
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of P-chirogenic phosphoramidate prodrugs of nucleoside analogues (ProTides) via copper catalysed reaction†
F.
Pertusati
* and
C.
McGuigan
School of Pharmacy and Pharmaceutical Sciences, Redwood Building, King Edwards VII Avenue, CF10 3NB, Cardiff, Wales, UK. E-mail: pertusatif1@cf.ac.uk
First published on 2nd April 2015
Abstract
The first copper-catalysed diastereoselective synthesis of P-chiral phosphoramidate prodrugs (ProTides) is reported. This procedure allows the synthesis of diastereomeric-enriched mixtures of ProTides. Application of this methodology to the asymmetric phosphorylation of purine and pyrimidine nucleoside analogues is presented.
Nucleoside analogues (NAs) are an important class of molecules accounting for half of all antiviral drugs currently on the market and a number of anticancer agents that are widely used.1 All NAs require phosphorylation to be active. Unfortunately, many nucleoside analogues are not phosphorylated effectively in vivo, and thus their therapeutic potential is often quite limited. Using various approaches, the ionizable phosphate group can be masked by derivatization, thus generating prodrugs with increased lipophilicity.2 Among different prodrug strategies is the ProTide (pronucleotide) approach, which we introduced. The ProTide of a nucleoside phosphate is a phosphoramidate prodrug consisting of an amino acid ester, promoiety linked via a P–N bond to a nucleoside aryl phosphate. The ProTide technology was successfully and extensively applied to a wide variety of nucleoside phosphates.3 Several leading pharmaceutical companies have applied this technology for anti-viral and anticancer treatments. Gilead, has just launched on the market its anti-HCV ProTide, Sofosbuvir 1 (PSI-7977)4 whereas Nucana Biomed has taken to trial a gemcitabine ProTide, (NUC-1031, 2)5 for patients with advanced solid tumours (Fig. 1). Gilead has exploited this technology to create an advanced anti-HIV drug (GS 7340), an acyclic phosphonate analogue now in Phase III clinical trial.6 To further highlight the importance of phosphoramidates we note that this functional group is also present in important biological active molecules such as Phosmidosine7 and Agrocin 84.8 Two different synthetic strategies for the preparation of phosphoramidate prodrugs are commonly used.9 In the first, tert-butyl magnesium chloride (tBuMgCl) is used as a base in the coupling reaction between a nucleoside and the phosphorochloridate bearing the desired promoieties. In the second approach, N-methyl imidazole (NMI) is used in place of tert-butyl magnesium chloride to promote the coupling. According to the two above-mentioned non-stereoselective procedures, phosphoroamidate prodrugs are generally prepared as 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of diastereoisomers (Rp and Sp) because of the newly formed chiral center at the phosphorus atom.
:1 mixtures of diastereoisomers (Rp and Sp) because of the newly formed chiral center at the phosphorus atom.
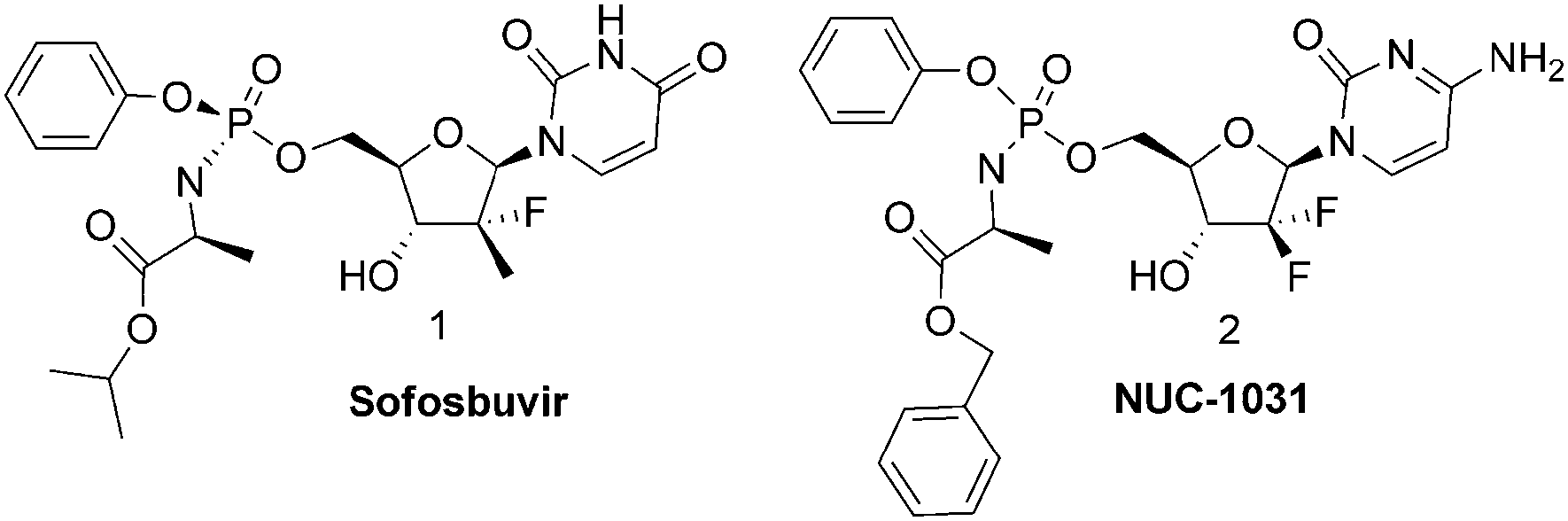 | ||
| Fig. 1 Structure of important phosphoroamidate prodrugs. | ||
Although stereoisomers have the same chemical structure they may exhibit differences in their pharmacology, toxicology, pharmacokinetics and metabolism. Up to now the phosphoramidate prodrugs cannot be easily prepared in the form of single diastereoisomers because of the lack of control of the stereochemistry at the phosphorus center during the synthesis. When it has been possible to separate the diastereoisomers, the biological activity has often been found dependent on the configuration on the phosphorus atom.10 For example, Sp diastereoisomer of the GS7340 is 10 fold more potent against HIV than the Rp diastereoisomer.10a Likewise, in HCV replicon assay Rp and Sp (more lipophilic) isomers of Sofosbuvir produced HCV activity with EC90 values of 7.5 μM and 0.42 μM respectively, thus demonstrating approximately 18 fold difference in activity between the two diastereoisomers.10b This has lead Gilead to launch Sofosbuvir as a single diastereoisomer, at considerable difficulty and expense. To select the optimal isomer for clinical development, the two single diastereomers of 1′-cyano-2′-C-methyl 4-aza-7,9-dideaza adenosine phosphoramidate, were profiled separately. The Sp isomer was found 6-fold more potent in the replicon assay and two times more efficient in the triphosphate formation in primary human hepatocytes.10c Finally, we were also able to demonstrate that phosphoramidates3a,b and phosphonoamidates3g diastereoisomers are processed at different rates in enzymatic assay by carboxypeptidase.
