
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An ultrasensitive impedimetric glycan biosensor with controlled glycan density for detection of lectins and influenza hemagglutinins†
A.
Hushegyi
,
T.
Bertok
,
P.
Damborsky
,
J.
Katrlik
and
J.
Tkac
*
Department of Glycobiotechnology, Institute of Chemistry, Slovak Academy of Sciences, Dubravska cesta 9, 845 38 Bratislava, Slovakia. E-mail: Jan.Tkac@savba.sk; Tel: +421 2 5941 0263
First published on 25th March 2015
Abstract
An impedimetric glycan biosensor with optimised glycan density was applied for the detection of lectins and influenza hemagglutinins down to attomolar concentrations (aM).
Glycans, complex carbohydrates attached to proteins, lipids or present on the cell surfaces, play an important role in many cellular processes including immune response, tumour metastasis, infection by bacteria/viruses, inflammation, molecular recognition and cell signaling.1 The importance of involvement of glycans in the cellular processes is recognized and understood at an amazing pace and such knowledge has a great potential for developing novel therapeutic and diagnostic tools for numerous diseases.2 Glycan biochips based on a fluorescent reading technology developed for highly parallel DNA and protein analysis3 became a “must have” tool for multiplexed analysis of binding of glycans with proteins or intact cells/viruses.4
Glycan biochips relying on a fluorescent signal reading have some limitations including a need to fluorescently label either glycan or a glycan-binding molecule with a risk that such modification can alter a biorecognition process between a glycan binding protein and surface confined glycan molecules.5 Moreover, bleaching of a fluorophore can lead to false negative results or can alter quantitative output from such assays. Traditionally glycans are not printed on the microarray slide with controlled density, which can influence the robustness and reliability of the results obtained by application of fluorescent glycan biochips. Additionally, the detection limit of fluorescent glycan biochips is not particularly impressive and thus, low abundant analyte molecules cannot be detected.6 Thus, various alternative and novel assay platforms working in a label-free mode of operation including electrochemical impedance spectroscopy (EIS) and other electrochemical techniques; field-effect transistor (FET)-based sensing; quartz crystal microbalance; surface plasmon resonance; microcantilever arrays; nanopore sensing; quenching of intrinsic fluorescence of single walled carbon nanotubes; etc. integrated with immobilised glycans and lectins (glycan recognising proteins) have been intensively sought to overcome such limitations as reviewed recently.7 It was recently summarised that the nanoscale controlled immobilisation process for glycans and lectins is required to achieve high selectivity and sensitivity assays by these newly developed devices.6,8 Moreover, from a diverse range of detection techniques applied so far, especially electrochemical based ones including EIS belong to the most sensitive ones.9 For example glycan biosensors with electrochemical detection (cyclic voltammetry and differential pulse voltammetry) exhibit a detection limit down to 0.5 nM,10 FET-based glycan biosensors offer low nM level,11 1 fM12 or aM13 detection limits depending on construction formats and EIS-based glycan biosensors were able to detect glycan binding proteins down to 0.12 nM.14 For comparison glycan biosensors based on surface plasmon resonance or localised surface plasmon resonance can detect analytes down to 0.2 nM15 and cantilever array based glycan devices can detect analytes down to 91 pM.16
Impedimetric antibody and lectin based biosensors can detect proteins and glycoproteins down to aM level,9b but so far impedimetric glycan biosensors could not detect such low levels of analytes.14 In our previous work we focused on the development of ultrasensitive impedimetric lectin biosensors with controlled architecture at the nanoscale in glycoprofiling (glycoprotein analysis) of some diseases.17 In this work we wanted to apply protocols previously developed for immobilisation of lectins for the development of ultrasensitive glycan biosensors. We show for the first time that aM detection limits by impedimetric glycan biosensors are achievable. This ultrasensitive analysis was possible by application of an optimised immobilisation process with controlled density of glycans achieved by formation of a mixed self-assembled monolayer to tune the density of functional carboxylic groups for analysis of three different analytes i.e. Maackia amurensis agglutinin (MAA) and two hemagglutinins H1N1 and H5N1.
