of cathepsin L-related biological effects. The results of this study corroborate the suitability of GP models for quantitatively predicting the target affinities of computationally designed NCEs, thereby complementing publically available prediction software. Our GP approach and similar in silico “polypharmacology” platforms may find multiple uses in chemical biology and pharmaceutical research by unveiling ligand–target associations that allow to identify and study biochemical pathways and processes.
ETH Zürich, Novartis Pharma and the OPO Foundation funded this research. The Chemical Computing Group provided an MOE software license.
Notes and references
- T. Rodrigues and G. Schneider, Synlett, 2014, 170 CAS.
- T. Rodrigues, T. Kudoh, F. Roudnicky, Y. F. Lim, Y. C. Lin, C. P. Koch, M. Seno, M. Detmar and G. Schneider, Angew. Chem., Int. Ed., 2013, 52, 10006 CrossRef CAS PubMed.
- T. Rodrigues, F. Roudnicky, C. P. Koch, T. Kudoh, D. Reker, M. Detmar and G. Schneider, Chem. Sci., 2013, 4, 1229 RSC.
- B. Spankuch, S. Keppner, L. Lange, T. Rodrigues, H. Zettl, C. P. Koch, M. Reutlinger, M. Hartenfeller, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2013, 52, 4676 CrossRef PubMed.
- J. Besnard, G. F. Ruda, V. Setola, K. Abecassis, R. M. Rodriguiz, X. P. Huang, S. Norval, M. F. Sassano, A. I. Shin, L. A. Webster, F. R. Simeons, L. Stojanovski, A. Prat, N. G. Seidah, D. B. Constam, G. R. Bickerton, K. D. Read, W. C. Wetsel, I. H. Gilbert, B. L. Roth and A. L. Hopkins, Nature, 2012, 492, 215 CrossRef CAS PubMed.
- D. Reker, T. Rodrigues, P. Schneider and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4067 CrossRef CAS PubMed.
- E. Lounkine, M. J. Keiser, S. Whitebread, D. Mikhailov, J. Hamon, J. L. Jenkins, P. Lavan, E. Weber, A. K. Doak, S. Cote, B. K. Shoichet and L. Urban, Nature, 2012, 486, 361 CAS.
- M. Reutlinger, T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 4244 CrossRef CAS PubMed.
- M. J. Keiser, V. Setola, J. J. Irwin, C. Laggner, A. I. Abbas, S. J. Hufeisen, N. H. Jensen, M. B. Kuijer, R. C. Matos, T. B. Tran, R. Whaley, R. A. Glennon, J. Hert, K. L. Thomas, D. D. Edwards, B. K. Shoichet and B. L. Roth, Nature, 2009, 462, 175 CrossRef CAS PubMed.
- M. Reutlinger, T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 582 CrossRef CAS PubMed.
- G. V. Paolini, R. H. Shapland, W. P. van Hoorn, J. S. Mason and A. L. Hopkins, Nat. Biotechnol., 2006, 24, 805 CrossRef CAS PubMed.
- T. Rodrigues, N. Hauser, D. Reker, M. Reutlinger, T. Wunderlin, J. Hamon, G. Koch and G. Schneider, Angew. Chem., Int. Ed., 2015, 54, 1551–1555 CrossRef CAS PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993 CrossRef CAS PubMed.
- A. A. Antolin, X. Jalencas, J. Yelamos and J. Mestres, ACS Chem. Biol., 2012, 7, 1962 CrossRef CAS PubMed.
- M. Dunkel, S. Gunther, J. Ahmed, B. Wittig and R. Preissner, Nucleic Acids Res., 2008, 36, W55 CrossRef CAS PubMed.
- A. Lagunin, A. Stepanchikova, D. Filimonov and V. Poroikov, Bioinformatics, 2000, 16, 747 CrossRef CAS PubMed.
- D. Reker, A. M. Perna, T. Rodrigues, P. Schneider, M. Reutlinger, B. Mönch, A. Koeberle, C. Lamers, M. Gabler, H. Steinmetz, R. Müller, M. Schubert-Zsilavevz, O. Werz and G. Schneider, Nat. Chem., 2014, 6, 1072 CrossRef CAS PubMed.
- M. Hartenfeller, H. Zettl, M. Walter, M. Rupp, F. Reisen, E. Proschak, S. Weggen, H. Stark and G. Schneider, PLoS Comput. Biol., 2012, 8, e1002380 CAS.
- De Novo Molecular Design, ed. G. Schneider, Wiley VCH, Weinheim, 2013 Search PubMed.
- M. Hartenfeller, S. Renner and E. Jacoby, in De Novo Molecular Design, ed. G. Schneider, Reaction-Driven De Novo Design: a Keystone for Automated Design of Target Family-Oriented Libraries, Wiley VCH, Weinheim, 2013, pp. 245–266 Search PubMed.
- G. Pochetti, R. Montanari, C. Gege, C. Chevrier, A. G. Taveras and F. Mazza, J. Med. Chem., 2009, 52, 1040 CrossRef CAS PubMed.