Separation of diastereoisomers by crystallization proved to be an inefficient method to use especially at the end of a multistep synthesis such as in the case of Sofosbuvir (a crystal of pure diastereoisomer was necessary). Chromatographic separation is often impossible or in general is a very difficult task to achieve, where reverse-phase chromatography provided successful results only in few cases.11 A multistep approach to obtain ProTides in a diastereoselective fashion has also been developed by the Meier group, using a chiral auxiliary on the phosphoramidating reagent.12 This methodology still requires the synthesis and chromatographic purification of the chiral auxiliary, which is generally unstable and cannot readily be isolated or recycled. This method is also limited by modest yields and was only documented on a nucleoside substrate lacking competing 2′,3′-hydroxyl groups. Ross and co-workers13 developed another synthetic method in which a diastereomerically pure phosphoramidating agent bearing a p-nitrophenyl or pentafluorophenyl as leaving groups was used. However, these reagents require purification by non-conventional supercritical fluid chromatography coupled with expensive chiral stationary phases. According to our experience they are also unstable and decompose easily, with racemization of the phosphorus stereocenter occurring. These features make all these methods not very efficient and especially high-priced, time consuming and hard to scale up. We were in search of a catalytic methodology that will allow generation of the ProTide motif in a diastereoisomeric fashion on a variety of biologically active molecules. Screening the literature, we came across with the work of Jones14 who developed a simple and effective method for the catalytic phosphorylation of alcohols using a chlorophosphate as phosphorylating agent in the presence of titanium catalyst. Since the common synthetic route toward ProTides implies the use of chlorophosphoroamidates9 (analogous to Jones chlorophosphate) we decided to test the possibility to yield ProTides with this approach having the intention next to tune the procedure to our advantage for the achievement of their diastereoselective synthesis. 2′-C-methyl-6-O-methyl guanosine 3 and 4a were selected respectively as model nucleoside and as phosphate source.15 The synthesis of the resulting phosphoramidate 5a (Rp:Sp 1:1 mixture) which reached phase two clinical trials against HCV virus, has been previously reported.15 When we treated a THF solution of 3 with phosphochloridate 4a in the presence of 0.2 equivalent of titanium chloride at room temperature, 5a was obtained in low yield (12%) and without any diastereoselectivity as revealed by RP-HPLC analysis of the crude reaction mixture (entry 1, Table 1). Not at all satisfied with the result achieved we turned our attention to another Jones report16 on the phosphoryl transfer from chiral N-phosphoryl-5,5-diphenyl oxazolidinone to alcohol under copper(II) triflate catalysis. Since in these studies the phosphorylation was reported to be successful on two examples of nucleosides, we decided to evaluate the catalytic activity of Cu(OTf)2 in the synthesis of ProTide 5a. We reacted nucleoside 3 with chloridate 4a in the presence of 0.2 equivalent of Cu(OTf)2, 1.5 equivalent of triethylamine and 0.1 equivalent of N,N′-ethylene bis-(benzaldimine) (BEN) as metal ligand in THF solution at r.t. The desired product 5a was obtained after 12 hours in 14% yield with a diastereomeric ratio 1:2.5 (RP-HPLC of crude mixture) in favour of the Sp isomer (Table 1, entry 2). Intrigued by the first indication of diastereoselectivity we decided to investigate the importance of the presence of BEN as metal ligand. When BEN was excluded from the reaction mixture, ProTide 5a was obtained in 37% yield and in a 1:7 Rp:Sp diastereomeric ratio (RP-HPLC) (Scheme 1, Table 1, entry 3). The yield was moderate but the diastereoisomeric excess was striking. Encouraged by these results we proceeded to further investigate the reaction conditions in order to find those optimal in terms of conversion and diastereoselectivity. A comprehensive screen of different parameters such as base, metal catalyst and solvent were thus performed (Table 1).