Polycrystalline gold electrodes were extensively cleaned to remove organic contaminants and gold oxide layer deposited during electrochemical cleaning as described previously18 with details provided in the ESI.† Gold electrodes were patterned by a mixed self-assembled monolayer (SAM), prepared from a mixture of two thiols, which is an effective way to control the density of functional ligands on surfaces (initially developed for immobilisation of lectins).17b A functional thiol (11-mercaptoundecanoic acid, MUA) was diluted by 6-mercaptohexanol (MH) by changing the amount of MUA in a mixture from 5% to 50%. In the next step a –COOH group of MUA was activated by the EDC–NHC mixture to form an active ester.17b Then, amine-terminated glycan (Fig. 1a and b) was added for covalent immobilisation of a glycan. Finally, the glycan biosensor was incubated with MAA (Fig. 1c) to find out which glycan density is optimal for binding of MAA. Faradaic EIS investigation described in the ESI,† showed that charge transfer resistance Rct, read as a diameter of a semicircle from a Nyquist plot (Fig. 2a), of the SAM was 42.1 kΩ and this value decreased to 17.6 kΩ, after EDC/NHS treatment due to an effective decrease of the negative charge on the SAM. The glycan immobilisation Rct increased to 36.4 kΩ, and further to the value of 55.5 kΩ after incubation of the glycan biosensor with MAA (770 pM) (Fig. 2a). The optimisation study aimed to find the optimal composition of a mixed SAM revealed that the glycan biosensor can detect 770 pM MAA with the highest response, when SAM was prepared from a thiol mixture containing 33.3% of MUA (Fig. 2b).
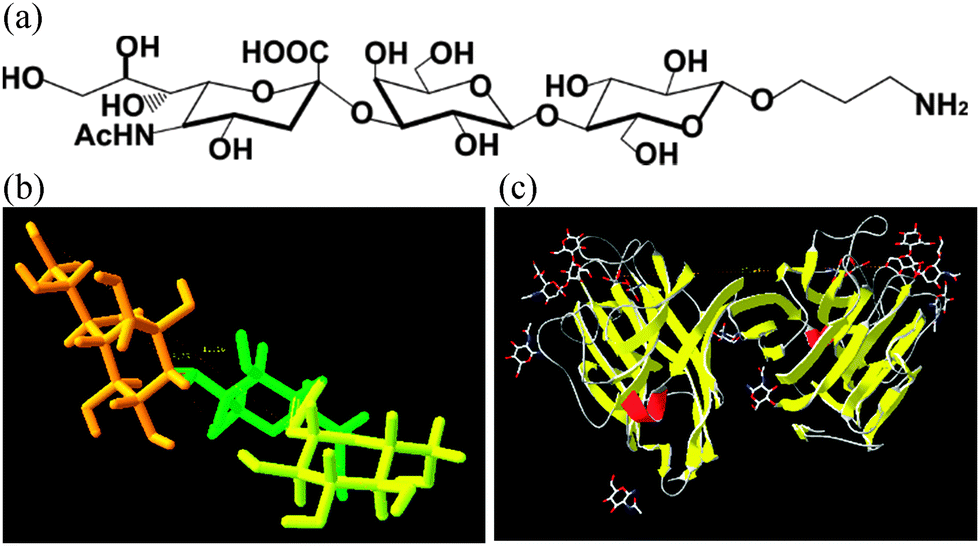 | ||
| Fig. 1 (a) Chemical structure of the amine glycan derivative of 2,3-sialyllactose with a formula Neu5Acα2-3Galβ1-4Glcβ-O-(CH2)3-NH2 applied for construction of a glycan biosensor, (b) a 3-D structure of 2,3-sialyllactose and (c) a structure of Maackia amurensis agglutinin (lectin) docked with 2,3-sialyllactose (pdb code: 1DBN). | ||
 | ||
| Fig. 2 (a) Impedimetric characterisation of the build-up process for preparation of the glycan biosensor with the formation of a mixed SAM composed of 11-meracptoundecanoic acid (MUA) and 6-mercaptohexanol (SAM), after activation by a coupling reagent (EDC/NHS), after covalent immobilisation of 2,3-sialyllactose (glycan) and after interaction with Maackia amurensis agglutinin (MAA lectin) and (b) optimisation of the MUA ratio within a mixed SAM for preparation of a glycan biosensor evaluated by MAA lectin interaction. | ||
Atomic force microscopy (AFM) analysis of the glycan modified surface showed the presence of individual features not seen on a SAM modified gold surface (Fig. 3) separated by an average distance of (2.7 ± 0.3) nm (Fig. S1, ESI†), which are attributed to individual glycan chains. The glycan patterned surface exhibited a mean square roughness Rq = 0.65 nm, the value of which is significantly larger compared to the plain mixed SAM with an Rq = 0.39 nm (Fig. 3). When the glycan surface was finally incubated with MAA Rq increased to the value of 6.45 nm, confirming the ability of a glycan bioreceptive layer to bind its analyte MAA (Fig. 3d). A difference in the topology between a glycan bound layer and plain SAM is clearer compared to a recently published study19 and allowed us to calculate glycan density as (22.7 ± 2.7) pmol cm−2 ((1.4 ± 0.2) × 1013 glycan molecules per cm2).
