
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-driven self-assembly: the case of redox-active discrete architectures
V.
Croué
,
S.
Goeb
* and
M.
Sallé
*
Université d'Angers, CNRS UMR 6200, Laboratoire MOLTECH-Anjou, 2 bd Lavoisier, 49045 ANGERS cedex, France. E-mail: sebastien.goeb@univ-angers.fr; marc.salle@univ-angers.fr; Fax: +33 241 735 405; Tel: +33 241 735 439
First published on 20th March 2015
Abstract
The metal-driven self-assembly strategy is well-established for the construction of discrete architectures featuring a cavity. The resulting molecular rings and cages are potentially useful as hosts for complementary guests. Recent years have seen growing interest in the introduction of given functionalities, including redox properties which, among others, allow modulating the ionic charge of the cavity. Depending on which subunit is electroactive, various situations can be encountered, with a global redox activity which is ligand- and/or metal-centered and which involves or not electronic interactions between the constituting units. In this feature article, we propose to survey those different situations by exploring some recent examples of the growing family of redox-active self-assembled rings and cages.
 From left to right: V. Croué, M. Sallé and S. Goeb | Vincent Croué (left) was born in Angers (France) in 1989. He obtained his Master's Degree from the University of Angers in Molecular Engineering and Nanotechnologies in 2012 working on TTF derivatives for metal-directed self-assembly. Since then, he has been a PhD Student in Organic Chemistry in Prof. Marc Sallé‘s team. His research interests mainly concern discrete redox active self-assembled cages incorporating extended-TTF units. |
Dr Sébastien Goeb (right) was born in Schiltigheim (France) in 1979. He studied chemistry at the University of Strasbourg and obtained his PhD degree in Organic Chemistry in 2006 under the supervision of Drs Raymond Ziessel and Antoinette De Nicola. He then carried out two successive one year postdoctoral stays respectively with Prof. Felix N. Castellano at the University of Bowling Green (USA) and with Dr Jean-Luc Parrain at the Institut des Sciences Moléculaires de Marseille (France). He became Chargé de Recherche CNRS in the laboratory MOLTECH-Anjou in 2008. His research interests focus on the organic chemistry of photo- and/or redox-active supramolecular discrete architectures. |
Prof. Marc Sallé (middle) was born in Toulon (France) in 1963. He studied chemistry at the Ecole Nationale Supérieure de Chimie de Rennes (ENSCR) and at the University of Paris VI. He received his PhD degree in Organic Chemistry from the University of Angers in 1991 under the supervision of Professor Alain Gorgues. For this work he was awarded the Dina Surdin prize of year 1992. After a postdoctoral stay with Prof. Martin R. Bryce at the University of Durham (UK), he became Maître de Conférences at the University of Angers, where he was promoted to a professor in 1998. He has been a junior member of the Institut Universitaire de France (IUF). His research interests concern the organic chemistry of molecular materials and supramolecular chemistry associated with electro(photo)-active molecular systems. Prof. Marc Sallé is currently the director of the Laboratory MOLTECH-Anjou (Angers, France). |
Introduction
The self-assembly methodology, more particularly when assisted by metals, constitutes a very efficient synthetic approach1 to obtain more and more sophisticated macrocyclic structures, otherwise very challenging to prepare through classical step-by-step covalent synthesis. In order to target applications,2 the resulting discrete structures, be they polygons or cages, may integrate various functionalities. In particular, there is great interest in responsive coordination-driven self-assembled architectures currently, which can be constructed through applying external stimuli, as for example irradiation with light.3 On these grounds, there is growing interest in assemblies responsive to a redox-stimulus and the number of electro-active metalla-rings/cages has been increasing drastically in the very recent years. The use of redox-active precursors in the self-assembly process can in principle produce discrete metalla-supramolecular assemblies which are redox-active. Various situations can be encountered, e.g. electroactivity which is brought about either by the organic ligand part (naked or appended with a redox-active unit), by the metal or by a co-ligand. Moreover, through-space or through-bond interactions between the different components can potentially occur. An immediate consequence with such a variety of situations lies on the possibility to switch, providing a suitable design, the ionic charge surrounding the cavity, by a simple tuning of the applied redox potential. It is worth noting that in addition to being redox-active, such systems intrinsically present a cavity which can therefore be used to bind a complementary guest.4 This combination of properties is of particular importance for applications such as sensing or controlling encapsulation/delivery processes of various guests. Finally, the supramolecular assembly approach may constitute a promising future strategy to address the design of organic materials (e.g. organic photovoltaics and molecular electronic devices). Indeed, mastering the geometry of multicomponent redox-active systems offers a unique opportunity to fine-tune electronic interactions within the material,5 which includes control over through-space electron hopping or delocalization and which is of prime importance for optimizing electron transport in organic materials.Altogether, the diversity of the redox-active structures which can be attained by the metal-driven self-assembly methodology opens fascinating perspectives for various research topics, ranging from drug delivery, guest sensing, catalysis or even molecular electronics. We propose in this feature article to cover the most recent advances in this field.
Metalla-rings/cages involving electron-poor ligands
2,4,6-Tris(4-pyridyl)-1,3,5-triazine-based cages
A prototypal self-assembled host system corresponds to the M6L4 cage 1 (Fig. 1a) extensively developed by M. Fujita and colleagues, which integrates triazine panels (2,4,6-tris(4-pyridyl)-1,3,5-triazine) and square-planar Pd(II) or Pt(II) as metal corners.1u,6 Because of the three coordinated pyridine moieties, the tridentate triazine core is highly electron-poor. The redox properties of this assembly have been studied in order to address the photoinduced host–guest electron-transfer occurring between panels and alkanes encapsulated within the cavity.7 In particular, this cage is able to form a clathrate complex with four adamantane molecules (1a⊃(adamentane)4). Upon irradiation, the colourless solution of 1a⊃(adamentane)4 turns blue, indicating the generation of radical species. The photolysis of the complex produces two oxidized derivatives (1-adamantyl peroxide and 1-adamentanol) in a total yield of ∼24% which indicates that only one among the four adamentanes has reacted.8 The proposed reaction mechanism involves the generation of the host anion-radical and guest cation-radical via a guest-to-host photoinduced electron transfer (PET), with only one triazine panel involved. The cyclic voltammetry of cage 1b was investigated and exhibits two redox systems (Fig. 1b). A reversible redox wave corresponding to a one-electron process, as confirmed by coulometric experiments and as supported by DFT calculations, is observed at E = −1.02 V vs. Ag/Ag+, followed by pseudoreversible redox waves from E = −1.40 to −2.00 V, assigned to three electrons in total. It is worth noting that in the case of cages 1a,b, the four triazine panels are not simultaneously reduced which is due to the electronic communication occurring between the side walls of the cage through the Pt ion centers. | ||
| Fig. 1 (a) Structure of self-assembled molecular hosts 1a–b; (b) cyclic voltammogram of 1b: C = 0.5 mM in CH3CN (Bu4NPF6 (50 mM)), scan rate 100 mV s−1, (c) structure of AdPt. | ||
Such a propensity of cage 1b to bind adamantane was very recently explored by J. T. Lippard and coworkers for drug delivery purposes.9 The authors developed a well-defined Pt drug delivery system based on the encapsulation of four adamantyl groups inside the cavity, each of them being appended with a Pt(IV) prodrug unit (AdPt). The cyclic voltammetry of assembly 1b·AdPt4 in water shows cathodic signals assigned to the triazine core (1b), as well as a less negative one corresponding to the irreversible reduction of Pt(IV). This scheme is confirmed by a chemical reduction of 1b·AdPt4 with ascorbic acid, which gives rise to a cisplatin release, beside 1-adamantylamine and succinic acid, providing therefore a proof of concept for a controlled delivery of cisplatin through reduction.
Finally, one can remind that tris(pyridyl)-triazine cages 1a,b present a good ability to encapsulate a wide variety of organic guests.1u This property was explored in the peculiar case of redox-active guests such as organic radicals of the nitronyl nitroxide type10 or ferrocene derivatives,11 whose redox properties are significantly modified upon encapsulation.
Rhenium-based rings and cages
The redox properties of the same tridentate triazine panel, in association with indigo anions and Re(I), were recently studied by C.-Y. Lin, K.-L. Lu and coworkers in the case of the neutral self-assembled triangular metalloprism 2 (Fig. 2a), which exhibits strong NIR absorption.12 The presence of electron-accepting indigo and triazine units in the same structure makes this metalloprism highly electron-poor and easy to reduce. Actually, 2 can be electrochemically switched to several polyanionic states through stepwise reduction processes involving up to eight electrons, giving rise to an efficient control over the corresponding optical properties. Four reversible reduction waves are observed from cyclic voltammetry experiments (Fig. 2b). Remarkably, this study coupled with EPR analysis and DFT calculations indicates that the stepwise reduction sequence is essentially ligand-centered. The first redox system at E = −0.29 V vs. Ag/AgNO3 is ascribed to the simultaneous one-electron reduction of three indigo ligands giving rise to 23˙−, the latter species corresponding to a metal-bound indigo radical anion exhibiting a small dπ-Re-indigo orbital mixing. The second and third reductions (E = −0.73 and −0.98 V) are tentatively assigned to one electron reductions of each triazine ligand, which is supported by EPR experiments. The splitting of these two reduction waves may be ascribed to a stabilization of a mixed-valence state between the facing tris(pyridyl)triazine units,13 a behavior which is not observed between the indigo pillars which are distant from each other. The fourth reduction process (E = −1.23 V) corresponds to the second one-step three-electron-reduction process at the indigo-pillared ligands. Interestingly, the metalloprism 2 can be also reversibly oxidized through a three-electron process, corresponding to the oxidation of each indigo dianion. It appears from these data that the structure 2 behaves as an electron reservoir endowed with programmed and well-defined redox-properties.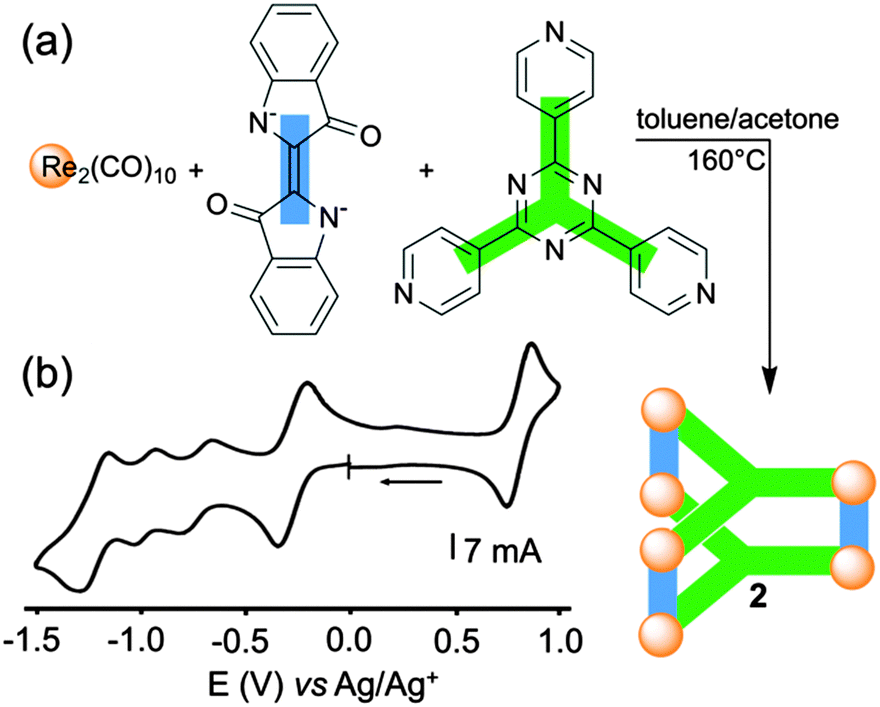 | ||
| Fig. 2 (a) Orthogonal-bonding approach for the preparation of metalloprism 2; (b) cyclic voltammogram of 2: C = 1 mM in THF (Bu4NPF6 (0.1 M)), scan rate 100 mV s−1. | ||
Another rhenium-based family of redox-active polygons corresponds to rectangles 3a–f depicted by J. T. Hupp and coworkers (Fig. 3a).14 The latter incorporate four Re(I) corners which are organized into two pairs by doubly chelating planar 2,2′-bisbenzimidazolate ligands and which are bridged by rigid or semirigid ditopic symmetrical (LL) ligands. These systems were designed to study the balance between intramolecular through-space electronic communication (between facing LL ligands) vs. coordination-bond-mediated interactions. Spectroelectrochemical studies conducted at the −1 charge state of the rectangles (i.e. mixed-valence state with respect to the ligands), demonstrated that those systems are dominated by electronic interactions of the former type. A cyclic voltammetry study of rectangles 3a–f was carried out and shows reversible reduction waves that are assigned to stepwise one-electron reductions of the symmetrical LL ligands within the rectangle (for a representative example, see the case of compound 3d, Fig. 3b). No oxidative wave is observed and the reduction potentials appear substantially shifted to less negative potentials in comparison with free ligands, which is due to the electrostatic stabilization promoted by the rhenium cations over the negative reduced forms of the ligands.