| Entry | MXn (equiv.) | Base | Solvent | Conversion to 5aa (%) | R p/Sp d.r.a |
|---|---|---|---|---|---|
| Reaction conditions: 3 100 mg, MXn 0.2 equiv.; 4a 1 equiv.; solvent 10 mL; base 1.5 equivalent; ambient temperature, 12 h.a Determined by RP-HPLC analysis of the crude mixture.‡b Isolated yield.c N,N′-Ethylene bis-benzaldimine, (BEN) used as ligand (2 mol%). | |||||
| 1 | TiCl4 | NEt3 | THF | 12 | 1:2.5 |
| 2c | Cu(OTf)2 | NEt3 | THF | 14 | 1:2.5 |
| 3 | Cu(OTf)2 | NEt3 | THF | 37 (28)b | 1:6.2 |
| 4 | Cu(OTf)2 | DBU | THF | 5 | 1:3.3 |
| 5 | Cu(OTf)2 | DMAP | THF | 4 | 1:2.5 |
| 6 | Cu(OTf)2 | NMI | THF | 1 | 1:1 |
| 7 | Cu(OTf)2 | i-Pr2NH | THF | 34 | 1:3 |
| 8 | Cu(OTf)2 | DIPEA | THF | 47 | 1:2.5 |
| 9 | Cu(OTf)2 | — | THF | 0 | — |
| 10 | Yb(OTf)3 | NEt3 | THF | 20 | 1:2 |
| 11 | Fe(OTf)3 | NEt3 | THF | 15 | 1:1.7 |
| 12 | La(OTf)3 | NEt3 | THF | 20 | 1.1:1 |
| 13 | AgOTf | NEt3 | THF | 0 | — |
| 14 | Cu(OAc)2·H2O | NEt3 | THF | 40 | 1:2.5 |
| 15 | CuI | NEt3 | THF | 22 | 1:3.2 |
| 16 | Cu(OAc)2·H2O | NEt3 | THF | 39 | 1:2 |
| 17 | Cu(SO4)2·H2O | NEt3 | THF | 10 | 1:3.8 |
| 18 | CuOAc | NEt3 | THF | 9 | 1:5 |
| 19 | — | NEt3 | THF | 0 | — |
| 20 | Cu(OTf)2 | NEt3 | Ethylene glycol | 0 | — |
| 21 | Cu(OTf)2 | NEt3 | 1,4 Dioxane | 38 | 1:2.3 |
| 22 | Cu(OTf)2 | NEt3 | DME | 14 | 1:5 |
| 23 | Cu(OTf)2 | NEt3 | CH3CN | Traces | 1 |
| 24 | Cu(OTf)2 | NEt3 | Toluene | Traces | 1:1.5 |
| 25 | Cu(OTf)2 | NEt3 | Pyridine | NR | — |
| 26 | Cu(OTf)2 | NEt3 | CHCl3 | Traces | 1:1.8 |
| 27 | Cu(OTf)2 | NEt3 | CH2Cl2 | Traces | 1:1.6 |
| 28 | Cu(OTf)2 | DIPEA | DME | 40 (35) |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8 :8
|
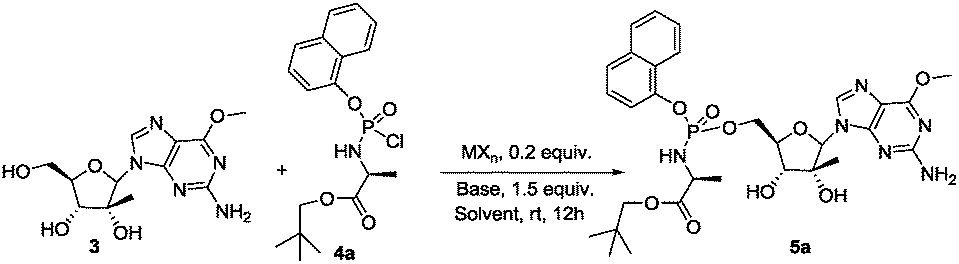 | ||
| Scheme 1 Synthesis of 5a under transition metal catalysed conditions. | ||
First, the nature of the base was probed (Table 1, entries 3–9). It can be observed that heterocyclic bases such as DBU, DMAP and NMI (Table 1, entries 3–5), were ineffective in promoting the reaction and returned the desired compounds only in traces amount. Most of the other bases screened were efficient in terms of product yield, with the best results obtained with alkyl amines. When the reaction yield was considered, diisopropyl ethylamine (DIPEA) was the best amine yielding the phosphoroamidate in 47% yield even if with a poor diastereomeric ratio (Table 1, entry 8). The best diastereomeric ratio was instead obtained with NEt3 in absence of ligand (Table 1, entry 3). It is interesting to note that no reaction occur in absence of the base (Table 1, entry 9). Because of these results triethylamine was selected as standard base for further screenings. Several metal salts were also evaluated (Table 1, entries 10 and 19). Since the absence of metal ligand seems to be beneficial to the reaction we decided to assess all these metal salts under ligand-free conditions. We began our investigation by screening ytterbium, iron, and lanthanum triflates. In all these