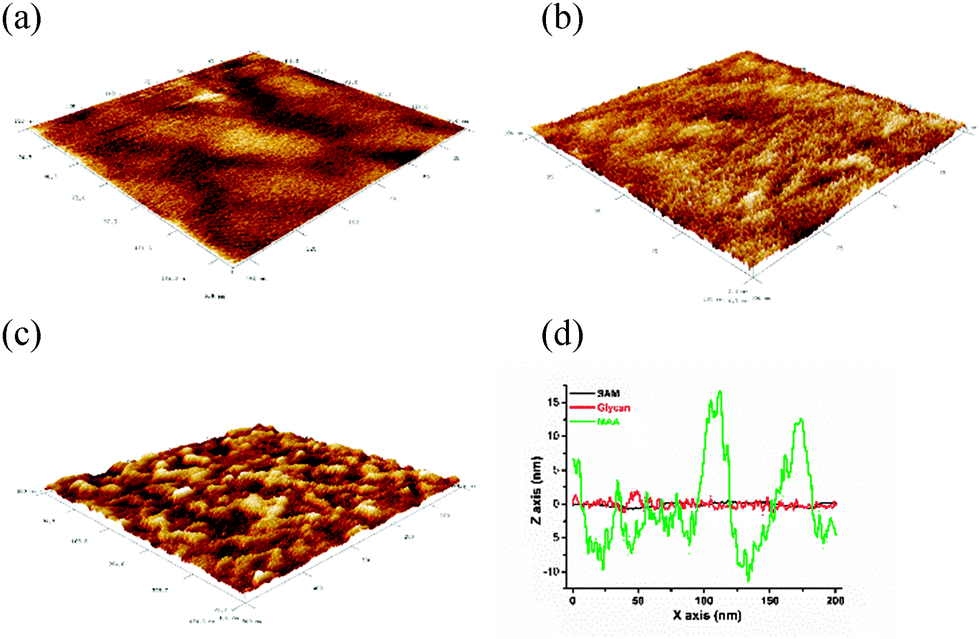 | ||
| Fig. 3 Atomic force microscopy (AFM) images of (a) mixed SAM composed of 11-meracptoundecanoic acid and 6-mercaptohexanol, (b) 2,3-sialyllactose covalently immobilised on SAM, (c) incubation of Maackia amurensis agglutinin (MAA) with a glycan surface and (d) AFM profile showing surface roughness for a mixed SAM (SAM), a glycan bound surface (glycan) and after incubation of the glycan interface with MAA lectin (MAA). | ||
When an independent method i.e. SPR was applied to calculate glycan density, the SPR response of 440 μRIU (Fig. S2, ESI†) corresponds to a glycan density of 61.8 pmol cm−2 (3.7 × 1013 glycan molecules per cm2). There are two facts worth mentioning regarding SPR assays: (1) a correlation factor of 1000 μRIU = 1 ng mm−2 provided by the manufacturer for proteins can be different for glycan molecules; and (2) a higher driving force for delivery of EDC/NHS or the glycan molecule was applied during the SPR experimental run under flow conditions compared to static immobilisation of glycan on an AFM gold chip. Nevertheless, both values of glycan density obtained here fall well within the range of 7.2 × 1012 cm−2–5.2 × 1013 cm−2 obtained in a previous study for diluted mannosyl moieties.19 Taking into account the diameter of 2,3-sialyllactose (Fig. S3, ESI†), the theoretical full glycan monolayer should have a density of 1.4 × 1014 cm−2, in agreement with the experimental value of 1.2 × 1014 cm−2 obtained for mannose,19 suggesting that optimal glycan density for lectin binding on the surface in this study is close to 10% of theoretical coverage. Since the distance between the binding sites in MAA is approx. 5.0 nm (Fig. S4, ESI†), the highest MAA binding at optimal glycan density (glycan separation of 2.7 nm) does not allow bivalent interaction. This is in agreement with a previous study, which concluded that the highest lectin binding was achieved on glycan surfaces allowing monovalent interactions.19 From the structure of hemagglutinin H1N1 (a protein from influenza viruses) (Fig. S5, ESI†) a protein diameter with a binding site bound to the glycan modified surface of ∼3.5 nm could be read, a value smaller than for MAA (∼5.3 nm, Fig. S4, ESI†) and thus binding of HA to the glycan modified surface should be less constrained compared to monovalent MAA binding.