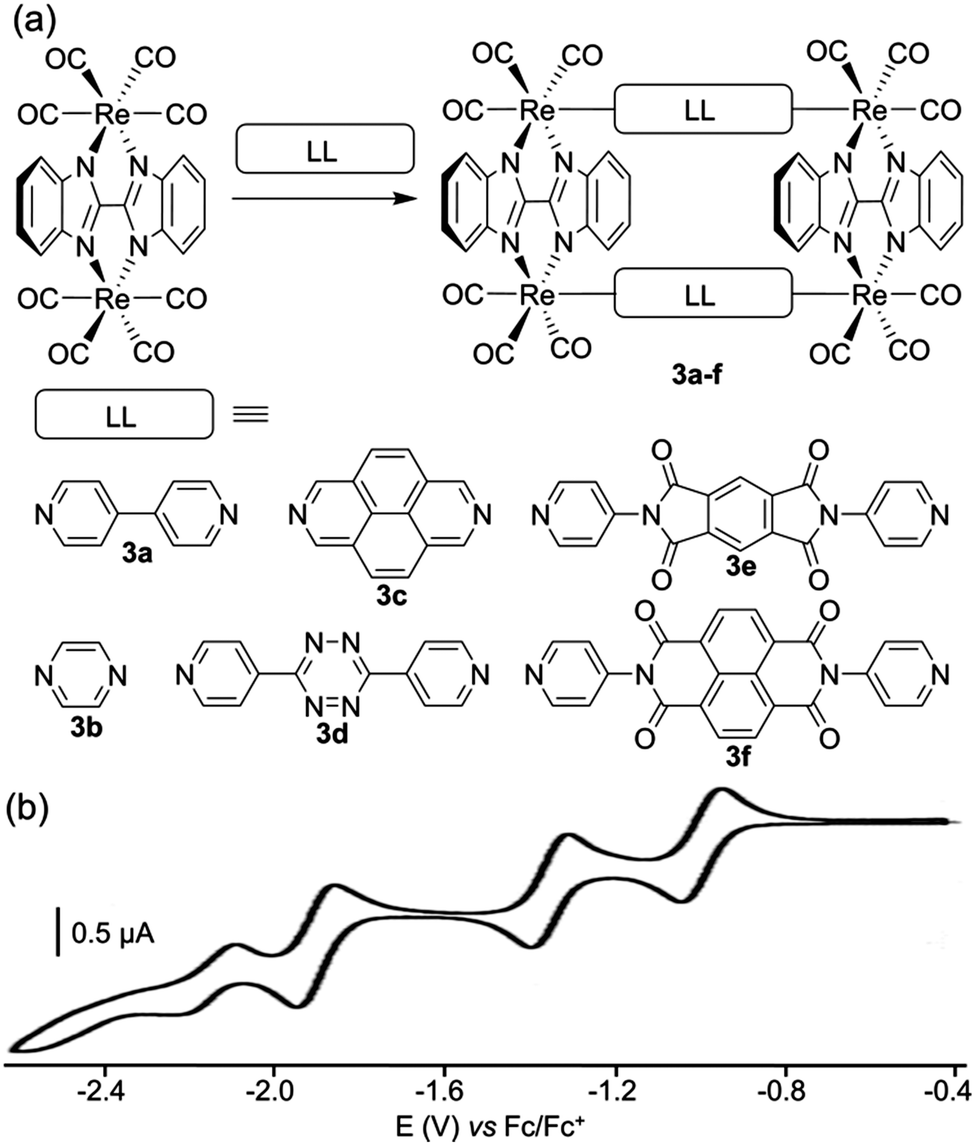 | ||
| Fig. 3 (a) Rhenium-based molecular rectangles 3a–f; (b) cyclic voltammogram of 3d in THF (Bu4NPF6 (0.1 M)). | ||
Perylene bisimide-based rings and cages
F. Würthner and colleagues have developed a series of self-assembled discrete architectures based on the photo- and electro-active perylene bisimide (PBI) unit. A prototypal example corresponds to metalla-square 4,15 constructed from a tetraaryl-substituted N,N′-4-pyridyl-substituted PBI ligand and a square planar Pt(II) complex (Fig. 4a). The UV/Vis and fluorescent properties of square 4 are not significantly changed compared to those of the PBI-ligand precursor, which is explained by the lack of electronic communication through the imide substituent. The cyclic voltammetry of square 4 (Fig. 4b) reveals two successive reversible reduction waves at E = −1.01 and −1.14 V vs. Fc/Fc+, which are slightly shifted compared to the free ligand PBI-ligand precursor (E = −1.08 and −1.23 V respectively). They correspond to the successive radical anion and dianion formation for each perylene unit, which behave independently contrary to the above-presented triazine-based cages, which is also confirmed by the spectroelectrochemical study conducted on 4 that exhibits redox processes which are exclusively ligand-centered. Finally, a reversible oxidation process is observed for the metalla-square 4 at E = +0.93 V vs. Fc/Fc+. Therefore, this electrochemical study demonstrate that the ionic state of square 4 can be in principle reversibly tuned from 0 to +12, in three steps of four electrons. Ultimately, this CV study also confirms the electrochemical inertness of the Pt(II) phosphane corner in the E = −1.30 V to +0.90 V range (vs. Fc/Fc+), this complex being one of the most commonly used for the construction of discrete self-assembled metalla-rings and cages via the coordination-driven approach. As demonstrated by the same group, the PBI panels can also be functionalized with pendant redox-active units such as in metalla-square 5 (Fig. 4a) decorated with 16 redox ferrocene moieties.16 Consequently, in addition to the typical reduction of each PBI unit to the radical anion and dianion states (E = −0.98 and −1.13 V vs. Fc/Fc+ respectively), compound 5 also exhibits oxidation of the ferrocenyl groups (Fig. 4c). Noteworthy, contrary to the reduction of the four PBI panels which occurs simultaneously in 5, the peripheral ferrocene units give rise to a splitting of the ferrocene/ferricinium redox wave, a consequence of through-space interactions occurring between ferrocene units in such a confined space.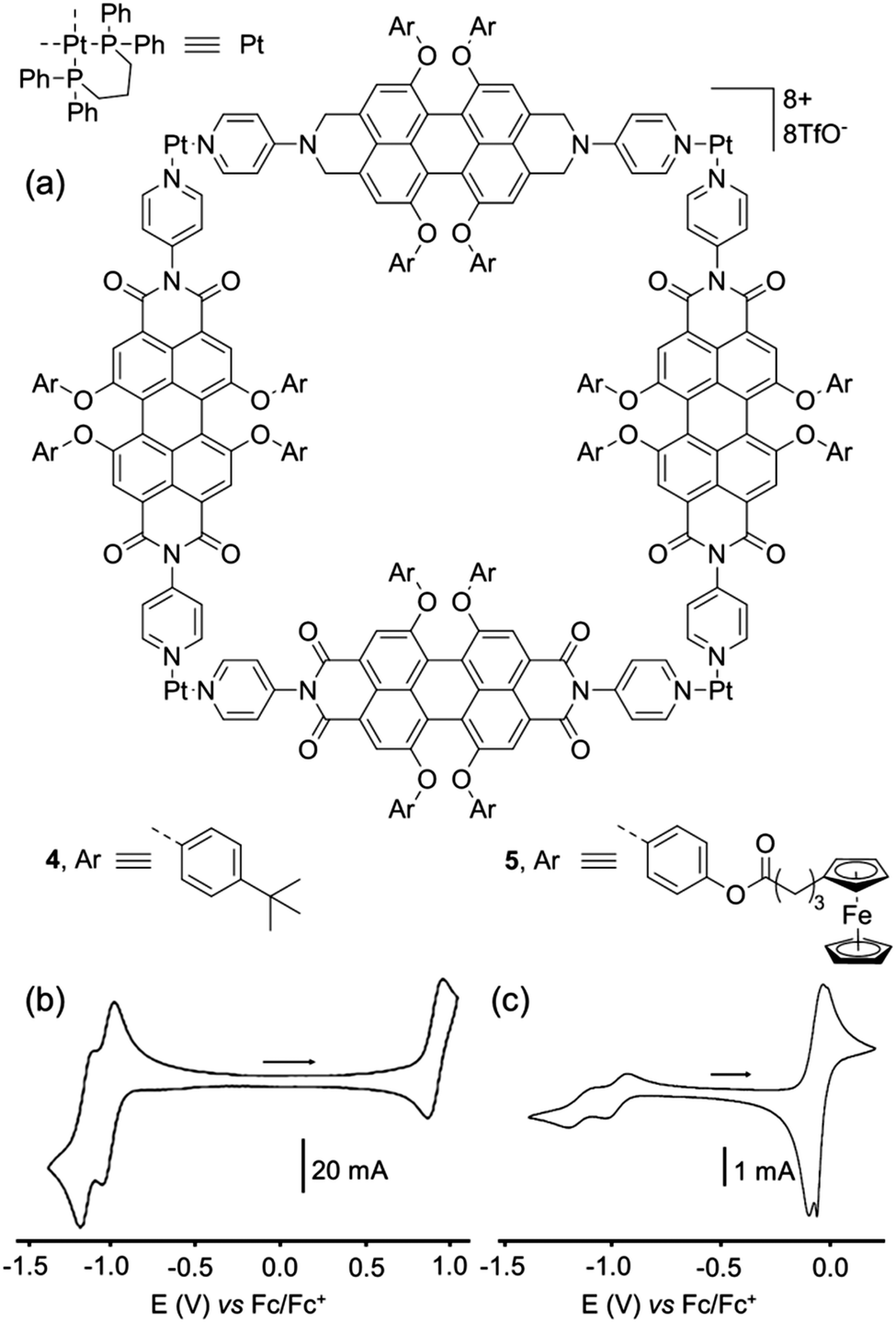 | ||
| Fig. 4 (a) Metallosupramolecular squares 4 and 5. Cyclic voltammogram of 4 (b) and 5 (c) in CH2Cl2 (Bu4NPF6 (0.1 M)), scan rate 100 mV s−1. | ||
Though presenting remarkable photophysical and electrochemical characteristics as well as defined cavities, no guest encapsulation from the above-described PBI-based metalla-macrocycles was mentioned by the authors. It is only very recently that the same group designed a robust PBI-based metalla-assembly presenting host–guest properties.17 The very large M4L6 tetrahedron 6 (Fig. 5a) is assembled from an octahedral Fe(II) ion and a PBI ligand bearing 2,2′-bipyridine units on the imide positions. The resulting Fe4(PBI)6 tetrahedron exhibits a particularly rich electrochemical activity manifested by multiple redox processes. As seen above, PBI derivatives are electron-accepting moieties that can be successively reduced into radical-anion and dianion species. This is observed in the CV of 6 which exhibits reversible redox waves at E = −0.94 and −1.11 V vs. Fc/Fc+ respectively (Fig. 5b). Moreover, assembly 6 incorporates four electroactive Fe(bpy)32+ centers which are known to undergo reversible electrochemical oxidation and reduction processes. The reductions of the bpy units on each metal center are not simultaneous and occur according to three successive one-electron reduction processes, giving rise to a total of five reversible reduction waves for assembly 6. In addition, two oxidation waves are observed at E = +0.73 and +0.95 V vs. Fc/Fc+, assigned to the successive Fe2+ and PBI oxidation. It is worth noting that tetrahedron 6 is redox-stable for an impressive broad range of charges (from −16 up to +18), which corresponds to a total of 34 electrons which can be switched, simply by controlling the electrochemical potential. The authors attribute this efficient charge balance to the highly porous character of the large tetrahedron 6, which allows a suitable mobility of counterions during the redox switches. Another consequence of the large size of the cavity lies on the ability of 6 to encapsulate C60. Moreover, the size complementarity is strengthened by the well-established electronic affinity between C60 and the PBI moiety. Actually, the authors have shown by ESI-TOF mass spectrometry that not only one, but two fullerene units can be encapsulated within tetrahedron 6, which is one of the largest cavity ever described for the M4L6 family.