cases 5a was formed in low yield and poor diastereoselectivity. Interestingly, the use of lanthanum triflate, in contrast with all other metal triflates, although in minimum amount, led to the inversion of the diastereoselectivity slightly in favour of Rp isomer. To evaluate the effect of the oxidation state of the copper as well as the effect of the counter ion on the outcome of the reaction we screened also other copper salts. Table 1 report some of the results obtained. Cu(OTf)2 was found to be the most suitable catalyst (Table 1, entry 3). Increasing the catalyst loading to 1 equivalent did not affect either the yield or diastereoselectivity. Our control experiment without catalyst yielded no product 5a (Table 1, entry 19).
| Entry | Cpds | R | R1 | R2 | Conversion to 5a–ea (%) | R p/Sp d.r.a |
|---|---|---|---|---|---|---|
| Reaction conditions: 3 100 mg, Cu(OTf)2 0.2 equiv.; 4a–e 1 equiv.; DME 20 mL; DIPEA 1.5 equivalent; r.t., 12 h.a Determined by RP-HPLC of the crude mixture.b Isolated yield. | ||||||
| 1 | 4a | CH3 | Naph | CH2tBu | 40 (38)b | 1:8.3 |
| 2 | 4b | CH3 | Ph | CH2tBu | 54 (27)b | 1:3 |
| 3 | 4c | CH3 | Naph | Bn | 72 (66)b | 1:6 |
| 4 | 4e | CH3 | Naph | CH(CH3)2 | 18 (12)b | 1:5.4 |
| 5 | 4d | CH(CH3)2 | Naph | Bn | 43 (35)b | 1:7.5 |
Finally, an assessment of the effect of the reaction medium on stereoselectivity and yield was performed (Table 1, entries 20–28). The use of ethereal solvents (except ethylene glycol) provided synthetically useful quantities of 5a maintaining a certain degree of diastereoselectivity. On the contrary, alkyl halide and aromatics solvents as well as pyridine and acetonitrile were less successful, yielding only traces of the desired phosphoramidate product 5a. Since in the base screening DIPEA was providing the best results in terms of yield we decided to assess DIPEA in combination with DME (Table 1, entry 28). To our delight in this conditions phosphoramidate 5a was obtained in 40% yield and in a 1:8.3 Rp/Sp d.r. as assessed by RP-HPLC analysis of the crude reaction mixture (Fig. 2). 5a (Rp/Sp d.r. 1:8) was isolated by column chromatography in 35% yield, and its identity was confirmed by comparison with literature data.‡
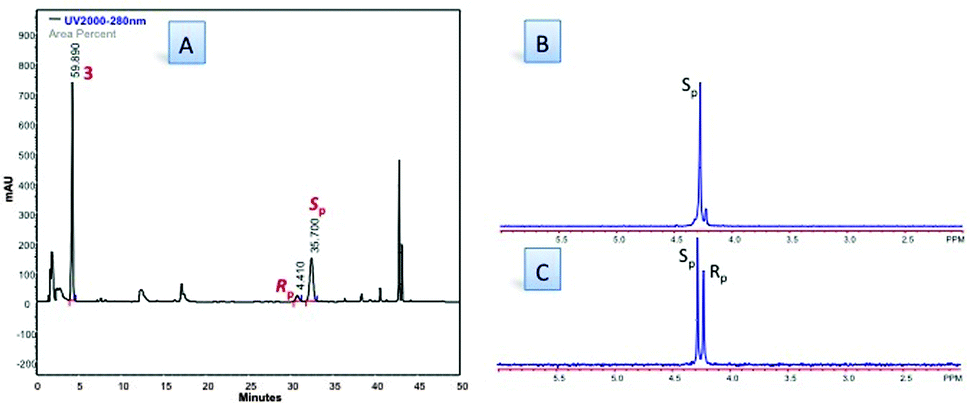 | ||
| Fig. 2 (A) HPLC traces of crude reaction mixture (Table 1, entry 28). HPLC conditions: 90:10 H2O/CH3CN to 0:100 H2O/CH3CN in 30 min. Flow = 1 mL min−1; λ = 280 nm; (B) 31PNMR (CD3OD, 202 MHz) of 5a (Rp:Sp d.r. = 1.2:8.8); (C) 31PNMR (CD3OD, 202 MHz) of 5a (Rp:Sp d.r. = 1:1). | ||
The optimum reaction time was found in all cases to be 8–12 h, with lower conversion being observed with shorter reaction times. Variation of reaction temperature did not improve our results. Optimal conditions were found with Cu(OTf)2 (0.2 equiv.), DIPEA (1.5 equiv.) in DME at room temperature for 12 h. The nature of the phosphorochloridate on the reaction outcome was then investigated (Scheme 2 and Table 2).