The glycan biosensor developed was then calibrated with two different glycan binding proteins – lectins (MAA and Datura stramonium lectin – DSL) and hemagglutinins H1N1 and H5N1. Results indicated that the glycan biosensor containing sialyllactose with α-2,3-bound sialic acid specifically interact with MAA, which is preferentially binding glycans with terminal α-2,3-bound sialic acid. The glycan biosensor could detect this lectin in the concentration window of 8 aM – 80 nM (i.e. 10 orders of magnitude) (Fig. 4a). DSL binding to β-1,4-linked N-acetylglucosamine oligomers, being a negative control, did not exhibit any significant binding until 8 pM and only then the response toward 2,3-sialyllactose modified interface increased (Fig. 4a). Serum albumins are quite often applied as probes to investigate non-specific interactions with bioreceptive layers,20 this is why an additional control experiment was performed by the investigation of binding of human serum albumin (HSA) to the glycan modified surface (HSA with an isoelectric point pI = 4.421 comparable to pI = 4.7 for MAA lectin22) without any significant binding in a concentration window of 140 aM – 14 nM (Fig. 4a), suggesting a specific biorecognition of surface confined 2,3-sialyllactose by MAA. H5N1 could be detected in a concentration window of 140 aM – 14 nM (i.e. 9 orders of magnitude, if a background value of ΔRct (%) at a concentration of 1.4 fM is not taken into account), while the response towards H1N1 was negligible up to 14 fM and only then linearly increased up to a concentration of 14 nM (6 orders of magnitude) (Fig. 4b). The lowest detection limit for determination of lectin with an EIS-based glycan biosensor so far is 0.12 nM.14c A field-effect transistor (FET)-based device relying on silicon nanowires could detect lectins down to 2 nM11b or 1 fM.12 Other techniques exhibit detection limits for lectins comparable to glycan biochips.6 So far hemagglutinins have not been determined using EIS-based glycan biosensors, but FET-based analysis was successfully applied. In this particular case H1N1 and H5N1 could be detected in a concentration window of 50 aM – 5 nM and 500 zM – 500 pM, respectively.13
 | ||
| Fig. 4 Calibration of the glycan biosensor by (a) two different lectins Maackia amurensis agglutinin (MAA), Datura stramonium lectin (DSL) and human serum albumin (HSA); and (b) two different influenza hemagglutinins H5N1 (black) and H1N1 (red). | ||
It is quite interesting that an interface modified by α-2,3-linked sialic acid in sialyllactose bind in our study besides H5N1 also H1N1, which should recognise α-2,6-linked sialic acid in sialyllactose.23 Hemagglutinins from avian influenza viruses (i.e. H5N1) preferentially bind to α-2,3-linked sialic acid terminated glycans, while hemagglutinins from human influenza viruses (i.e. H1N1) preferentially bind to α-2,6-linked sialic acid terminated glycans, but such interactions are more complex than previously thought.24 Other attributes such as glycan topology (i.e. cone-like vs. umbrella-like structure), the effect of neighbouring glycan and proper spacing of minimal glycan epitopes can play significant roles in the viral recognition of host cellular glycans via hemagglutinins.23b,25 Interrogation of glycan biochips with hemagglutinins isolated from different influenza viruses revealed that some human H1N1 hemagglutinins can bind to α-2,3-linked sialic acid terminated glycans and avian H5N1 hemagglutinins can bind to α-2,6-linked sialic acid terminated glycans.26 Another recent study showed that H1N1 hemagglutinin isolated from the viral strain New Caledonia 20/99 (used in this study) not only binds effectively