 | ||
| Fig. 5 (a) Self-assembly of metallosupramolecular tetrahedron 6 and C60 encapsulation; (b) cyclic voltammogram of 6: C = 0.1 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 200 mV s−1. | ||
Metalla-rings/cages involving electron-rich ligands
Rings and cages incorporating ferrocene subunit(s)
The design and the synthesis of molecular systems constituted from several redox-active units within the same assembly is attractive for various applications such as photochemical devices or information storage. In this context, in addition to the PBI-based assembly 5 described above, several electro-active self-assembled rings and cages decorated with multiple Fc units were reported, notably by the groups of P. J. Stang, H.-B. Yang and coworkers. Using the straightforward coordination-driven self-assembly strategy, the authors were able to build both hexagonal18 and triangular19 supramolecular metalla-cycles incorporating ferrocenyl moieties either inside18a or outside18b,c,19 the cavity. The general synthetic methodology involves the rational combination of ferrocenyl skeletons presenting 120° (target = hexagons) or 60° (target = triangles) coordination sites, with complementary linear units. For example, the synthesis depicted below (Fig. 6a), involves a linear 4,4′-bipyridine ligand with bis-metallic complexes bearing one (target = 8)18b or two (target = 7)19 pendant ferrocenyl group(s). For the preparation of hexagons, an alternative approach consists of the combination of two complementary 120° oriented electro-active building blocks (9).18b Electrochemical properties of the resulting multi-ferrocenyl complexes were studied by cyclic voltammetry. All hexagonal assemblies exhibit a single reversible redox wave with several electrons transferred during the oxidation process indicating that all ferrocene units react independently (see example of assemblies 8 and 9, Fig. 6c and d). Due to the large cavity size, this phenomenon occurs even when the ferrocenyl units point towards the center of the cavity.18a Remarkably, the redox-active character of the pendant Fc probes provides a unique opportunity to address a quantitative approach which can be supported by electrochemical studies. In particular, the diffusion coefficient (D) of the metalla-cycles could be estimated from the Randles–Sevcik equation by measuring the peak intensity at different scan rates or by potential-step chronoamperometry. The determined D values suitably correlate with the corresponding hydrodynamic diameters of the self-assembled architectures, as expected from the inverse proportionality between D and the metalla-macrocycle size. Therefore, the Fc units offer here an original way to probe electrochemically the size of the edifice, which is in principle complementary to DOSY NMR experiments. Finally, unlike hexagonal assemblies 8 and 9, it has to be noted that in the case of compound 7,19 the presence of two spatially close Fc units at vicinal positions of each phenanthrene moiety results in a splitting of the associated redox wave in the cyclic voltammogram (Fig. 6b). Such behaviour is assigned to a through-space electronic communication between the two proximal Fc units. | ||
| Fig. 6 (a) Synthesis of self-assemblies 7, 8 and 9; cyclic voltammogram of 7 (b), 8 (c), 9 (d): C = 0.20 mM in CH2Cl2 (Bu4NPF6 (0.15 M)), scan rate 25–250 mV s−1. | ||
With the aim of producing supramolecules that combine multiple properties, the same authors recently depicted multifunctional systems associating both an electro-active unit (i.e. Fc) and a crown-ether binding site in a same self-assembled macrocycle (Scheme 1).20 The single redox wave corresponding to the ferrocene unit in compound 10 remains reversible, and allows for the determination of the corresponding D values. Its binding properties with a dibenzyl ammonium salt were studied by 1H NMR and cyclic voltammetry. No change in the half-wave potential was observed upon complexation of the dibenzyl ammonium guest, indicating the lack of interaction between pendant Fc and crown-ether subunits. Nevertheless, the voltammogram shape of complex 11 compared to free 10 indicates as expected a lower D value upon complexation. Finally, efforts towards the synthesis of electroactive dendritic metalla-assemblies have to be mentioned.21 Based on a similar exo-functionalization approach as for the metalla-cycles depicted above, the same groups prepared a series of metallodendrimers of controlled size and shape, bearing both Fc and dendron functional groups. For instance, the reaction between 120° dendritic donors and 120° ferrocenyl acceptors resulted in hexagonal, hexafunctionnalized metallocycles 12a–c (Scheme 2).21d Their cyclic voltammograms reveal a unique redox wave corresponding to the reversible oxidation of the Fc units. Again, the authors could extract the values of the diffusion coefficients D from the CV experiments with, as expected, decreasing values when increasing the molecular weight.
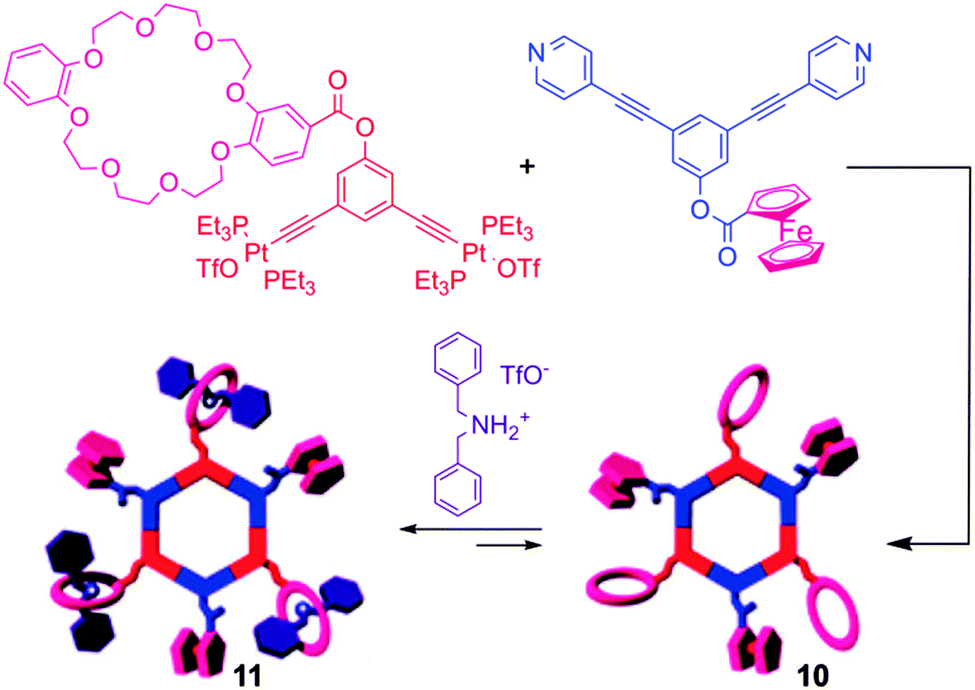 | ||
| Scheme 1 Synthesis of self-assembly 10 and tris[2]pseudorotaxane 11. | ||
 | ||
| Scheme 2 Synthesis of dendritic multiferrocenyl hexagons 12a–c. | ||
A step further lies on the extension of this strategy from metalla-rings bearing Fc units to analogous 3D metalla-cages.22 The cuboctahedral complex 13 (Scheme 3) was obtained from a 120° Fc acceptor complex and a tritopic pyridyl ligand. Its redox properties were explored in detail. Each Fc unit exhibits a similar half-wave potential and no decomposition of the assembly is observed during the redox process. The D values extracted from the electrochemical measurements are similar to those obtained from the DOSY NMR experiment. Remarkably, the corresponding external diameter calculated from the Stokes-Einstein23 equation (6.7 nm) is close to the value obtained from the molecular force field model structure (6.9 nm). Finally, the diffusion coefficient allows assessing of the presence of twelve redox units on the metalla-cage, indicating that all the ferrocenyl moieties are oxidized simultaneously. In addition to these self-assembled rings and cages decorated with pendant redox active Fc units, P. J. Stang and coworkers described a rectangular assembly 14 which incorporates the Fc moiety as a constituting part of the ring skeleton (Fig. 7a).24 The electronic properties of 14 were studied by cyclic voltammetry (Fig. 7b) and show two reversible waves at E = +0.12 and +0.33 V vs. Fc/Fc+. The first redox process was assigned to a two-electron exchange centered on both Fc units and the second one, to the simultaneous oxidation of anthracenyl units. Interestingly, the authors could determine from the spectroelectrochemical analysis that the intermediate 142+ coexists in equilibrium with the neutral and the tetracationic forms.
 | ||
| Scheme 3 Synthesis of cuboctahedral assembly 13. | ||
 | ||
| Fig. 7 (a) Rectangular assembly 14; (b) cyclic voltammogram of 14: CH2Cl2 (Bu4NPF6 (0.1 M)), scan rate 200 mV s−1. | ||
Rings and cages incorporating the tetrathiafulvalene core
The tetrathiafulvalene (TTF) moiety has received a great deal of attention as a key building block in the preparation of conducting and semiconducting materials.25 Its remarkable electrochemical behaviour is characterized by two reversible oxidation steps at low potentials, which produce stable oxidized species TTF˙+ and TTF2+. This property has promoted this unit to a key building block for the elaboration of numerous molecular and supramolecular switchable systems.26 Besides offering an extra redox state compared to ferrocene, another key issue for TTF lies on its planar π-conjugated geometry. Therefore, unlike ferrocene, TTF and its derivatives can constitute themselves the side walls of the cavity upon a self-assembly process, giving rise to metalla-rings or -cages endowed with electron rich internal cavities.J.-L. Zuo and coworkers described neutral complexes 15 and 16 (Fig. 8a).27 The latter was obtained in one step by the reaction of a mixture of cis and trans bis(pyrazole)-TTF ligands with Re(CO)5Cl. Their XRD analysis revealed that the TTF skeleton is bended in 15 and planar in metalla-ring 16. The high geometric constraints occurring in such small size self-assembled systems have of course a strong impact on the electronic integrity of the TTF unit (Fig. 8b). In both cases, the usual well-defined two-step redox behaviour of TTF cannot be recovered, due to the strong geometric constraint applied to the TTF core upon oxidation in the case of 15 and to an electronic coupling between the facing TTF redox units in the case of 16.
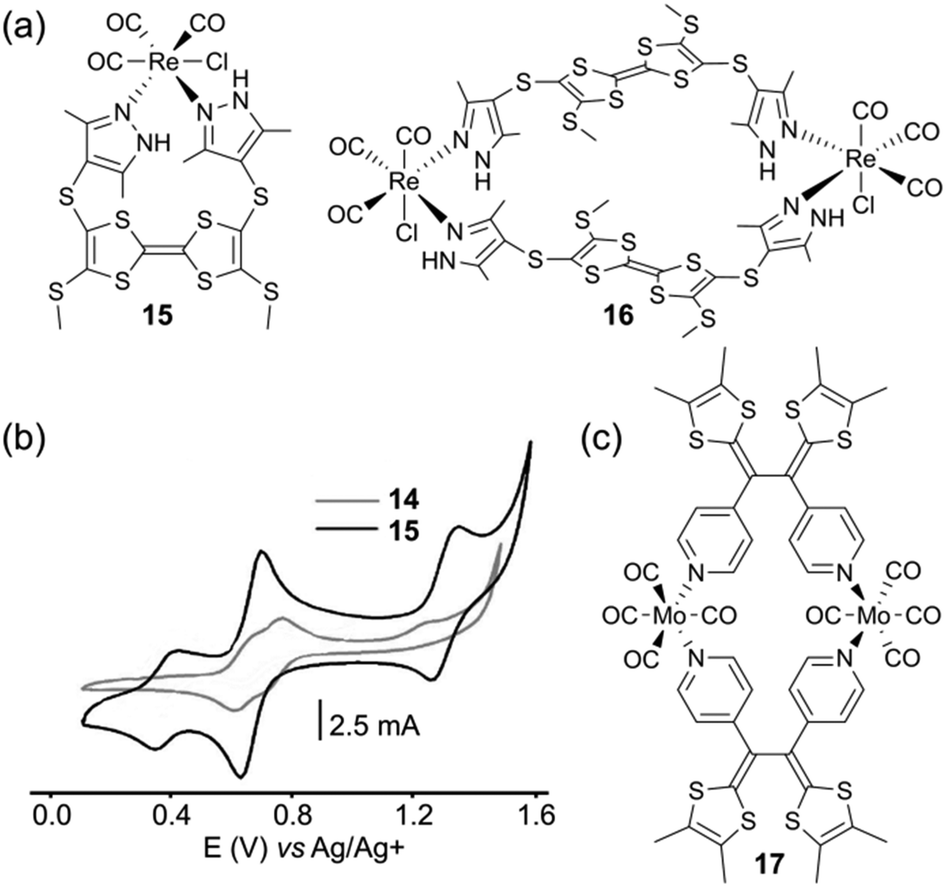 | ||
| Fig. 8 (a) Assemblies 15 and 16; (b) cyclic voltammograms of 15 and 16: C = 0.5 mM in CH2Cl2 (Bu4NClO4), scan rate 100 mV s−1; (c) assembly 17. | ||
Another metalla-ring involving a metal carbonyl complex and a TTF derivative, namely vinylogous tetrathiafulvalene (TTFV), was described by D. Lorcy and coworkers (Fig. 8c).28 The latter is constructed from the self-assembly of a non-planar bispyridyl TTFV ligand with hexacarbonyl molybdenum in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The electrochemical properties of metalla-square 17 which incorporates four redox-active units were studied by cyclic voltammetry. The system appears to be oxidized according to two successive processes at E = +0.48 and +0.74 V vs. SCE. The first one is attributed to the 0/•+ oxidation of the organic part, which is slightly shifted compared to the TTFV ligand precursor, the second wave being ascribed to the superimposition of both metal center oxidation with the •+/2+ ligand centered oxidation.
:1 molar ratio. The electrochemical properties of metalla-square 17 which incorporates four redox-active units were studied by cyclic voltammetry. The system appears to be oxidized according to two successive processes at E = +0.48 and +0.74 V vs. SCE. The first one is attributed to the 0/•+ oxidation of the organic part, which is slightly shifted compared to the TTFV ligand precursor, the second wave being ascribed to the superimposition of both metal center oxidation with the •+/2+ ligand centered oxidation.
In the course of the studies conducted at Angers, related to the synthesis of electron-rich supramolecular discrete systems, we described recently several examples of metalla-rings and -cages whose cavities are made from TTF or its derivatives. For this purpose, several di- and tetra-topic ligands either designed from the parent TTF skeleton (18) or two of its well-known derivatives, namely bis(pyrollo)TTF (BPTTF)29 (19, 20) and the so-called extended-TTF (exTTF)30 (21) were synthesized (Scheme 4).