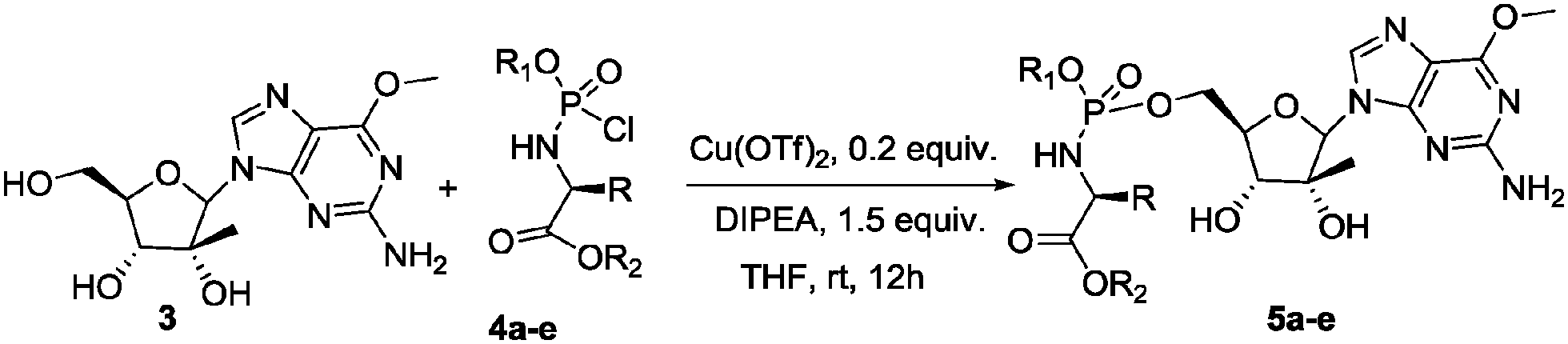 | ||
| Scheme 2 Synthesis of phosphoramidates 5a–e under transition metal catalysed conditions. | ||
From the data in Table 2 it appears immediately clear that bulky substituents on the phosphochloridate partner exert a certain effect on the stereoselectivity. In fact, high diastereoselectivity is obtained with neopentyloxy-L-alanyl phosphorochloridate 4a bearing a naphthyl group as aryloxy moiety, whereas a dramatic decrease in diastereoselectivity is observed with 4b bearing the less bulky phenyl group (Table 2, entries 1 vs. 2). Moreover, if the naphthyloxy phosphorochloridate 4c affords the phosphoroamidate 5b with a moderate diastereoisomeric ratio, the analogue phosphochloridate 4d bearing the more sterically hindered L-valinyl benzyl ester yielded 5d with an improved diastereoselectivity (Table 2 entries 3 vs. 5). Moderate diastereoselectivity was also obtained for 5e starting from phosphochloridate 4e. Compounds 5a–e were all isolated by column chromatography on silica gel and fully characterized (see ESI†). Finally, it is noteworthy to highlight that in all the above reactions, the 5′ phosphoramidate is formed exclusively (1H-NMR and MS analysis) despite the fact that two OH groups are not protected. This constitutes a great synthetic advantage over other available methods since it avoids tedious protection–deprotection steps typical of nucleoside chemistry.
Having a successful protocol in hand its application to other nucleosides was investigate. Given our interest in the anticancer field we decided to apply this protocol to Clofarabine 6 and Nelarabine 7. The results are collected in Scheme 3. Phosphoroamidate 9a was obtained with an excellent conversion (70% isolated yield) and remarkably with opposite diastereoselectivity (d.r. = 3.5:1).