- M. E. Schnute, P. M. O'Brien, J. Nahra, M. Morris, W. Howard Roark, C. E. Hanau, P. G. Ruminski, J. A. Scholten, T. R. Fletcher, B. C. Hamper, J. N. Carroll, W. C. Patt, H. S. Shieh, B. Collins, A. G. Pavlovsky, K. E. Palmquist, K. W. Aston, J. Hitchcock, M. D. Rogers, J. McDonald, A. R. Johnson, G. E. Munie, A. J. Wittwer, C. F. Man, S. L. Settle, O. Nemirovskiy, L. E. Vickery, A. Agawal, R. D. Dyer and T. Sunyer, Bioorg. Med. Chem. Lett., 2010, 20, 576 CrossRef CAS PubMed.
- R. Morales, S. Perrier, J. M. Florent, J. Beltra, S. Dufour, I. De Mendez, P. Manceau, A. Tertre, F. Moreau, D. Compere, A. C. Dublanchet and M. O'Gara, J. Mol. Biol., 2004, 341, 1063 CrossRef CAS PubMed.
- Z. R. Wasserman, Chem. Biol., 2005, 12, 143 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100 CrossRef CAS PubMed.
- M. Reutlinger, C. P. Koch, D. Reker, N. Todoroff, P. Schneider, T. Rodrigues and G. Schneider, Mol. Inf., 2013, 32, 133 CrossRef CAS PubMed.
- E. Degerman, T. R. Landstrom, J. Wijkander, L. S. Holst, F. Ahmad, P. Belfrage and V. Manganiello, Methods, 1998, 14, 43 CrossRef CAS.
- W. J. Coates, H. D. Prain, M. L. Reeves and B. H. Warrington, J. Med. Chem., 1990, 33, 1735 CrossRef CAS.
- S. D. Edmondson, A. Mastracchio, J. He, C. C. Chung, M. J. Forrest, S. Hofsess, E. MacIntyre, J. Metzger, N. O'Connor, K. Patel, X. Tong, M. R. Tota, L. H. Van der Ploeg, J. P. Varnerin, M. H. Fisher, M. J. Wyvratt, A. E. Weber and E. R. Parmee, Bioorg. Med. Chem. Lett., 2003, 13, 3983 CrossRef CAS PubMed.
- S. G. Gordon, M. W. Miller and A. B. Saunders, J. Am. Anim. Hosp. Assoc., 2006, 42, 90 CrossRef PubMed.
- S. Hashida, E. Kominami and N. Katunuma, J. Biochem., 1982, 91, 1373 CAS.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Nat. Biotechnol., 2007, 25, 197 CrossRef CAS PubMed.
- A. Brik and C. H. Wong, Org. Biomol. Chem., 2003, 1, 5 CAS.
- J. M. Lankelma, D. M. Voorend, T. Barwari, J. Koetsveld, A. H. Van der Spek, A. P. De Porto, G. Van Rooijen and C. J. Van Noorden, Life Sci., 2010, 86, 225 CrossRef CAS PubMed.
- M. M. Mohamed and B. F. Sloane, Nat. Rev. Cancer, 2006, 6, 764 CrossRef CAS PubMed.
- R. E. Burden, P. Snoddy, R. J. Buick, J. A. Johnston, B. Walker and C. J. Scott, Mol. Cancer Ther., 2008, 7, 538 CrossRef CAS PubMed.
- H. Varma, N. M. Gangadhar, R. R. Letso, A. J. Wolpaw, R. Sriramaratnam and B. R. Stockwell, Exp. Cell Res., 2013, 319, 1759 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719 CrossRef CAS PubMed.
- D. Lagorce, O. Sperandio, H. Galons, M. A. Miteva and B. O. Villoutreix, BMC Bioinf., 2008, 9, 396 CrossRef PubMed.
- W. P. Walters and M. A. Murcko, Adv. Drug Delivery Rev., 2002, 54, 255 CrossRef CAS.
- R. C. Kamboj, S. Pal, N. Raghav and H. Singh, Biochimie, 1993, 75, 873 CrossRef CAS.
- M. Novinec, M. Korenc, A. Caflisch, R. Ranganathan, B. Lenarcic and A. Baici, Nat. Commun., 2014, 5, 3287 Search PubMed.
- Y. Choe, F. Leonetti, D. C. Greenbaum, F. Lecaille, M. Bogyo, D. Bromme, J. A. Ellman and C. S. Craik, J. Biol. Chem., 2006, 281, 12824 CrossRef CAS PubMed.
- M. L. Biniossek, D. K. Nagler, C. Becker-Pauly and O. Schilling, J. Proteome Res., 2011, 10, 5363 CrossRef CAS PubMed.
- J. Reiser, B. Adair and T. Reinheckel, J. Clin. Invest., 2010, 120, 3421 CrossRef CAS PubMed.
- C. S. Hsieh, P. deRoos, K. Honey, C. Beers and A. Y. Rudensky, J. Immunol., 2002, 168, 2618 CrossRef CAS.
- S. Conus and H. U. Simon, Swiss Med. Wkly., 2010, 140, w13042 Search PubMed.
- M. Sun, M. Ouzounian, G. de Couto, M. Chen, R. Yan, M. Fukuoka, G. Li, M. Moon, Y. Liu, A. Gramolini, G. J. Wells and P. P. Liu, J. Am. Heart Assoc., 2013, 2, e000191 Search PubMed.
- J. Dennemarker, T. Lohmuller, J. Mayerle, M. Tacke, M. M. Lerch, L. M. Coussens, C. Peters and T. Reinheckel, Oncogene, 2010, 29, 1611 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, synthesis and biochemical testing. See DOI: 10.1039/c5cc01376c |
| This journal is © The Royal Society of Chemistry 2015 |