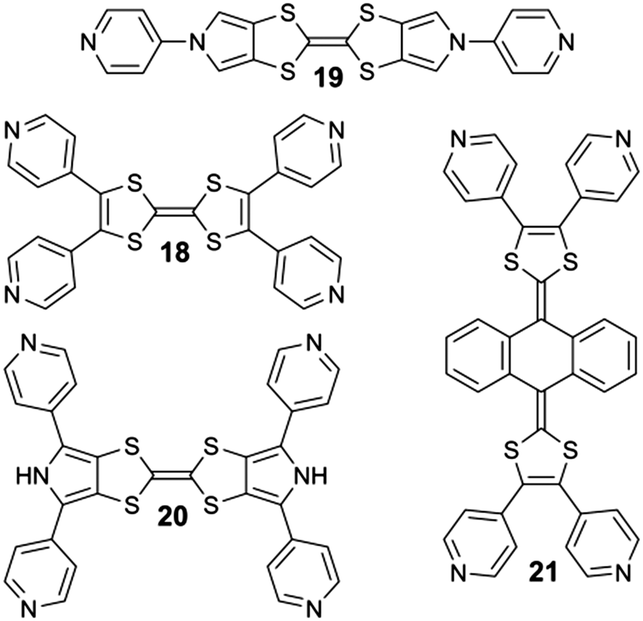 | ||
| Scheme 4 Di- and tetra-topic TTF-based ligands 18, 19, 20 and 21 prepared at Angers. | ||
As a common feature, all of these ligands integrate a central sulfur-rich electron-rich framework. The first example of a metal-driven self-assembled system issued from the parent TTF corresponds to the octanuclear metalla-cage 22, which was prepared in collaboration with B. Therrien and colleagues (Fig. 9a). Reaction between tetratopic ligand 18 and dinuclear p-cymene oxalato ruthenium acceptor Ru1 afforded metalla-cube 22, whereas the reaction with dinuclear p-cymene quinonato complex Ru2 afforded tetranuclear plate 23.31 The selective formation of these two different architectures results from geometrical complementary interactions between 18 and the respective Ru1 and Ru2 counterparts. Two pyridyl nitrogen atoms located on the same dithiol unit in ligand 18 are separated by 6.7 Å, whereas both ruthenium centers are at a distance of 5.5 Å in complex Ru1 and at 7.9 Å in complex Ru2. Therefore, the good size matching between ligand 18 and Ru2 affords tetranuclear plate 23 whereas the smaller ruthenium acceptor Ru1 generates cage 22. Both assemblies were studied by cyclic voltammetry (Fig. 9b). Two characteristic redox waves corresponding to the TTF moiety are observed in plate 23 (E = +0.28 and +0.58 V vs. Fc/Fc+), shifted to higher potential in comparison to ligand 18 (E = +0.22 and +0.55 V vs. Fc/Fc+) as a consequence of the coordination to the metal centers. The electrochemical behaviour observed for metalla-cage 22 is significantly different since only one oxidation wave is observed, at E = +0.44 V vs. Fc/Fc+. This peculiar behaviour is assigned to the rigidity of the metalla-assembly which may alter the kinetics associated with the conformational changes which usually occur upon the electrochemical oxidation of TTF derivatives. It is indeed well-established that the TTF parent system undergoes severe conformational changes upon oxidation to heteroaromatic 1,3-dithiolium cations TTF˙+ and TTF2+. Whereas neutral TTF corresponds to a distorded boat-like structure, TTF˙+ is planar and TTF2+ is significantly twisted around the central C–C bond.25a,32 Consequently, if structural elements located on the periphery of the TTF skeleton prevent those conformational changes upon oxidation, the usual well-defined reversible two-step redox behaviour of TTF is not observed anymore. This is observed for instance with geometrically constrained tetrathiafulvalenophanes33 or TTF-based cage molecules,34 and is also observed in the case of metalla-cage 22. Another family of electro-active discrete self-assembled cages could be synthesized from ligand 18. The reaction of the latter with cis-blocked Pd(II) or Pt(II) complexes (i.e. cis-M(dppf)(OTf)2; dppf = 1,1′-bis(diphenylphosphino)ferrocene; OTf = trifluoromethane-sulfonate) in a 1:2 molar ratio afforded metalla-architectures 24a (Pt) or 24b (Pd).35 Both present an estimated hydrodynamic radii of ca. 16 Å as extracted from DOSY NMR experiments which, in addition to other spectroscopic data, well agrees with a M8L4 cubic geometry (Fig. 10a). Cyclic voltammetry experiments conducted on metalla-cages 24a and 24b show three reversible redox processes. Two are assigned to the TTF side walls and the third one to the corner ferrocene units. Interestingly, the latter can be used as an internal reference to address the number of electrons exchanged. The relative peak intensities of the three systems (i.e. 1/1/2) observed from the deconvoluted cyclic voltammogram (Fig. 10b) agree with two successive independent oxidations of the four TTF units at E = +0.30 and +0.60 V and of the eight ferrocenes at E = +0.80 V vs. Fc/Fc+, giving rise in total, to the reversible generation of sixteen positive charges on the periphery of the cavity. Molecular squares prepared by the coordination-driven self-assembly strategy are typically issued from the interaction between a linear ditopic ligand and a cis-blocked square planar metallic center. Depending on the length of the linear ligand or on its flexibility, the reaction can also lead to triangular systems beside the molecular square, both polygons being usually difficult to separate. Thus, the reaction of the linear N,N′-dipyridyl(BPTTF) ligand3619 with an equimolar ratio of cis-Pt(dppp)(OTf)2, (dppp = 1,3-bis(diphenylphosphino)propane), produces a mixture of triangle 25 and square 26 (Fig. 11a). Remarkably, in this case, each polygon could be easily isolated by selective precipitation.37 Both metalla-rings 25 and 26 were fully characterized by different methods.
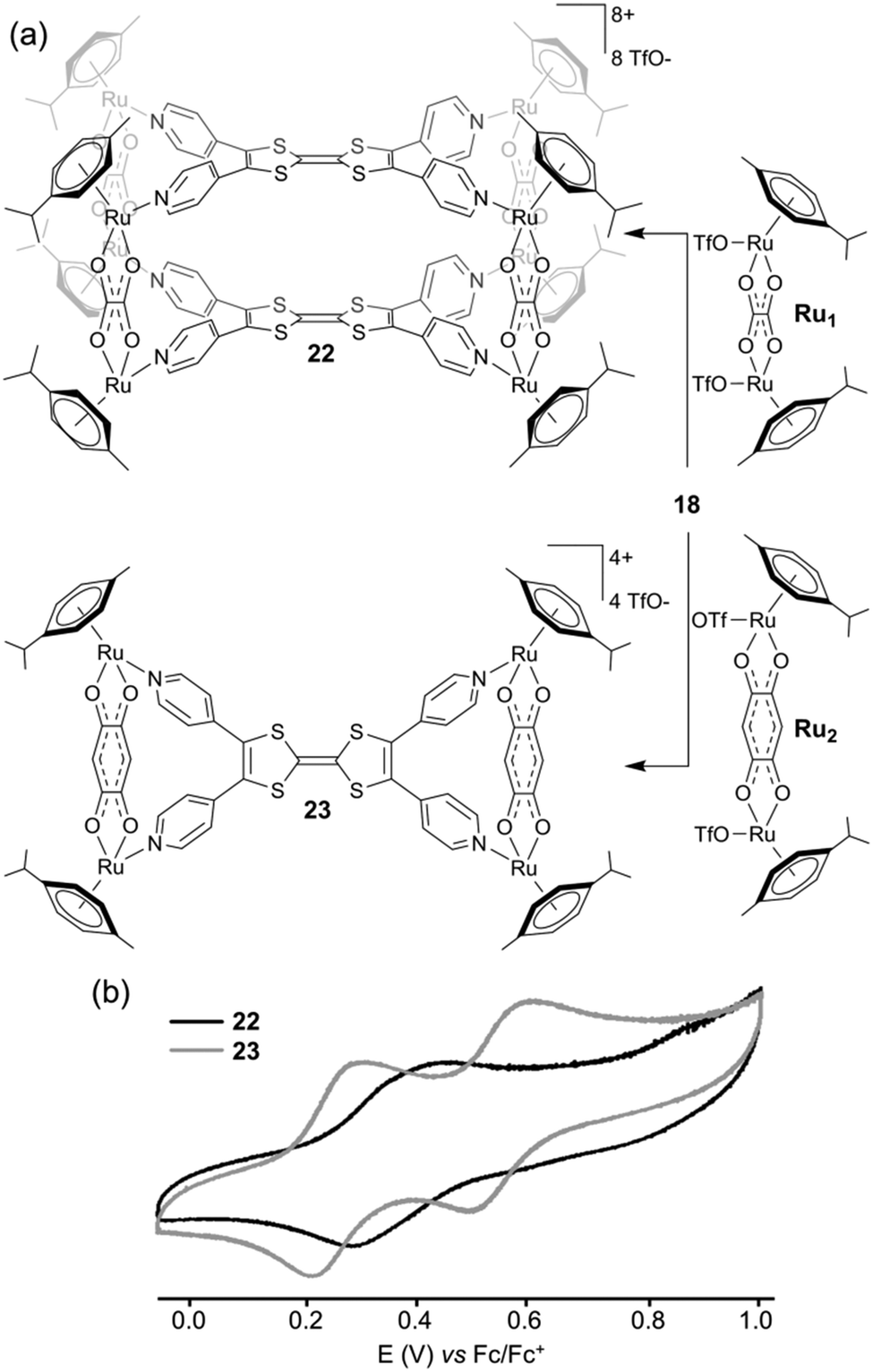 | ||
| Fig. 9 (a) Synthesis of metalla-cage 22 and metalla-plate 23; (b) cyclic voltammogram of 22 and 23: C = 0.5 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 50 mV s−1. | ||
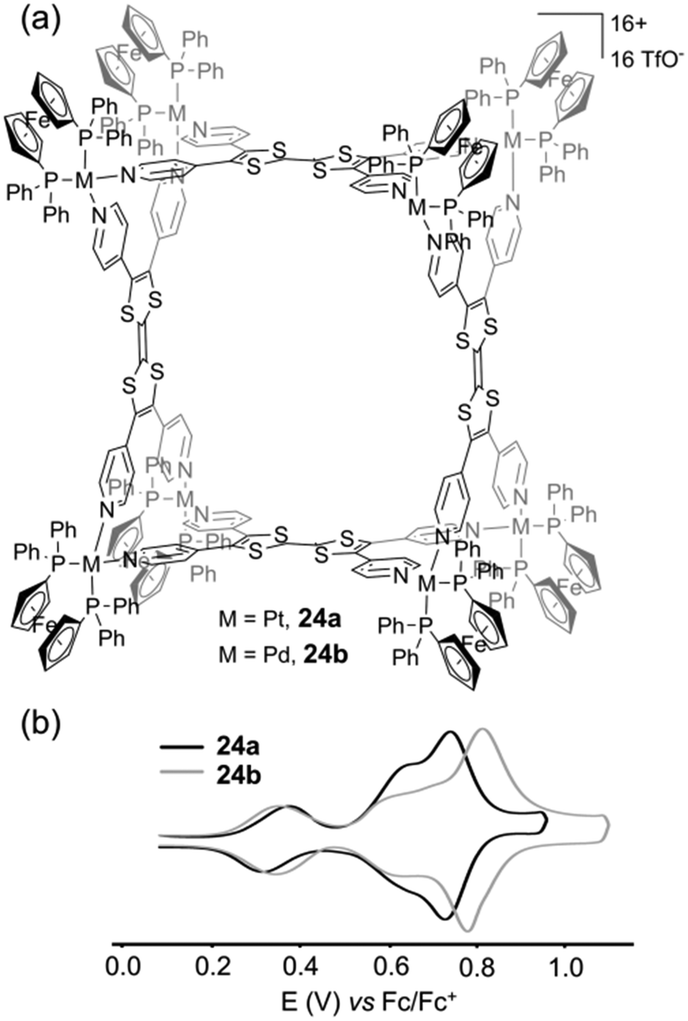 | ||
| Fig. 10 (a) Cubic assemblies 24a and 24b; (b) deconvoluted cyclic voltammogram of 24a and 24b: C = 0.5 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 20 mV s−1. | ||
 | ||
| Fig. 11 (a) Triangle 25 and square 26; (b) deconvoluted cyclic voltammogram of 25 and 26 in the presence of ferrocene (3.0 and 4.0 equiv. respectively): C = 0.5 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 20 mV s−1; (c) Job's plot at 407 nm, triangle 25vs. C60 (C = 1.3 × 10−5 M in CS2/CH2Cl2 (4/1)). | ||
Noteworthy, the ratio between their respective experimental radii (Rtriangle/Rsquare) extracted from DOSY NMR reaches a value of 0.80, in excellent agreement with the theoretical value. As usually observed for BPTTF derivatives, triangle 25 and square 26 exhibit two reversible oxidation waves (E = +0.70 and +1.00 V vs. Fc/Fc+) (Fig. 11b). In order to address the number of electrons exchanged in each process, ferrocene was added to the electrochemical solutions and used as an internal coulometric reference (i.e. three equivalents for triangle 25 and four equivalents for square 26). Analysis of the resulting respective peak intensities from the deconvoluted CV (Fig. 11b) suggests that the BPTTF side walls behave independently and that the metalla-rings are fully oxidized to their respective 256+ and 268+ stable states. Given the π-donating character of BPTTF derivatives, a peculiar feature of such polygons lies on the strong electronic density which is spread over the walls of the cavity, which in principle is favourable to bind to a complementary electron-poor guest within the cavity. On the other hand, the preparation of molecular and supramolecular hosts able to bind fullerene derivatives is a subject of intense interest.38 In this context, the binding affinity of 25 and 26 for electro-deficient C60 or C70 was monitored by UV-Vis titrations. The titration spectra as well as a Job's plot analysis (Fig. 11c) demonstrate a good binding affinity of triangle 25 for C60 (or C70) in a 1:1 stoichiometry. This result is supported by a good electronic complementarity of both components as well as by a suitable size matching between the cavity of 25 and a fullerene guest. The latter property is not satisfied in the case of square 26 whose cavity is much larger and which indeed does not exhibit any affinity for those fullerenes in UV-vis titration experiments.