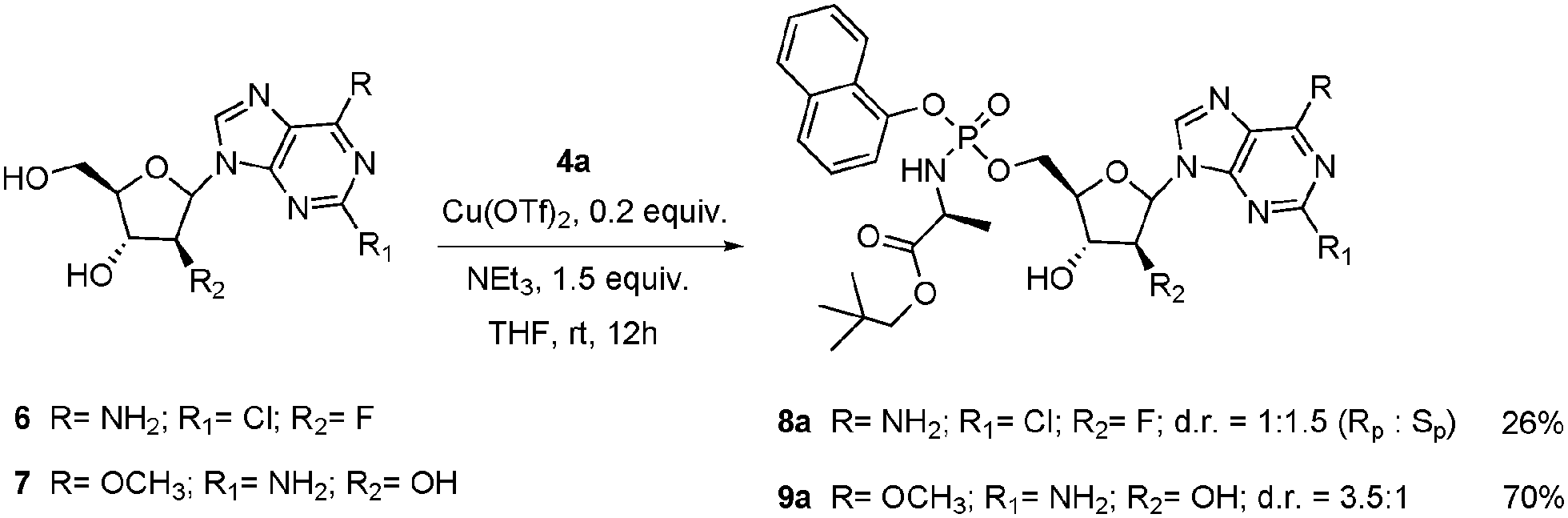 | ||
| Scheme 3 Scope of purine nucleosides, ProTides 8a and 9a. Isolated yield after column chromatography. | ||
To further expand the scope of the present methodology and especially because the recent approval of the anti-HCV drug Sofosbuvir (as single diastereoisomer)3 we decided to apply our protocol to pyrimidine nucleosides. 2′-Deoxy-2′-fluorouridine 10 was selected as model nucleoside (Scheme 4). Disappointingly, when 10 was reacted with phosphochloridate 4e under the optimized conditions, the phosphoroamidate 11e was recovered with a moderate conversion (38% by crude mixture HPLC analysis) with no remarkable diastereoselectivity (Table 3, entry 1). This prompted us to investigate further the reaction conditions in case of pyrimidine nucleosides.
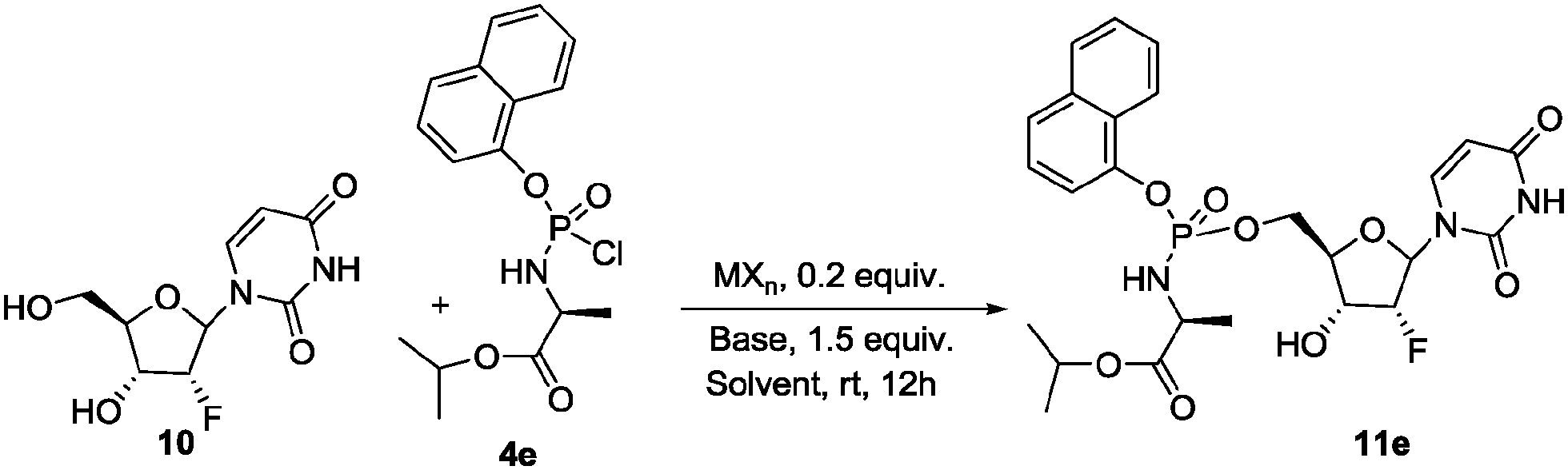 | ||
| Scheme 4 Synthesis of 11e under transition metal catalysed conditions. | ||
| Entry | MXn | Base | Solvent | Conversion to 11ea (%) | d.r.a |
|---|---|---|---|---|---|
| Reaction conditions: 10 100 mg, 4a 1 equiv.; solvent 20 mL.a Determined by RP-HPLC of the crude mixture.b Isolated yield.c 3′-O-regioisomer was observed. | |||||
| 1 | Cu(OTf)2 | DIPEA | DME | 42 | 1:1 |
| 2 | Yb(OTf)3 | DIPEA | DME | 35c | 1:1.7 |
| 3 | Fe(OTf)3 | NEt3 | THF | Traces | — |
| 4 | La(OTf)3 | NEt3 | THF | 50 | 1:2 |
| 5 | Cu(OAc)2 | DIPEA | DME | Traces | — |
| 6 | CuSO4 | NEt3 | THF | 60 | 1:2.8 |
| 7 | CuOAc | DIPEA | DME | 35 |
1:6.3
|
| 8 | CuI | NEt3 | THF | 41 (28)b | 1:2.1 |
| 9 | CuCl | NEt3 | THF | 53 | 1:1.5 |
| 10 | Cu(CF3CO2)2 | DIPEA | DME | 60 | 1:4.2 |
| 11 | Cu(OTf)·C6H6 | DIPEA | THF | 40 | 1:2.2 |
| 12 | (MeCN)4·CuOTf | NEt3 | THF | 60 | 1:2.4 |
Cu(OTf)2 under a variety of conditions (solvent, base, etc.) was unproductive, returning the desired phosphoroamidate 11e in poor diastereoselective manner. Similar results were obtained screening other metal triflates (Table 3, entries 2–4). We therefore returned our attention to other copper salts-complexes. The results are collected in Table 3. Copper(I) acetate proved to be the best catalyst in terms of diatereoselectivity returning 11e in 35% yield and 1:6.3 d.r. (Table 3, entry 7).§ The use of Cu(CF3CO2)2 enhance the yield at the expense of the d.r. (Table 3, entry 10).