to various glycans with α-2,3-linked sialic acid termination, but more interestingly enhancement of binding was observed, when galactose next to terminal sialic acid was modified by a negatively charged sulfo group.27 The most recent study showed that the binding preference of intact H1N1 influenza viruses (A/Puerto Rico/8/34) to 2,3-sialyllactose or 2,6-sialyllactose modified glycan microarrays exhibits binding specificity depending on glycan density i.e. at low printed glycan density (from 0.1 mM glycan solution) the H1N1 influenza virus exhibits preferential binding to 2,6-sialyllactose, while at higher printed glycan density (1–10 mM of glycan in solution), the same virus preferentially binds to 2,3-sialyllactose.28 This study is in agreement with the conclusion of another study that alternative moieties including sulfonated ones may be important in the recognition of host glycan receptors by influenza viruses.29 Thus, a more systematic investigation of glycan density, topology and the presence of various charged species within glycan interfaces on binding of viral agglutinins is needed for the determination of various different viral strains. Impedimetric glycan biosensors based on gold surfaces patterned by thiols terminated in numerous functionalities in a flexible way can shed light on interaction between glycan epitopes and viral proteins.14b Moreover, impedimetric glycan biosensors could be useful for analysis of activities of glycan processing enzymes, not yet performed.30
We have developed the most sensitive impedimetric glycan biosensor for analysis of lectins with the detection limit 7 orders of magnitude lower compared to the best described glycan biosensor. The glycan biosensor described here is the first impedimetric glycan biosensor for analysis of hemagglutinins with detection limits down to the attomolar level with sensitivity similar to the best glycan biosensor based on FET described so far.13 Moreover, this study provides ultrasharp visualisation of glycans immobilised on surfaces by AFM.
Funding from the Slovak research and development agency APVV 0282-11 and VEGA 2/0162/14 is acknowledged. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Program (FP/2007-2013)/ERC Grant Agreement no. 311532.
Notes and references
- (a) N. C. Reichardt, M. Martín-Lomas and S. Penadés, Chem. Soc. Rev., 2013, 42, 4358 RSC; (b) S. Park, J. C. Gildersleeve, O. Blixt and I. Shin, Chem. Soc. Rev., 2013, 42, 4310 RSC; (c) A. Varki, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 2009 Search PubMed; (d) S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525 CrossRef CAS PubMed.
- (a) W. R. Alley, B. F. Mann and M. V. Novotny, Chem. Rev., 2013, 113, 2668 CrossRef CAS PubMed; (b) D. R. Burton, P. Poignard, R. L. Stanfield and I. A. Wilson, Science, 2012, 337, 183 CrossRef CAS PubMed; (c) M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 37, DOI:10.1126/science.1235681; (d) A. Rouvinski, P. Guardado-Calvo, G. Barba-Spaeth, S. Duquerroy, M.-C. Vaney, C. M. Kikuti, M. E. N. Sanchez, W. Dejnirattisai, W. Wongwiwat, A. Haouz, C. Girard-Blanc, S. Petres, W. E. Shepard, P. Despres, F. Arenzana-Seisdedos, P. Dussart, J. Mongkolsapaya, G. R. Screaton and F. A. Rey, Nature, 2015 DOI:10.1038/nature14130; (e) M. S. Macauley, B. M. Arlian, C. D. Rillahan, P.-C. Pang, N. Bortell, M. C. G. Marcondes, S. M. Haslam, A. Dell and J. C. Paulson, J. Biol. Chem., 2014, 289, 35149 CrossRef CAS PubMed; (f) S. Bournazos, F. Klein, J. Pietzsch, M. S. Seaman, M. C. Nussenzweig and J. V. Ravetch, Cell, 2014, 158, 1243 CrossRef CAS PubMed; (g) R. A. Medina and A. García-Sastre, Nat. Rev. Microbiol., 2011, 9, 590 CrossRef CAS PubMed.