With the aim to switch from electron-rich metalla-rings to metalla-cages, the tetratopic BPTTF-based ligand 20 (Scheme 4) was designed. The latter was reacted with square planar cis-Pt(PEt3)2(OTf)2 complex and the reaction converged in less than two hours into a single discrete prismatic cage 27 (Fig. 12a).39 The M6L3 formula was confirmed by ESI-MS mass spectroscopy and by an X-ray diffraction experiment (Fig. 12b). Cyclic voltammetry of metalla-cage 27 (Fig. 12c) reveals the presence of two successive oxidation waves, located at fairly low potentials (E = +0.43 and +0.62 V vs. Fc/Fc+) but poorly reversible due to a combination of steric and electronic factors. The guest binding capability of prism 27 was studied by UV-Vis spectroscopy with an electro-deficient compound (TCNQ-F4). The progressive addition of the guest to a solution of electron-rich M6L3 cage 27 leads to the formation of an inclusion complex with a 1:1 stoichiometry as extracted from a Job's plot analysis.
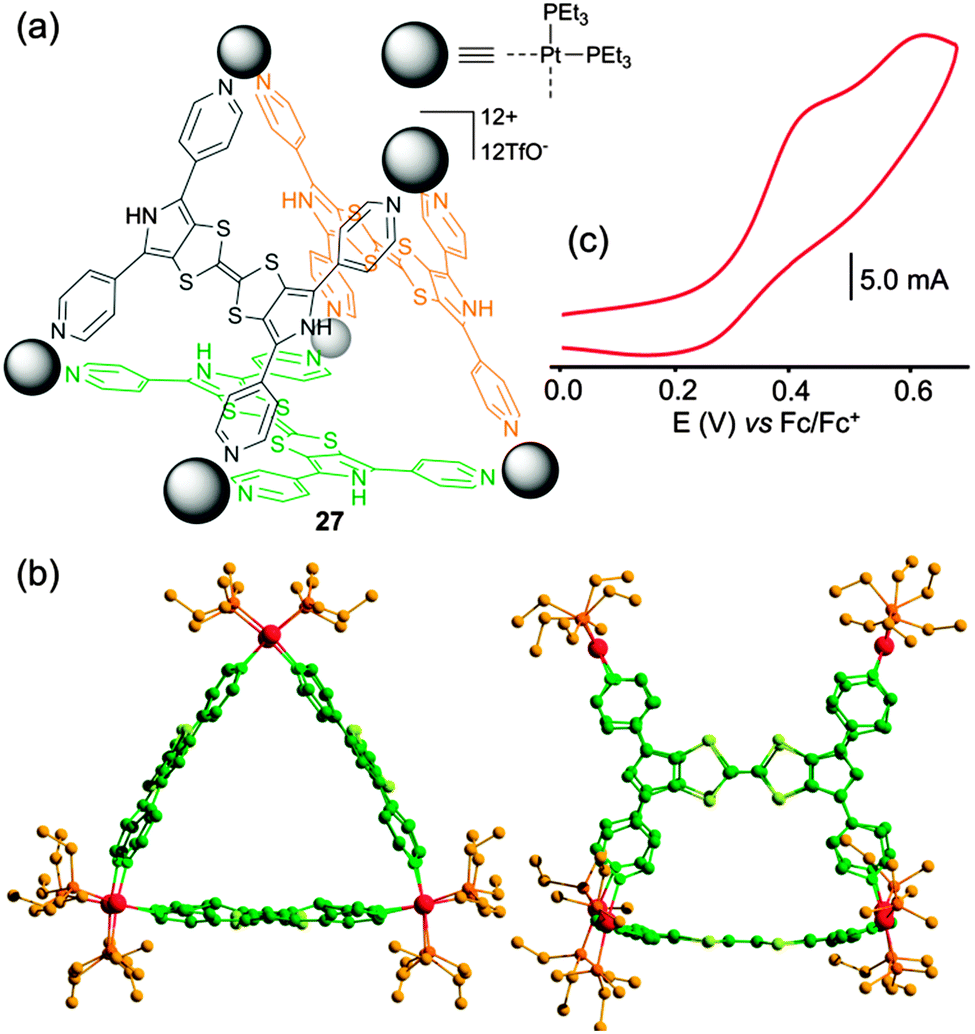 | ||
| Fig. 12 (a) Metalla-prism 27; (b) X-ray crystal structure of 27; (c) cyclic voltammogram of 27: C = 0.6 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 50 mV s−1. | ||
A third family of discrete metalla-assemblies that we have been developing40 is based on the exTTF framework. This π-donating skeleton exhibits unique electronic and geometrical properties that have been widely explored by N. Martín and coworkers in the construction of covalent hosts prone to encapsulate electro-deficient fullerene guests.38a
On this basis, tetrapyridyl-exTTF ligand 21 (Scheme 4) was synthesized in one step from pristine exTTF through a palladium catalyzed C–H arylation. A mixture of ligand 21 and cis-Pd(dppf)(OTf)2 in a 1:2 molar ratio afforded in only five minutes one discrete assembly 28a (Fig. 13a).40 Both species, ligand 21 and resulting M4L2 assembly 28a (Fig. 13b) could be characterized by X-ray diffraction. Ligand 21 displays the usual exTTF butterfly shape with a 86° dihedral angle between both 1,3-dithiol-2-ylidene mean plans. This curvature is amplified in the M4L2 container 28a with a dihedral angle of 56° which gives rise to an ovoid cavity whose size and shape are suitable for the inclusion of a planar guest. In particular, the formation of a 1:1 host–guest inclusion complex with perylene (Ka ≈ 3.9 × 103) could be demonstrated by 1H NMR, DOSY NMR and ESI-FTICR. Electrochemical properties of ligand 21 and container 28a were studied by cyclic voltammetry (Fig. 13c). In addition to a first oxidation wave attributed to the two-electron oxidation steps of each exTTF scaffold (E = +0.57 V vs. Fc/Fc+), container 28a exhibits a second oxidation process centered on the four ferrocene units (E = +0.80 V). Compared to ligand 21, the oxidation of exTTF in the complex is shifted to higher potentials due to the coordination to the metal center and appears irreversible presumably because of the rigidity of the self-assembly which prevents redox-driven conformational changes which usually occur with exTTF derivatives.
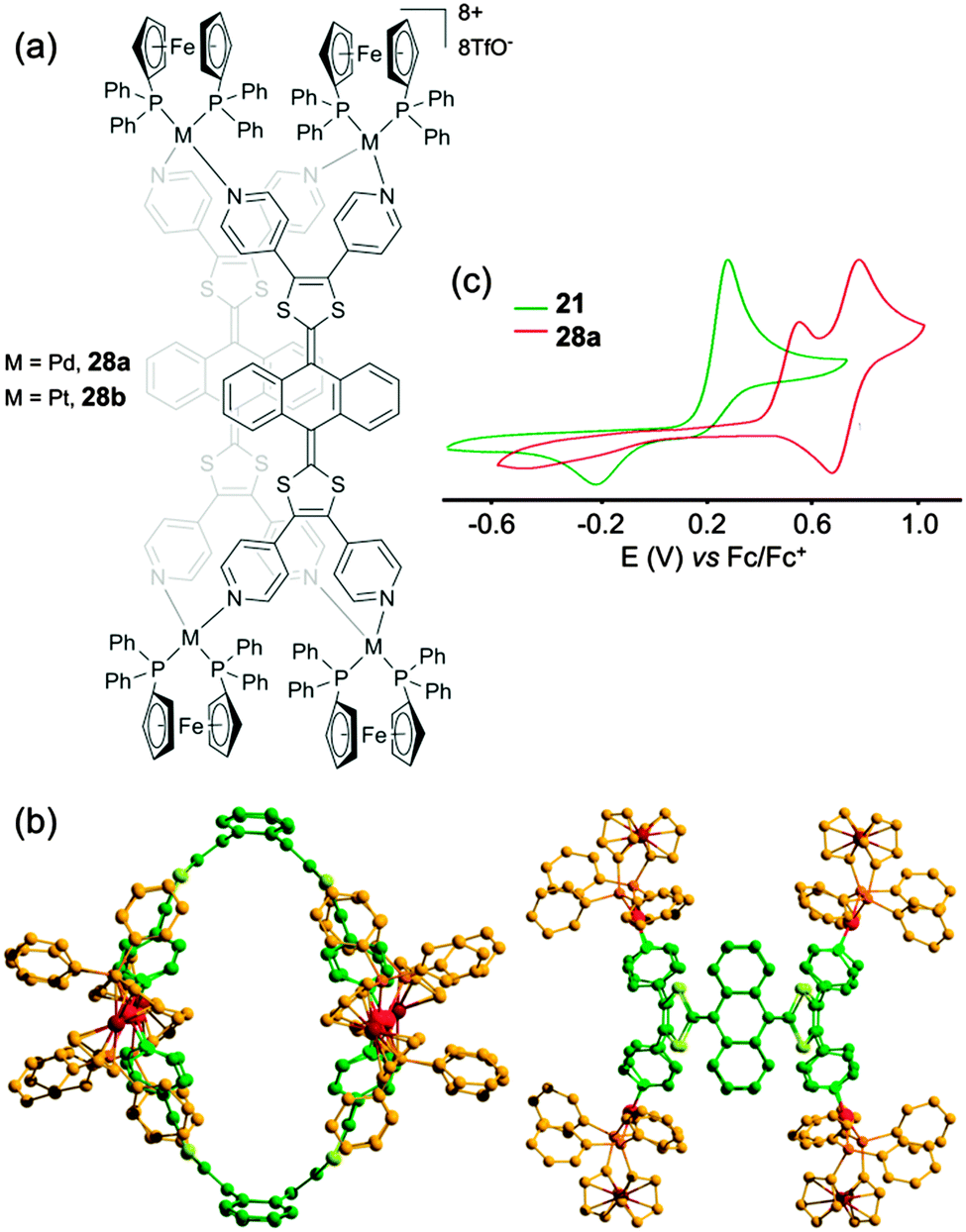 | ||
| Fig. 13 (a) Metalla-assemblies 28a and 28b; (b) X-ray crystal structure of cage 28a; (c) cyclic voltammogram of ligand 21 and cage 28a: C = 1.0 mM in CH3CN/CH2Cl2 (1/1) (21) or CH3CN (28a) (Bu4NPF6 (0.1 M)), scan rate 100 mV s−1. | ||
Miscellaneous
Self-assembled polygons/polyhedra are prepared from complementary ligands and metal complexes and the redox activity of the resulting assemblies is closely linked to the electroactivity of each partner. Basically, the redox character of the metalla-ring/cage can be mainly localized on the cavity walls (in general the ligand) as shown above through various examples, or on the corner (in general the metal), or on both partners. In the latter case, the ligand and the metal can behave independently or are sometimes electronically coupled as shown through some examples above.A promising development of self-assembled metalla-cages concerns their use as anti-cancer agents.1a,t,41 Based on the fact that various Ru(III) and arene Ru(II) complexes exhibit interesting cytotoxic effects, B. Therrien and colleagues have developed several families of water soluble cationic metalla-cages constructed from arene ruthenium complexes.1t,41a,c The authors have demonstrated the efficiency of such assemblies for increasing the solubility and cell permeability of drug-like molecules or photodynamic therapy (PDT) agents. Some of these metalla-assemblies were electrochemically studied. An illustrative example is given with the tetranuclear metalla-rectangle 29 (Fig. 14a),42 which is obtained from the reaction of a bipyridine ligand with a dinuclear arene ruthenium complex for which two Ru centers are bridged by a 1,4-benzoquinonato moiety. The redox properties of metalla-rectangle 29 have been studied by cyclic voltammetry (Fig. 14b). Compared to the dinuclear arene ruthenium precursor Ru2, rectangle 29 appears much easier to reduce, due to the presence of four positive charges in the metalla-assembly. Rectangle 29 undergoes three successive reduction waves at E = −0.68 (reversible, vs. Fc/Fc+), −1.39 (irr.) and −1.90 V (irr.) and no oxidation could be detected before electrolyte decomposition. Though the ligand orbitals are closely mixed with metal-based ones, these reduction processes can formally be assigned to electron injection occurring predominantly onto the quinonato bridging ligand.