In conclusion we have developed a catalytic system capable of delivery the Sp diastereoisomer of phosphoramidate prodrug of nucleoside analogues in a good diastereomeric enhanced fashion even if still in moderate yield. Cu(OTf)2 proved to be the catalyst of choice for purine nucleosides whereas CuOAc appeared to be superior for pyrimidine nucleosides. This methodology can be successfully applied to a diverse set of nucleosides and phosphorochloridates. Further studies of this procedure will focus on improving the yield while retaining or further enhancing the diastereoselectivity. Investigations for elucidating the mechanism are currently underway in our laboratories. Jonah Wilkes is acknowledged for the synthesis of one ProTide.
Notes and references
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discov., 2013, 12, 447–464 CrossRef CAS PubMed.
- (a) S. J. Hecker and M. D. Erion, J. Med. Chem., 2008, 51, 2328–2345 CrossRef CAS PubMed; (b) L. W. Peterson and C. E. McKenna, Expert Opin. Drug Delivery, 2009, 6, 405–420 CrossRef CAS.
- (a) C. McGuigan, P. Murziani, M. Slusarczyk, B. Gonczy, J. Vande Voorde, S. Liekens and J. Balzarini, J. Med. Chem., 2011, 54, 7247–7258 CrossRef CAS; (b) M. Slusarczyk, M. H. Lopez, J. Balzarini, M. Mason, G. W. Jiang, S. Blagden, E. Thompson, E. Ghazaly and C. McGuigan, J. Med. Chem., 2014, 57, 1531–1542 CrossRef CAS; (c) K. S. Toti, M. Derudas, F. Pertusati, D. Sinnaeve, F. Van den Broeck, L. Margamuljana, J. C. Martins, P. Herdewijn, J. Balzarini, C. McGuigan and S. Van Calenbergh, J. Org. Chem., 2014, 79, 5097–5112 CrossRef CAS; (d) C. McGuigan, M. Derudas, B. Gonczy, K. Hinsinger, S. Kandil, F. Pertusati, M. Serpi, R. Snoeck, G. Andrei, J. Balzarini, T. D. McHugh, A. Maitra, E. Akorli, D. Evangelopoulos and S. Bhakta, Bioorg. Med. Chem., 2014, 22, 2816–2824 CrossRef CAS PubMed; (e) W. Wu and R. F. Borch, Mol. Pharmacol., 2006, 3, 451–456 CrossRef CAS PubMed; (f) W. Wu, J. Sigmond, G. J. Peters and R. F. Borch, J. Med. Chem., 2007, 50, 3743–3746 CrossRef CAS PubMed; (g) F. Pertusati, K. Hinsinger, Á. S. Flynn, N. Powell, A. Tristram, J. Balzarini and C. McGuigan, Eur. J. Med. Chem., 2014, 78, 259–268 CrossRef CAS PubMed.