- (a) K. Drickamer and M. E. Taylor, Genome Biol., 2002, 3, 1034 CrossRef; (b) S. Fukui, T. Feizi, C. Galustian, A. M. Lawson and W. Chai, Nat. Biotechnol., 2002, 20, 1011 CrossRef CAS PubMed; (c) B. T. Houseman and M. Mrksich, Chem. Biol., 2002, 9, 443 CrossRef CAS; (d) S. Park and I. Shin, Angew. Chem., Int. Ed., 2002, 41, 3180 CrossRef CAS; (e) D. Wang, S. Liu, B. J. Trummer, C. Deng and A. Wang, Nat. Biotechnol., 2002, 20, 275 CrossRef CAS PubMed.
- (a) C. M. Arthur, R. D. Cummings and S. R. Stowell, Curr. Opin. Chem. Biol., 2014, 18, 55 CrossRef CAS PubMed; (b) O. Blixt and U. Westerlind, Curr. Opin. Chem. Biol., 2014, 18, 62 CrossRef CAS PubMed; (c) R. D. Cummings and J. M. Pierce, Chem. Biol., 2014, 21, 1 CrossRef CAS PubMed; (d) A. Geissner, C. Anish and P. H. Seeberger, Curr. Opin. Chem. Biol., 2014, 18, 38 CrossRef CAS PubMed; (e) S. A. Svarovsky and L. Joshi, Anal. Methods, 2014, 6, 3918 RSC; (f) J. E. Stencel-Baerenwald, K. Reiss, D. M. Reiter, T. Stehle and T. S. Dermody, Nat. Rev. Microbiol., 2014, 12, 739 CrossRef CAS PubMed; (g) N. Laurent, J. Voglmeir and S. L. Flitsch, Chem. Commun., 2008, 4400 RSC.
- (a) P. Gemeiner, D. Mislovičová, J. Tkáč, J. Švitel, V. Pätoprstý, E. Hrabárová, G. Kogan and T. Kožár, Biotechnol. Adv., 2009, 27, 1 CrossRef CAS PubMed; (b) J. Katrlík, J. Švitel, P. Gemeiner, T. Kožár and J. Tkac, Med. Res. Rev., 2010, 30, 394 Search PubMed.
- A. Hushegyi and J. Tkac, Anal. Methods, 2014, 6, 6610 RSC.
- (a) T. Bertók, J. Katrlík, P. Gemeiner and J. Tkac, Microchim. Acta, 2013, 180, 1 CrossRef; (b) J. Q. Gerlach, S. Cunningham, M. Kane and L. Joshi, Biochem. Soc. Trans., 2010, 38, 1333 CrossRef CAS PubMed; (c) B. Mu, J. Zhang, T. P. McNicholas, N. F. Reuel, S. Kruss and M. S. Strano, Acc. Chem. Res., 2014, 47, 979 CrossRef CAS PubMed; (d) N. F. Reuel, B. Grassbaugh, S. Kruss, J. Z. Mundy, C. Opel, A. O. Ogunniyi, K. Egodage, R. Wahl, B. Helk, J. Zhang, Z. I. Kalcioglu, K. Tvrdy, D. O. Bellisario, B. Mu, S. S. Blake, K. J. Van Vliet, J. C. Love, K. D. Wittrup and M. S. Strano, ACS Nano, 2013, 7, 7472 CrossRef CAS PubMed; (e) N. F. Reuel, B. Mu, J. Zhang, A. Hinckley and M. S. Strano, Chem. Soc. Rev., 2012, 41, 5744 RSC; (f) J. Tkac, T. Bertok, J. Nahalka and P. Gemeiner, Methods Mol. Biol., 2014, 1200, 421 CrossRef.
- L'. Kluková, T. Bertók, P. Kasák and J. Tkac, Anal. Methods, 2014, 6, 4922 RSC.
- (a) E. Palecek and M. Bartosik, Chem. Rev., 2012, 112, 3427 CrossRef CAS PubMed; (b) E. Paleček, J. Tkáč, M. Bartošík, T. Bertók, V. Ostatná and J. Paleček, Chem. Rev., 2015, 115, 2045 CrossRef PubMed.