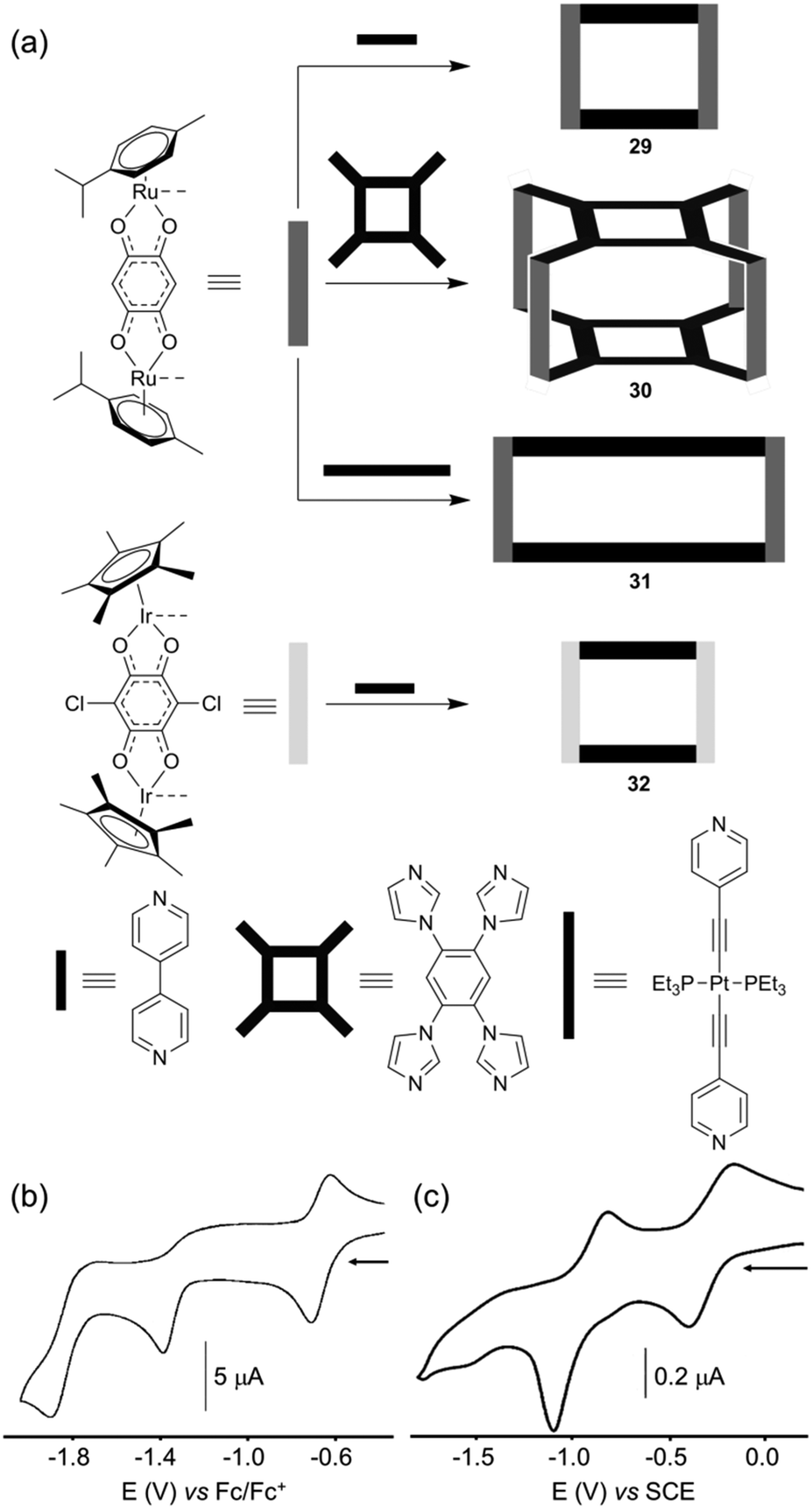 | ||
| Fig. 14 (a) Schematic representation and synthesis of metalla-assemblies 29–32; cyclic voltammogram of 29 (b) and 30 (c): (b) C = 0.5 mM in CH2Cl2 (Bu4NPF6 (0.1 M)), scan rate 100 mV s−1; (c) CH2Cl2 (NBu4PF6 (0.1 M)), scan rate 100 mV s−1. | ||
A similar electrochemical behaviour (Fig. 14c) could be observed by P. Mukherjee and colleagues, from a 3D macrocyclic cage 30 (Fig. 14a) featuring the same bis-bidentate quinonato Ru complex Ru2, which is associated in this case with a tetra-imidazole ligand.43 Note that 2D analogous assemblies based on bis-imidazole ligands were also electrochemically characterized.44 Finally, the highly modular character of the stepwise strategy using half-sandwich piano-stool complexes for the construction of discrete electroactive polygons is illustrated by the possibility to change either the bridging dipyridyl ligand to a longer one as in rectangle 31,45 or the metal as in 32 which integrates an iridium metal complex instead of a ruthenium one.1q,46
Beyond Ru-based cages and as mentioned above with the encapsulation of a Pt(IV) prodrug AdPt,9 metalla-cages incorporating square planar Pt(II) and Pd(II) complexes have also been used as drug delivery vectors. This is the case with metalla-cages 33a–d recently described by J. D. Crowley and colleagues (Scheme 5a) which integrate a variety of functional groups on the outer surface of the host cage. They were prepared according to a very efficient modular approach based on the CuAAC ‘click’ reaction.47 Using this strategy, various functions were appended on the periphery of the cage, some being redox-active. The cyclic voltammetry study of compounds 33a–d in DMF shows basically a superposition of the redox contribution of each electroactive partner, be they reducible or oxidable. They display irreversible reductions at negative potentials (ca. E = −1.7 and −1.9 V vs. Fc*/Fc*+) assigned to the 2,6-bis(ethynyl)pyridine ligand framework as well as redox processes corresponding to the appended functional groups. In particular, the ferrocenyl-substituted cage 33a displays the usual reversible oxidation process of ferrocene at E = +0.5 V vs. Fc*/Fc*+ and no electronic communication was observed between the four pendant Fc units, as expected from their large spatial separation.
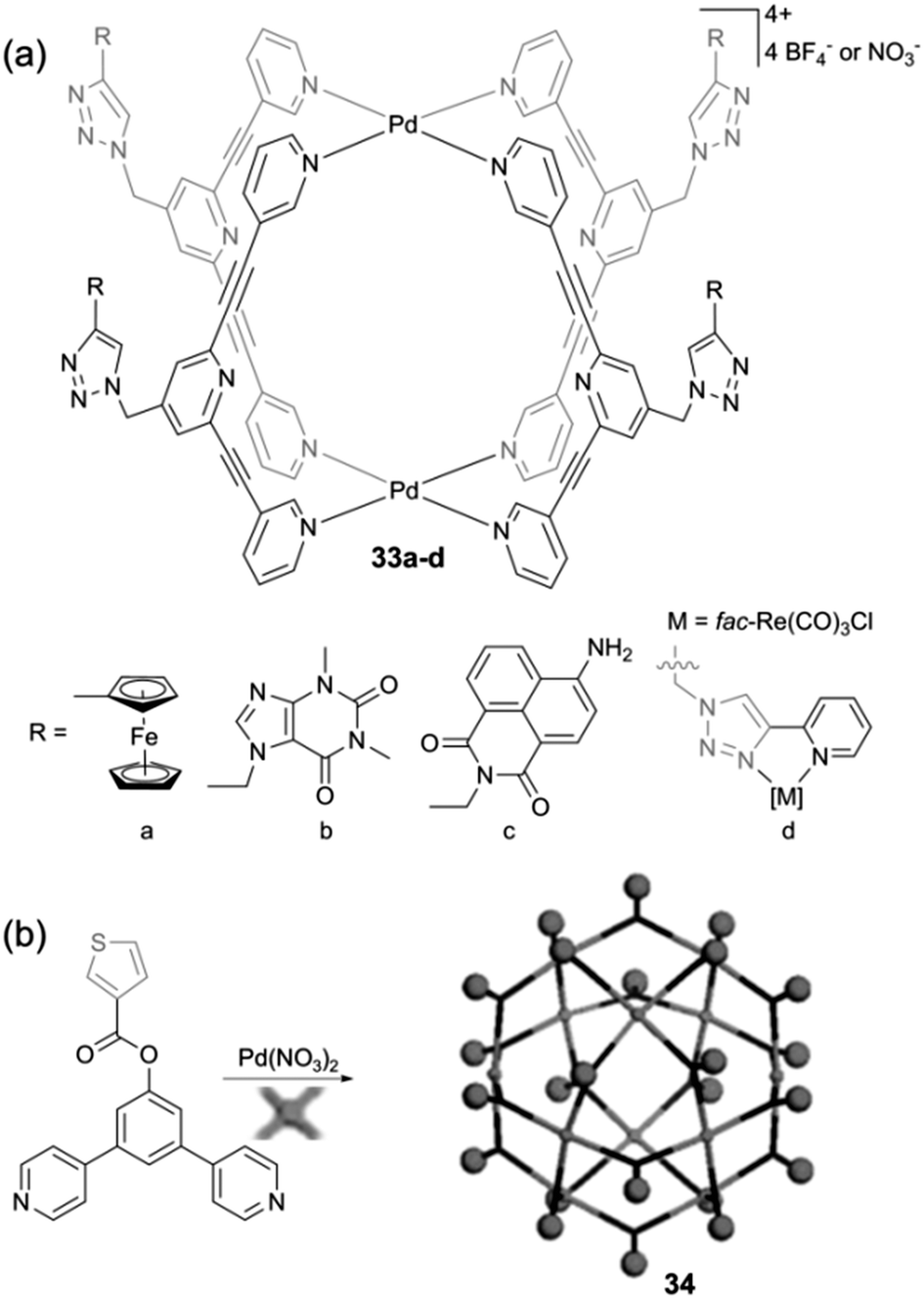 | ||
| Scheme 5 Metalla-assemblies 33a–d and 34. | ||
With a similar objective to decorate the outer surface of a metalla-structure, R. Yang et al. described the M12L24 sphere 34 (Scheme 5b) which integrates 24 thiophene units.48 The investigation of the electrochemical properties of 34 in DMSO is characterized by a reduction process at E = −1.06 V vs. Ag/AgCl. No information is provided regarding the oxidation and electropolymerization ability of this thiophene-rich system.
The case of square 35 (Scheme 6) depicted by A. J. Lees and colleagues49 is interesting since it encompasses three redox active centers, i.e. Ru(II)/Ru(III), Fc/Fc+ and the terpyridine ligand, all of them behaving independently. Two oxidation waves are observed and assigned to the Ru(II)/Ru(III) (E = +0.70 V vs. Fc/Fc+) and to the Fc corner (E = +0.88 V) respectively, whereas the reduction of the terpyridine fragment gives rise to two successive reduction processes. The comparison of the electrochemical behaviour of cyclen Ru-based triangle 36 and square 37 (Fig. 15a) described by J. R. Long and coworkers, provides an interesting case for using the electrochemistry tool in probing a molecular structure.50 Unlike triangle 25 and square 26 presented above and which exhibit similar cyclic voltammograms, quite different ones are observed for the triangular (cyclen)3[Ru3(4,4′-bpy)3](PF6)636 and the square (cyclen)4[Ru4-(4,4′-bpy)4](OTf)837. The former is oxidized following three distinct redox processes at E = +0.23, +0.37 and +0.48 V vs. Fc/Fc+ in acetonitrile, each of them corresponding to a Ru-centered one-electron exchange, whereas square 37 exhibits one single reversible four-electron process at E = +0.48 V and a high scan rate (1000 mV s−1). This difference is assigned to the more sterically constrained structure in the case of the triangle, which prevents any rotation of the pyridine rings with respect to each other and which favours electronic coupling by delocalization. Such a difference between homologous polygons nicely illustrates how the geometry of molecules built from the same components can have a direct impact on their electrochemical properties.