- “Approval of Sovaldi (Sofosbuvir) tablets for the treatment of chronic hepatitis C”. FDA. 7 Dec 2013. Retrieved 9 June 2014.
- http://clinicaltrials.gov/ct2/show/NCT01621854?term=nuc1031&rank=3 .
- http://clinicaltrials.gov/ct2/show/NCT01497899?term=gs7340&rank=3 .
- D. R. Phillips, M. Uramoto, K. Isono and J. A. McCloskey, J. Org. Chem., 1993, 58, 854–859 CrossRef CAS.
- M. E. Tate, P. J. Murphy, W. P. Roberts and A. Kerr, Nature, 1979, 280, 697 CrossRef CAS.
- M. Serpi, K. Madela, F. Pertusati and M. Slusarczyk, Synthesis of nucleotide prodrugs using the proTide approach in Curr. Protoc. Nucleic Acid Chem., John Wiley & Sons, Inc. , 2013, vol. 53, pp. 15.5.1–15.5.15 Search PubMed.
- (a) M. K. H. Chapman, E. Prisbe, J. Rohloff, M. Sparacino, T. Terhorst and R. Yu, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 621–625 CrossRef PubMed; (b) M. J. Sofia, D. Bao, W. Chang, J. Du, D. Nagarathnam, S. Rachakonda, P. G. Reddy, B. S. Ross, P. Wang, H.-R. Zhang, S. Bansal, C. Espiritu, M. Keilman, A. M. Lam, H. M. M. Steuer, C. Niu, M. J. Otto and P. A. Furman, J. Med. Chem., 2010, 53, 7202–7218 CrossRef CAS PubMed; (c) A. Cho, L. Zhang, J. Xu, R. Lee, T. Butler, S. Metobo, V. Aktoudianakis, W. Lew, H. Ye, M. Clarke, E. Doerffler, D. Byun, T. Wang, D. Babusis, A. C. Carey, P. German, D. Sauer, W. Zhong, S. Rossi, M. Fenaux, J. G. McHutchison, J. Perry, J. Feng, A. S. Ray and C. U. Kim, J. Med. Chem., 2013, 57, 1812–1825 CrossRef PubMed.
- (a) N. Mesplet, Y. Saito, P. Morin and L. A. Agrofoglio, J. Chromatogr. A, 2003, 983, 115–124 CrossRef CAS; (b) C. J. Allender, K. R. Brain, C. Ballatore, D. Cahard, A. Siddiqui and C. McGuigan, Anal. Chim. Acta, 2001, 435, 107–113 CrossRef CAS.
- (a) J. B. C. Arbelo Roman and C. Meier, J. Med. Chem., 2010, 53, 7675 CrossRef PubMed; (b) C. Arbelo Román, P. Wasserthal, J. Balzarini and C. Meier, Eur. J. Org. Chem., 2011, 4899 Search PubMed.
- P. G. Reddy, B.-K. Chun, H.-R. Zhang, S. Rachakonda, B. S. Ross and M. J. Sofia, J. Org. Chem., 2011, 76, 3782–3790 CrossRef CAS PubMed.
- S. Jones and D. Selitsianos, Org. Lett., 2002, 4, 3671–3673 CrossRef CAS PubMed.
- C. McGuigan, K. Madela, M. Aljarah, A. Gilles, A. Brancale, N. Zonta, S. Chamberlain, J. Vernachio, J. Hutchins, A. Hall, B. Ames, E. Gorovits, B. Ganguly, A. Kolykhalov, J. Wang, J. Muhammad, J. M. Patti and G. Henson, Bioorg. Med. Chem. Lett., 2010, 20, 4850–4854 CrossRef CAS PubMed.
- S. Jones and C. Smanmoo, Org. Lett., 2005, 7, 3271–3274 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization data and copies of NMR spectra, HPLC traces. See DOI: 10.1039/c5cc00448a |
| ‡ The absolute configuration of the Sp isomer of 5a was determined by comparison of the 31P and 1H NMR spectra of our sample with the 31P and 1H NMR spectra reported in the patent WO 2010/081082 were the absolute configuration at the phosphorus was assigned by vibrational circular dichroism. |
| § Separation of the RP and SP11e diastereoisomers was achieved by preparative HPLC (see ESI†). Unfortunately, no suitable crystals for X-ray analysis were obtained. |
| This journal is © The Royal Society of Chemistry 2015 |