- (a) X.-P. He, X.-W. Wang, X.-P. Jin, H. Zhou, X.-X. Shi, G.-R. Chen and Y.-T. Long, J. Am. Chem. Soc., 2011, 133, 3649 CrossRef CAS PubMed; (b) H. Zeng, J. Yu, Y. Jiang and X. Zeng, Biosens. Bioelectron., 2014, 55, 157 CrossRef CAS PubMed.
- (a) Y. Chen, H. Vedala, G. P. Kotchey, A. Audfray, S. Cecioni, A. Imberty, S. Vidal and A. Star, ACS Nano, 2012, 6, 760 CrossRef CAS PubMed; (b) H. Vedala, Y. Chen, S. Cecioni, A. Imberty, S. b. Vidal and A. Star, Nano Lett., 2010, 11, 170 CrossRef PubMed; (c) M. Ali, S. Nasir, P. Ramirez, J. Cervera, S. Mafe and W. Ensinger, J. Phys. Chem. C, 2013, 117, 18234 CrossRef CAS.
- G.-J. Zhang, M. J. Huang, J. A. J. Ang, Q. Yao and Y. Ning, Anal. Chem., 2013, 85, 4392 CrossRef CAS PubMed.
- S. Hideshima, H. Hinou, D. Ebihara, R. Sato, S. Kuroiwa, T. Nakanishi, S.-I. Nishimura and T. Osaka, Anal. Chem., 2013, 85, 5641 CrossRef CAS PubMed.
- (a) S. Szunerits, J. Niedziǒłka-Jönsson, R. Boukherroub, P. Woisel, J.-S. b. Baumann and A. Siriwardena, Anal. Chem., 2010, 82, 8203 CrossRef CAS PubMed; (b) O. A. Loaiza, P. J. Lamas-Ardisana, E. Jubete, E. Ochoteco, I. Loinaz, G. n. Cabañero, I. García and S. Penadés, Anal. Chem., 2011, 83, 2987 CrossRef CAS PubMed; (c) Z. Wang, C. Sun, G. Vegesna, H. Liu, Y. Liu, J. Li and X. Zeng, Biosens. Bioelectron., 2013, 46, 183 CrossRef CAS PubMed.
- (a) N. Maalouli, A. Barras, A. Siriwardena, M. Bouazaoui, R. Boukherroub and S. Szunerits, Analyst, 2013, 138, 805 RSC; (b) G. Bellapadrona, A. B. Tesler, D. Grunstein, L. H. Hossain, R. Kikkeri, P. H. Seeberger, A. Vaskevich and I. Rubinstein, Anal. Chem., 2012, 84, 232 CrossRef CAS PubMed; (c) T. Nagatsuka, H. Uzawa, K. Sato, S. Kondo, M. Izumi, K. Yokoyama, I. Ohsawa, Y. Seto, P. Neri, H. Mori, Y. Nishida, M. Saito and E. Tamiya, ACS Appl. Mater. Interfaces, 2013, 5, 4173 CAS.
- (a) K. Gruber, T. Horlacher, R. Castelli, A. Mader, P. H. Seeberger and B. A. Hermann, ACS Nano, 2011, 5, 3670 CrossRef CAS PubMed; (b) K. Gruber, B. Hermann and P. Seeberger, Angew. Chem., Int. Ed., 2011, 50, A46 CrossRef PubMed.
- (a) T. Bertok, P. Gemeiner, M. Mikula, P. Gemeiner and J. Tkac, Microchim. Acta, 2013, 180, 151 CrossRef CAS; (b) T. Bertok, L. Klukova, A. Sediva, P. Kasák, V. Semak, M. Micusik, M. Omastova, L. Chovanová, M. Vlček, R. Imrich, A. Vikartovska and J. Tkac, Anal. Chem., 2013, 85, 7324 CrossRef CAS PubMed; (c) T. Bertok, A. Sediva, J. Katrlik, P. Gemeiner, M. Mikula, M. Nosko and J. Tkac, Talanta, 2013, 108, 11 CrossRef CAS PubMed; (d) T. Bertok, A. Sediva, A. Vikartovska and J. Tkac, Int. J. Electrochem. Sci., 2014, 9, 890 Search PubMed; (e) L. Klukova, T. Bertok, M. Petrikova, A. Sediva, D. Mislovicova, J. Katrlik, A. Vikartovska, J. Filip, P. Kasak, A. Andicsová-Eckstein, J. Mosnáček, J. Lukáč, J. Rovenský, R. Imrich and J. Tkac, Anal. Chim. Acta, 2015, 853, 555 CrossRef CAS PubMed.
- J. Tkac and J. J. Davis, J. Electroanal. Chem., 2008, 621, 117 CrossRef CAS PubMed.
- J. Yang, J.-N. Chazalviel, A. Siriwardena, R. Boukherroub, F. Ozanam, S. Szunerits and A. C. Gouget-Laemmel, Anal. Chem., 2014, 86, 10340 CrossRef CAS PubMed.
- J. J. Davis, J. Tkac, R. Humphreys, A. T. Buxton, T. A. Lee and P. Ko Ferrigno, Anal. Chem., 2009, 81, 3314 CrossRef CAS PubMed.
- R. Liu, R. Pidikiti, C.-E. Ha, C. E. Petersen, N. V. Bhagavan and R. G. Eckenhoff, J. Biol. Chem., 2002, 277, 36373 CrossRef CAS PubMed.
- http://https://www.vectorlabs.com/data/brochures/K4-K7.pdf downloaded on 09.03.2015.
- (a) C. M. Nycholat, R. McBride, D. C. Ekiert, R. Xu, J. Rangarajan, W. Peng, N. Razi, M. Gilbert, W. Wakarchuk, I. A. Wilson and J. C. Paulson, Angew. Chem., Int. Ed., 2012, 51, 4860 CrossRef CAS PubMed; (b) Y. Watanabe, M. S. Ibrahim, Y. Suzuki and K. Ikuta, Trends Microbiol., 2012, 20, 11 CrossRef CAS PubMed.
- H. Shelton, G. Ayora-Talavera, J. Ren, S. Loureiro, R. J. Pickles, W. S. Barclay and I. M. Jones, J. Virol., 2011, 85, 1875 CrossRef CAS PubMed.
- (a) C. A. Bewley, Nat. Biotechnol., 2008, 26, 60 CrossRef CAS PubMed; (b) Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G.-J. Boons, Science, 2013, 341, 379 CrossRef CAS PubMed.
- A. Chandrasekaran, A. Srinivasan, R. Raman, K. Viswanathan, S. Raguram, T. M. Tumpey, V. Sasisekharan and R. Sasisekharan, Nat. Biotechnol., 2008, 26, 107 CrossRef CAS PubMed.
- K. C. Bradley, C. A. Jones, S. M. Tompkins, R. A. Tripp, R. J. Russell, M. R. Gramer, J. Heimburg-Molinaro, D. F. Smith, R. D. Cummings and D. A. Steinhauer, Virology, 2011, 413, 169 CrossRef CAS PubMed.
- M. L. Huang, M. Cohen, C. J. Fisher, R. T. Schooley, P. Gagneux and K. Godula, Chem. Commun., 2015, 51, 5326 RSC.
- L. Glaser, G. Conenello, J. Paulson and P. Palese, Virus Res., 2007, 126, 9 CrossRef CAS PubMed.
- (a) C. J. Gray, M. J. Weissenborn, C. E. Eyers and S. L. Flitsch, Chem. Soc. Rev., 2013, 42, 6378 RSC; (b) P. J. Halling, R. V. Ulijn and S. L. Flitsch, Curr. Opin. Biotechnol., 2005, 16, 385 CrossRef CAS PubMed; (c) N. Laurent, R. Haddoub and S. L. Flitsch, Trends Biotechnol., 2008, 26, 328 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A full description of experimental conditions, a detailed topological profile of the glycan patterned surface (Fig. S1), the SPR sensorgram (Fig. S2), structures of 2,3-sialyllactose glycan (Fig. S3), Maackia amurensis agglutinin docked with 2,3-sialyllactose (Fig. S4) and H1N1 hemagglutinin docked with 2,3-sialyllactose (Fig. S5). See DOI: 10.1039/c5cc00922g |
| This journal is © The Royal Society of Chemistry 2015 |