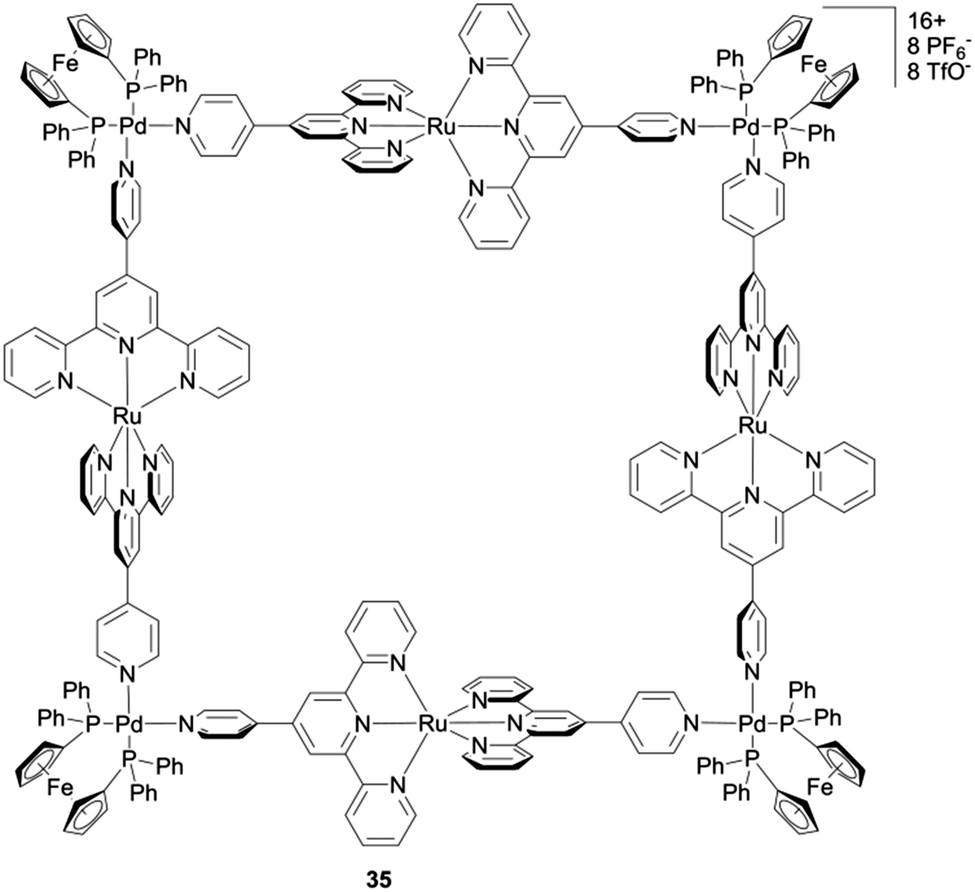 | ||
| Scheme 6 Metalla-square 35. | ||
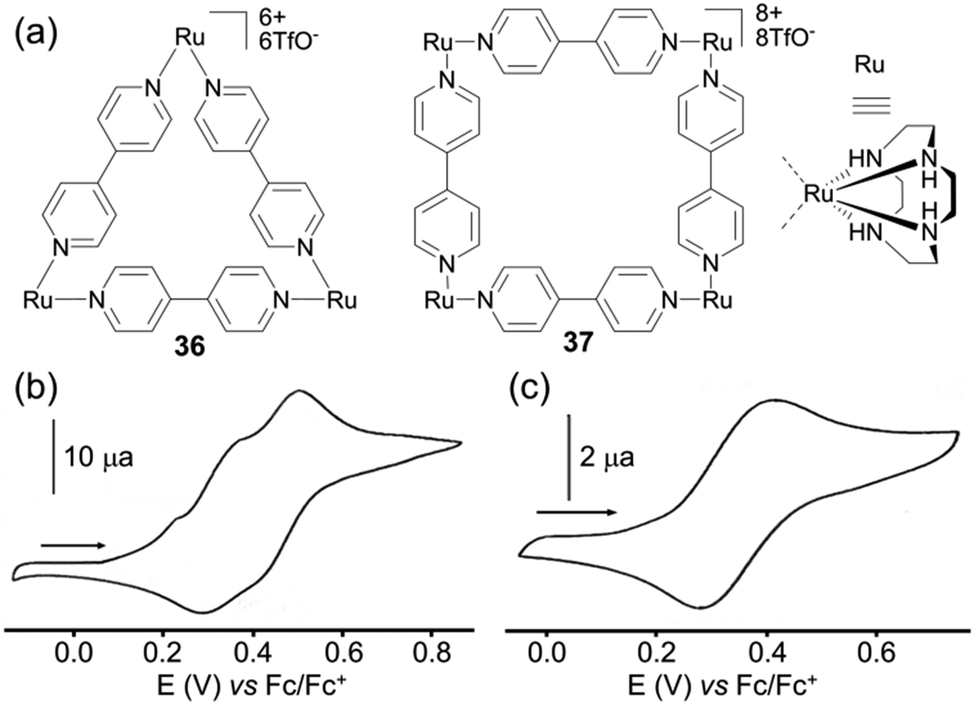 | ||
| Fig. 15 (a) Metalla-triangle 36 and metalla-square 37; cyclic voltammograms of 36 (b) and 37 (c), C = 1 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 100 mV s−1 (b) and (Bu4NClO4 (0.1 M)), scan rate 1000 mV s−1 (c). | ||
Because of their unusual topology and as illustrated with 36, metalla-triangles are appealing for the study of their redox properties. Another relevant example corresponds to Ru-based triangles 38a,b built from adenine ligands (Fig. 16). These metalla-rings, depicted by J. A. Thomas and coworkers,51 display a well-defined reversible electrochemical behaviour which is manifested by three Ru-based reversible oxidation processes, from which the authors could demonstrate the occurrence of two discernible mixed-valence states.
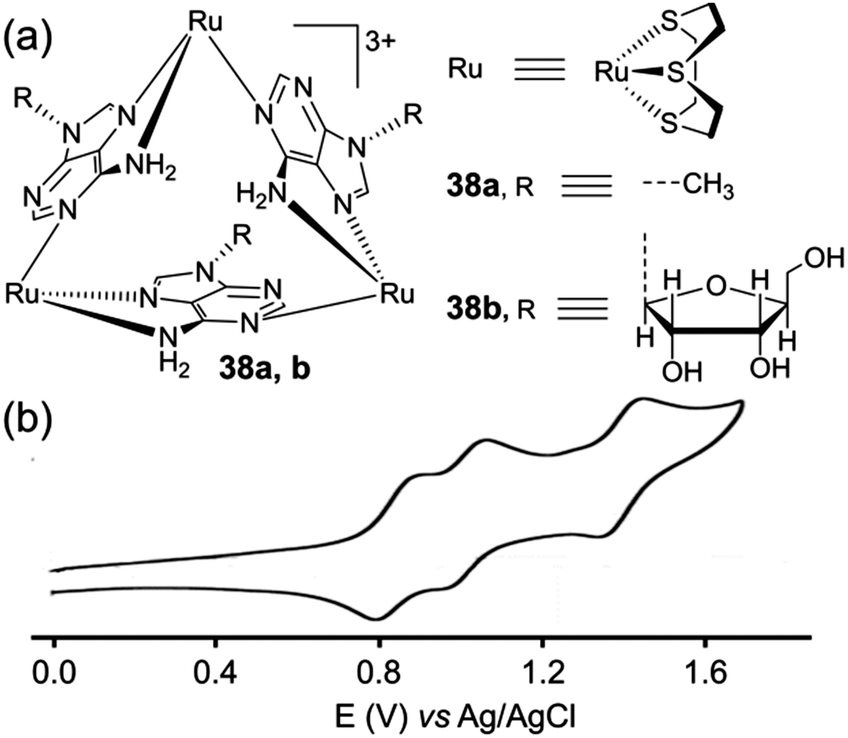 | ||
| Fig. 16 (a) Metalla-triangle 38a–b; (b) cyclic voltammograms of 38a, CH3CN (Bu4NPF6 (0.1 M)). | ||
The case of the phenothiazine heterocycle is interesting since this system can be reversibly oxidized into a radical-cation at E = +0.73 V vs. SCE (acetonitrile) with the possibility of further oxidation or disproportionation. This favourable redox behaviour prompted G. H. Clever and colleagues to describe recently a phenothiazine-based coordination cage 39a.1c,52 The latter corresponds to an interpenetrated double cage based on eight phenothiazine ligands and four square-planar-coordinated Pd(II) cations (Fig. 17). Remarkably, the corresponding S-monooxygenated sulfoxyde and S-dioxygenated sulfone cages 39b and 39c could be successively obtained from 39a through a stepwise chemical oxidation with Cu(NO3)2·3H2O or, in the case of 39b, upon standing in air for two months. The three double cages 39a–c exhibit significant different shapes in the solid state as shown by their respective X-ray diffraction structures (Fig. 17b–d).52,53 Interestingly, titration with the oxidant[Fe(III)(bipyridine)3]3+, followed by reaction with water, shows that the intermediate cation radical 39a8˙+ reacts much faster with water compared to the cation-radical generated from the phenothiazine ligand precursor. This is ascribed to a cage effect which, upon oxidation, favours an intramolecular disproportionation. This property results in neutral and dicationic phenothiazine facets in the double cage, the latter dications being known intermediates in the production of S-monooxygenated sulfoxyde upon reaction with water. Finally, an interesting case concerns metalla-rectangle 40 described by C. N. Hsu and colleagues54 This tetranuclear iron thiolate complex can be electrochemically reduced according to three successive reversible redox processes (E = −0.28, −0.84 and −0.93 V vs. Fc/Fc+) (Fig. 18). It appears that in this rectangle, the 1,4-diisocyanobenzene ligand does not assist the electron communication between both iron thiolate cores, which behave independently at the first two-electron redox process (404+/402+). The second and third processes are assigned to two one-electron exchanges corresponding to 402+/40+ and 40+/400 couples respectively, redox states for which a weak exchange interaction seems to occur through the 1,4-diisocyanobenzene ligand. The most striking feature of this system, which could be demonstrated thanks to the single-crystal X-ray structures of 404+ and 402+, comes from their different cavity size, which appears larger in the tetracationic form than in the reduced one. Therefore, this work provides a nice illustration of how the size of a self-assembled redox-active system can be controlled thanks to a redox stimulus, opening therefore interesting perspectives in terms of electrochemical-triggering of guest binding.
 | ||
| Fig. 17 X-ray single-crystal structures of double cages 39a (b), 39b (c), 39c (d) (Metal = Palladium) and their corresponding constitutive ligands (a). | ||
 | ||
| Fig. 18 (a) Redox-controlled cavity modulation of rectangular complex 40; (b) cyclic voltammogram of 40: C = 0.2 mM in CH3CN (Bu4NPF6 (0.1 M)), scan rate 100 mV s−1. | ||
Conclusions and outlooks
Thanks to the synthetic efficiency and to the modular character of the self-assembly approach, a wide diversity of discrete redox-rings/cages have been made available during the last decade. This diversity is supported by the cumulated inherent structural and electronic characteristics of the starting building blocks, i.e. ligands and metal complexes or can also result from synergetic effects between both components. The latter can be electronically coupled or not, giving rise to a large panoply of reducible or oxidable discrete cavities which differ in the structure or in the function. From this point of view, and as shown through various examples in this article, a key issue of such redox-active systems relies on the possibility to fine tune the charge state of the cavity, be it positive or negative, by a simple control over the applied electrical potential or of the oxidizing/reducing chemical agent. On this ground, it is worth mentioning that beyond potential applications in electrochemical sensing for environmental and medical sciences4,55 or for redox catalysis,1i a valuable extension of such control over the cavity charge lies in the possibility to trigger: (i) the cavity size, as in the case of metalla-rectangle 40,54 (ii) the binding affinity between the redox-active host cavity and a given guest, a key issue for delivery applications. From this point of view, the possibility to trigger the host–guest affinity, thanks to an external physical stimulus, is the subject of intense interest. Promising results have been already obtained for discrete self-assembled host cavities, as for example in the case of light-switchable metalla-cages.1c,3 Chemical stimuli such as addition of a competing ligand or the use of pH-sensitive components56 have also been used successfully. On the other hand, redox-stimuli are able to tune the charge state of the host cavity, that is, its ionic level, which attracts a strong interest. Such a system should allow in principle to bind, or conversely to expel the guest, simply by modifying the electrostatic host–guest interactions. This concept has already been demonstrated for instance with a molecular organocyanometalate box which permits a redox-switch of the complexation/decomplexation process of K+ and Cs+.57 Given the tremendous advances recently achieved regarding the design of new highly functionalized redox-responsive self-assembled hosts, promising perspectives are therefore offered for extending this concept to the binding control of various guests of interest, be they ionic or neutral.Acknowledgements
The authors gratefully acknowledge the MENRT for a PhD grant (VC).Notes and references
- For recent reviews, see: (a) N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Chem. Soc. Rev., 2015, 44, 9 RSC; (b) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419 RSC; (c) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848 RSC; (d) S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239 RSC; (e) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS PubMed; (f) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703 RSC; (g) A. Mishra, S. C. Kang and K.-W. Chi, Eur. J. Inorg. Chem., 2013, 5222 CrossRef CAS; (h) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (i) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC; (j) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015 CrossRef CAS PubMed; (k) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2012, 113, 734 CrossRef PubMed; (l) L. R. MacGillivray, Angew. Chem., Int. Ed., 2012, 51, 1110 CrossRef CAS PubMed; (m) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (n) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed; (o) S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555 RSC; (p) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760 CrossRef CAS PubMed; (q) Y.-F. Han, W.-G. Jia, W.-B. Yu and G.-X. Jin, Chem. Soc. Rev., 2009, 38, 3419 RSC; (r) B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554 CrossRef CAS PubMed; (s) P. J. Stang, J. Org. Chem., 2009, 74, 2 CrossRef CAS PubMed; (t) B. Therrien, Eur. J. Inorg. Chem., 2009, 2445 CrossRef CAS; (u) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (v) M. W. Cooke, D. Chartrand and G. S. Hanan, Coord. Chem. Rev., 2008, 252, 903 CrossRef CAS PubMed; (w) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825 CrossRef CAS PubMed; (x) B. H. Northrop, D. Chercka and P. J. Stang, Tetrahedron, 2008, 64, 11495 CrossRef CAS PubMed; (y) B. H. Northrop, H. B. Yang and P. J. Stang, Chem. Commun., 2008, 5896 RSC; (z) E. Zangrando, M. Casanova and E. Alessio, Chem. Rev., 2008, 108, 4979 CrossRef CAS PubMed.
- (a) C. H. Amijs, G. P. van Klink and G. van Koten, Dalton Trans., 2006, 308 RSC; (b) F. Wurthner, C.-C. You and C. R. Saha-Moller, Chem. Soc. Rev., 2004, 33, 133 RSC.
- (a) M. Han, R. Michel, B. He, Y. S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319 CrossRef CAS PubMed; (b) T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5133 CrossRef CAS PubMed.
- S. Goeb, D. Canevet and M. Sallé, Organic Synthesis and Molecular Engineering, John Wiley & Sons, Inc., 2013, p. 213 Search PubMed.
- S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100 CrossRef CAS PubMed.
- (a) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS; (b) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509 RSC.
- Y. Furutani, H. Kandori, M. Kawano, K. Nakabayashi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2009, 131, 4764 CrossRef CAS PubMed.
- M. Yoshizawa, S. Miyagi, M. Kawano, K. Ishiguro and M. Fujita, J. Am. Chem. Soc., 2004, 126, 9172 CrossRef CAS PubMed.
- Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189 RSC.
- K. Nakabayashi, M. Kawano, T. Kato, K. Furukawa, S. Ohkoshi, T. Hozumi and M. Fujita, Chem. – Asian J., 2007, 2, 164 CrossRef CAS PubMed.
- W.-Y. Sun, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 2002, 124, 11570 CrossRef CAS PubMed.
- D. Bhattacharya, C. H. Chang, Y. H. Cheng, L. L. Lai, H. Y. Lu, C. Y. Lin and K. L. Lu, Chem. – Eur. J., 2012, 18, 5275 CrossRef CAS PubMed.
- P. H. Dinolfo, V. Coropceanu, J.-L. Brédas and J. T. Hupp, J. Am. Chem. Soc., 2006, 128, 12592 CrossRef CAS PubMed.
- P. H. Dinolfo, M. E. Williams, C. L. Stern and J. T. Hupp, J. Am. Chem. Soc., 2004, 126, 12989 CrossRef CAS PubMed.
- (a) F. Würthner, A. Sautter, D. Schmid and P. J. A. Weber, Chem. – Eur. J., 2001, 7, 894 CrossRef; (b) F. Wurthner and A. Sautter, Chem. Commun., 2000, 445 RSC.
- C. C. You and F. Wurthner, J. Am. Chem. Soc., 2003, 125, 9716 CrossRef CAS PubMed.
- K. Mahata, P. D. Frischmann and F. Wurthner, J. Am. Chem. Soc., 2013, 135, 15656 CrossRef CAS PubMed.
- (a) L. J. Chen, Q. J. Li, J. M. He, H. W. Tan, Z. Abliz and H. B. Yang, J. Org. Chem., 2012, 77, 1148 CrossRef CAS PubMed; (b) K. Ghosh, Y. Zhao, H. B. Yang, B. H. Northrop, H. S. White and P. J. Stang, J. Org. Chem., 2008, 73, 8553 CrossRef CAS PubMed; (c) H. B. Yang, K. Ghosh, Y. Zhao, B. H. Northrop, M. M. Lyndon, D. C. Muddiman, H. S. White and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 839 CrossRef CAS PubMed.
- G.-Z. Zhao, Q.-J. Li, L.-J. Chen, H. Tan, C.-H. Wang, D. A. Lehman, D. C. Muddiman and H.-B. Yang, Organometallics, 2011, 30, 3637 CrossRef CAS.
- K. Ghosh, J. M. Hu, H. B. Yang, B. H. Northrop, H. S. White and P. J. Stang, J. Org. Chem., 2009, 74, 4828 CrossRef CAS PubMed.
- (a) Q. J. Li, G. Z. Zhao, L. J. Chen, H. W. Tan, C. H. Wang, D. X. Wang, D. A. Lehman, D. C. Muddiman and H. B. Yang, Organometallics, 2012, 31, 7241 CrossRef CAS; (b) Q. Han, Q. J. Li, J. He, B. Hu, H. Tan, Z. Abliz, C. H. Wang, Y. Yu and H. B. Yang, J. Org. Chem., 2011, 76, 9660 CrossRef CAS PubMed; (c) G. Z. Zhao, Q. J. Li, L. J. Chen, H. W. Tan, C. H. Wang, D. X. Wang and H. B. Yang, Organometallics, 2011, 30, 5141 CrossRef CAS; (d) G.-Z. Zhao, L.-J. Chen, C.-H. Wang, H.-B. Yang, K. Ghosh, Y.-R. Zheng, M. M. Lyndon, D. C. Muddiman and P. J. Stang, Organometallics, 2010, 29, 6137 CrossRef CAS.
- K. Ghosh, J. M. Hu, H. S. White and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 6695 CrossRef CAS PubMed.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520 CrossRef CAS PubMed.
- N. Das, A. M. Arif, P. J. Stang, M. Sieger, B. Sarkar, W. Kaim and J. Fiedler, Inorg. Chem., 2005, 44, 5798 CrossRef CAS PubMed.
- (a) N. Martin, Chem. Commun., 2013, 49, 7025 RSC; (b) M. R. Bryce, J. Mater. Chem., 2000, 10, 589 RSC; (c) P. Batail, Chem. Rev., 2004, 4887 CrossRef CAS.
- (a) D. Canevet, M. Salle, G. X. Zhang, D. Q. Zhang and D. B. Zhu, Chem. Commun., 2009, 2245 RSC; (b) J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef CAS.
- J. Xiong, W. Liu, Y. Wang, L. Cui, Y.-Z. Li and J.-L. Zuo, Organometallics, 2012, 31, 3938 CrossRef CAS.
- E. Gontier, N. Bellec, P. Brignou, A. Gohier, M. Guerro, T. Roisnel and D. Lorcy, Org. Lett., 2010, 12, 2386 CrossRef CAS PubMed.
- (a) J. Y. Balandier, M. Chas, P. I. Dron, S. Goeb, D. Canevet, A. Belyasmine, M. Allain and M. Sallé, J. Org. Chem., 2010, 75, 1589 CrossRef CAS PubMed; (b) J. Y. Balandier, A. Belyasmine and M. Salle, Synthesis, 2006, 2815 CAS; (c) J. O. Jeppesen and J. Becher, Eur. J. Org. Chem., 2003, 3245 CrossRef CAS; (d) J. O. Jeppesen, K. Takimiya, F. Jensen, T. Brimert, K. Nielsen, N. Thorup and J. Becher, J. Org. Chem., 2000, 65, 5794 CrossRef CAS PubMed.
- (a) F. G. Brunetti, J. L. Lopez, C. Atienza and N. Martin, J. Mater. Chem., 2012, 22, 4188 RSC; (b) A. J. Moore and M. R. Bryce, J. Chem. Soc., Perkin Trans. 1, 1991, 157 RSC; (c) M. R. Bryce, A. J. Moore, M. Hasan, G. J. Ashwell, A. T. Fraser, W. Clegg, M. B. Hursthouse and A. I. Karaulov, Angew. Chem., Int. Ed. Engl., 1990, 29, 1450 CrossRef; (d) Y. Yamashita, Y. Kobayashi and T. Miyashi, Angew. Chem., Int. Ed. Engl., 1989, 28, 1052 CrossRef.
- V. Vajpayee, S. Bivaud, S. Goeb, V. Croué, M. Allain, B. V. Popp, A. Garci, B. Therrien and M. Sallé, Organometallics, 2014, 33, 1651 CrossRef CAS.
- (a) S. B. Nielsen, M. B. Nielsen and H. J. A. Jensen, Phys. Chem. Chem. Phys., 2003, 5, 1376 RSC; (b) R. Andreu, J. Garin and J. Orduna, Tetrahedron, 2001, 57, 7883 CrossRef CAS.
- J. Lau, P. Blanchard, A. Riou, M. Jubault, M. P. Cava and J. Becher, J. Org. Chem., 1997, 62, 4936 CrossRef CAS.
- (a) T. K. Hansen, T. Jorgensen, F. Jensen, P. H. Thygesen, K. Christiansen, M. B. Hursthouse, M. E. Harman, M. A. Malik, B. Girmay, A. E. Underhill, M. Begtrup, J. D. Kilburn, K. Belmore, P. Roepstorff and J. Becher, J. Org. Chem., 1993, 58, 1359 CrossRef CAS; (b) T. Jorgensen, J. Becher, T. K. Hansen, K. Christiansen, P. Roepstorff, S. Larsen and A. Nygaard, Adv. Mater., 1991, 3, 486 CrossRef.
- S. Goeb, S. Bivaud, V. Croué, V. Vajpayee, M. Allain and M. Sallé, Materials, 2014, 7, 611 CrossRef CAS PubMed.
- J.-Y. Balandier, M. Chas, S. Goeb, P. I. Dron, D. Rondeau, A. Belyasmine, N. Gallego and M. Sallé, New J. Chem., 2011, 35, 165 RSC.
- S. Goeb, S. Bivaud, P. I. Dron, J.-Y. Balandier, M. Chas and M. Sallé, Chem. Commun., 2012, 48, 3106 RSC.
- (a) D. Canevet, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248 CrossRef CAS PubMed; (b) E. M. Perez and N. Martin, Chem. Soc. Rev., 2008, 37, 1512 RSC.
- (a) S. Bivaud, S. Goeb, J. Y. Balandier, M. Chas, M. Allain and M. Salle, Eur. J. Inorg. Chem., 2014, 2440 CrossRef CAS; (b) S. Bivaud, J. Y. Balandier, M. Chas, M. Allain, S. Goeb and M. Sallé, J. Am. Chem. Soc., 2012, 134, 11968 CrossRef CAS PubMed.
- S. Bivaud, S. Goeb, V. Croue, P. I. Dron, M. Allain and M. Sallé, J. Am. Chem. Soc., 2013, 135, 10018 CrossRef CAS PubMed.
- (a) B. Therrien, Nanomaterials in Drug Delivery, Imaging, and Tissue Engineering, John Wiley & Sons, Inc., 2013, p. 145 Search PubMed; (b) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K. W. Chi, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS PubMed; (c) B. Therrien, in Chemistry of Nanocontainers, ed. M. Albrecht and E. Hahn, Springer, Berlin, Heidelberg, 2012, vol. 319, p. 35 Search PubMed.
- J. Mattsson, P. Govindaswamy, A. K. Renfrew, P. J. Dyson, P. Štěpnička, G. Süss-Fink and B. Therrien, Organometallics, 2009, 28, 4350 CrossRef CAS.
- S. Shanmugaraju, D. Samanta and P. S. Mukherjee, Beilstein J. Org. Chem., 2012, 8, 313 CrossRef CAS PubMed.
- D. Samanta, S. Shanmugaraju, A. A. Adeyemo and P. S. Mukherjee, J. Organomet. Chem., 2014, 751, 703 CrossRef CAS PubMed.
- V. Vajpayee, Y. H. Song, Y. J. Yang, S. C. Kang, T. R. Cook, D. W. Kim, M. S. Lah, I. S. Kim, M. Wang, P. J. Stang and K. W. Chi, Organometallics, 2011, 30, 6482 CrossRef CAS PubMed.
- Y.-F. Han, Y.-J. Lin, W.-G. Jia and G.-X. Jin, Organometallics, 2008, 27, 4088 CrossRef CAS.
- (a) J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833 RSC; (b) J. E. M. Lewis, C. John McAdam, M. G. Gardiner and J. D. Crowley, Chem. Commun., 2013, 49, 3398 RSC.
- F. Jiang, N. Wang, Z. Du, J. Wang, Z. Lan and R. Yang, Chem. – Asian J., 2012, 7, 2230 CrossRef CAS PubMed.
- S. S. Sun and A. J. Lees, Inorg. Chem., 2001, 40, 3154 CrossRef CAS PubMed.
- L. A. Berben, M. C. Faia, N. R. Crawford and J. R. Long, Inorg. Chem., 2006, 45, 6378 CrossRef CAS PubMed.
- (a) N. Shan, J. D. Ingram, T. L. Easun, S. J. Vickers, H. Adams, M. D. Ward and J. A. Thomas, Dalton Trans., 2006, 2900 RSC; (b) N. Shan, S. J. Vickers, H. Adams, M. D. Ward and J. A. Thomas, Angew. Chem., Int. Ed., 2004, 43, 3938 CrossRef CAS PubMed.
- M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102 CrossRef CAS PubMed.
- M. Frank, L. Krause, R. Herbst-Irmer, D. Stalke and G. H. Clever, Dalton Trans., 2014, 43, 4587 RSC.
- P.-C. Lin, H.-Y. Chen, P.-Y. Chen, M.-H. Chiang, M. Y. Chiang, T.-S. Kuo and S. C. N. Hsu, Inorg. Chem., 2011, 50, 10825 CrossRef CAS PubMed.
- M. Lahav, R. Gabai, A. N. Shipway and I. Willner, Chem. Commun., 1999, 1937 RSC.
- (a) J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC; (b) I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457 RSC; (c) P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297 CrossRef CAS PubMed; (d) F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561 CrossRef CAS.
- J. L. Boyer, M. Ramesh, H. Yao, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2007, 129, 1931 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |