
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective photodeposition of Pt on a particulate Sc-La5Ti2CuS5O7 photocathode: evidence for one-dimensional charge transfer†
Guijun
Ma
ab,
Jingyuan
Liu
a,
Takashi
Hisatomi
ab,
Tsutomu
Minegishi
ab,
Yosuke
Moriya
ab,
Motoki
Iwase
ab,
Hiroshi
Nishiyama
bc,
Masao
Katayama
ab,
Taro
Yamada
bc and
Kazunari
Domen
*ab
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp
bJapan Technological Research Association of Artificial Photosynthetic Chemical Process (ARPChem), 5-1-5 Kashiwanoha, Kashiwa-shi, 277-8589 Chiba, Japan
cDepartment of Chemical System Engineering, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-shi, 277-8589 Chiba, Japan
First published on 4th February 2015
Abstract
Photodeposition of Pt on the Sc-doped La5Ti2CuS5O7 (Sc-LTC) photocathode, a visible-light-responsive semiconducting oxysulfide, is accomplished in a solution containing H2PtCl6 and K2C2O4. Pt particles are selectively deposited on the top surface of rod-like Sc-LTC particles as a result of one-dimensional transfer of photogenerated electrons.
Photoelectrochemical (PEC) H2 production from water utilizing solar energy has attracted much attention in recent years because of concerns about environmental pollution and energy shortage. A number of semiconductors with visible-light activity have been developed as photoelectrodes for water splitting, because only 4.6% of the solar energy distribution consists of UV light (λ < 400 nm) whereas visible light (400 nm < λ < 800 nm) constitutes 54.3% of the distribution.1–3 The process of PEC water splitting can be divided into three main steps:1–4 (1) photon absorption by semiconductors for excitation of electron–hole pairs, (2) migration of minority and majority carriers to, respectively, the photoelectrode/electrolyte interface and the counter electrode/electrolyte interface via a backside electrode, and (3) (photo)electrochemical hydrogen and oxygen evolution reactions on the surfaces of the (photo)electrodes. Undesirable charge recombination competes with the above steps. It is therefore essential to improve the charge separation efficiency and charge mobility in a semiconductor photoelectrode in order to suppress charge recombination and improve the efficiency of PEC water splitting.
The diffusion length of minority carriers, which reflects their lifetime and diffusion coefficient, is a critical factor for PEC properties. A semiconductor photoelectrode having a short charge diffusion length has to be small so that excited minority carriers can reach the surface before recombination. For example, most hematite (α-Fe2O3) photoanodes are prepared at the nanoscale because the hole diffusion length for hematite is 2–4 nm, which is much shorter than that for TiO2 (∼1 μm) or WO3 (∼100 nm).5–7 However, the thinness of the photoelectrode inevitably results in inadequate light absorption. To improve the charge separation efficiency and charge mobility, one idea is to produce functional surface states (e.g., surface microstructure) having an atypical energy potential so that the photogenerated electrons and/or holes are transferred in different directions. Many highly efficient photocatalysts have been reported that are based on this mechanism, such as NaTaO3:La,8 Cd0.5Zn0.5S9 and anatase/rutile.10 Another idea is to selectively load cocatalysts on the reduction and/or oxidation site of a photocatalyst, because it is possible that randomly loaded cocatalysts will act as recombination centers for photogenerated electrons and holes.11–14 Taking BiVO4 as a model semiconductor, Li et al. reported the enhancement of the photocatalytic or photoelectrochemical water oxidation reaction by controllable deposition of oxidation/reduction cocatalysts on the corresponding oxidation/reduction facets.11
La5Ti2CuS5O7 (LTC), with an absorption edge wavelength of 650 nm, equal to a band gap of 1.9 eV, was reported by our group as a visible-light-responsive oxysulfide photocatalyst and photocathode for water splitting.15–17 The photocatalytic activity for both H2 and O2 evolution under sacrificial reagents implies that the valence and conduction band edges of LTC straddle the hydrogen and oxygen evolution redox potentials.15,16 It was recently found that the PEC performance of LTC photocathodes was significantly increased by doping with the appropriate amount of Sc (abbreviated as Sc-LTC, hereafter).17 Thus, we developed a method to load Pt as a hydrogen evolution catalyst on the edge of rod-like Sc-LTC particles selectively via PEC reduction in an electrolyte solution containing H2PtCl6 and K2C2O4. As a result, the hypothetic half-cell solar-to-hydrogen conversion efficiency (HC-STH) near the onset potential of the photocathodic current was improved. Tracking of the Pt nanoparticles loaded on the surface suggested that the Sc-LTC photocathode was capable of one-dimensional charge transfer.
Sc-LTC powder was prepared by a solid-state reaction (see ESI†). Scanning electron microscopy (SEM) images showed that the produced Sc-LTC comprised highly crystallized rod material with diameters and lengths ranging from 0.7 to 1.4 μm and from 2 to 6 μm (Fig. S1 in the ESI†), respectively, reflecting the crystal structure. Sc-LTC particles were embedded into a Au thin film by the particle transfer (PT) method (Fig. S2 in the ESI†).18 This Sc-LTC/Au assembly functioned as a photocathode. Pt was loaded on Sc-LTC/Au by PEC reduction in a three-electrode system. The electrolyte solution used for photodeposition was an aqueous Na2SO4 solution (0.1 M, 100 mL) containing K2C2O4 (0.1 M) and H2PtCl6 (3.5 × 10−6 M for subsequent PEC measurements and 10 × 10−6 M for SEM and EDX measurements). Fig. S3 in the ESI† shows that the Pt deposition process was accomplished in 1 h. For comparison, a sputtering method was employed to deposit Pt particles with an optimized nominal thickness of 1 nm on Sc-LTC/Au.17 Pt loaded by in situ PEC reduction and sputtering will hereafter be abbreviated as Pt-photo and Pt-sputter, respectively. PEC hydrogen evolution reactions were carried out using Pt-photo and Pt-sputter Sc-LTC/Au photocathodes in a fresh aqueous Na2SO4 solution (0.1 M, 100 mL) whose pH was adjusted to 10 with a diluted aqueous NaOH solution. The photocathode samples were illuminated with a solar simulator.
Fig. 1(A) shows current–potential curves of Sc-LTC/Au photocathodes with and without Pt as a catalyst for PEC hydrogen evolution under simulated solar illumination (AM 1.5G). Compared with the unmodified sample, the photocathodic current was significantly increased on both Pt-photo and Pt-sputter Sc-LTC/Au photocathodes, and the former showed a higher cathodic current as the electrode potential was varied from 0.0 to 0.9 V vs. RHE. This difference leveled off, especially at potentials more negative than 0 V vs. RHE, the thermodynamic potential for H2 evolution from water. It is noted that, for a photocathode, a higher photocathodic current at a more positive potential is particularly meaningful for HC-STH and for applications in p/n PEC cells employing another photoanode to accomplish overall water splitting. As shown in Fig. 1(B), the maximum HC-STH was estimated to be 0.07% (at 0.65 V) and 0.05% (at 0.45 V) for Pt-photo and Pt-sputter Sc-LTC/Au photocathodes, respectively.
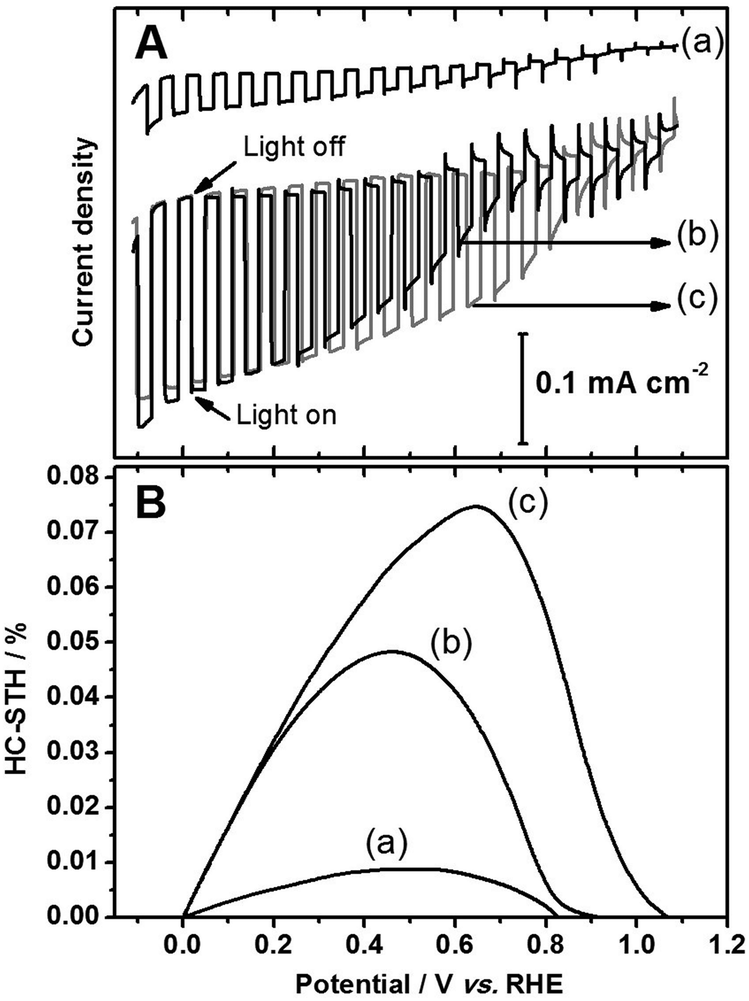 | ||
| Fig. 1 Current–potential curves (A) and HC-STH (B) for (a) Sc-LTC/Au, (b) Pt/Sc-LTC/Au-sputter and (c) Pt/Sc-LTC/Au-photo electrodes under solar light irradiation (AM 1.5G). The electrolyte is a 0.1 M aqueous Na2SO4 solution (pH = 10). | ||
In previous research, PEC loading of Pt was accomplished by using PtCl62− ions as a precursor in the absence of reducing reagents such as K2C2O4.19–21 However, in our study, Pt could be loaded on Sc-LTC/Au photocathodes slowly and the addition of K2C2O4 accelerated the photodeposition. This is presumably due to kinetic reasons rather than thermodynamics, because the potential of the conduction band edge of the Sc-LTC semiconductor is more negative than the hydrogen evolution potential and thus the redox potentials of PtCl62− and possible intermediates (see ESI†). K2C2O4 was reported to produce strongly reducing CO2˙− radicals via oxidation by electrolysis or photocatalysis.22–24 The potential of the CO2˙−/CO2 couple was reported to be about −1.96 V vs. SHE. Therefore, CO2˙− is capable of electron transfer to many metal ions including PtCl62−. It is believed that in the present three-electrode PEC reaction system, the CO2˙− radicals produced on a counter electrode could reduce PtCl62− ions into more reactive intermediates.23 Such intermediates could be reduced to Pt metal more readily by photogenerated electrons on Sc-LTC photocathodes. Note that, through the same process as that used for Pt loading, other noble metals, such as Rh, Ir, Pd and Ru, were loaded on Sc-LTC as a catalyst; however, these did not lead to as significant an enhancement in PEC H2 evolution as Pt did.
Tracking the Pt particles deposited on the surface of Sc-LTC can provide information about the reduction sites of this material, namely, the transfer direction of photogenerated electrons. Fig. 2 shows SEM images of Sc-LTC/Au electrodes before and after PEC deposition of Pt. It is found that small Pt particles (most <10 nm) were deposited on the edge of the Sc-LTC rod upon photoreduction of PtCl62− ions, while no such particles were observed on the lateral side of the rod. In addition, Pt particles were selectively loaded on a step when the Sc-LTC rod was broken into half and a step was exposed (Fig. 2d and Fig. S4, ESI†). EDX analysis of Pt was carried out on different sites of a Sc-LTC rod loaded with excess Pt (Fig. 3) to confirm the site-selective photoreduction of Pt nanoparticles. A line scan is plotted over a distance of ca. 1.3 μm to show the distribution of Pt from the middle to top of the Sc-LTC rod. It can be seen from the top right of Fig. 3 that the Pt signals detected on the body of the Sc-LTC rod was within the noise level, whereas a significant increase was observed at the edge of the rod. The Pt concentrations were compared by collecting EDX signals from different parts of the Sc-LTC rod. As tabulated in the table in Fig. 3, the Pt concentration at point a (the edge of a Sc-LTC particle) was higher than the whole area measured, whereas only a negligible Pt signal was detected at point b (the lateral part of the Sc-LTC particle). These observations strongly suggest that, for the rod-like Sc-LTC semiconductor particles, photogenerated electrons transfer along the long axis of the rod (i.e., one-dimensionally) until reaching any transection plane (top or broken parts) where reduction reactions occur.
 | ||
| Fig. 2 SEM characterization of Sc-LTC/Au (a and b) and Pt/Sc-LTC/Au (c and d) photoelectrodes. Pt is loaded by photodeposition. All the scale bars are 300 nm. | ||
 | ||
| Fig. 3 EDX analysis of Pt distribution on a Pt/Sc-LTC/Au photoelectrode. Pt is loaded by in situ photodeposition. Top right: plot of a line scan. Bottom right: spot and overall content. The scale bar is 1 μm. | ||
It may be concerned that the site-selective photodeposition of Pt was due to the selective adsorption of K2C2O4 or PtCl62− species on different crystal faces of Sc-LTC rods. To exclude such possibilities, photodeposition of Pt and Pd on Sc-LTC was carried out in electrolyte solutions containing PtCl62− (without K2C2O4), (NH4)2PdCl4 and K2C2O4, respectively. SEM and EDX analyses (Fig. S5 and S6, ESI†) show that, in both cases, Pt and Pd particles were selectively deposited on the top faces of Sc-LTC rods, which confirms that the site-selective photodeposition is caused by the selectivity of charge transfer rather than the selectivity of absorption.
It should be noted that Pt-photo was deposited at the edge of LTC rods although these rods were as long as 6 μm. In the crystal structure of LTC, TiOxS6−x (x = 4 or 5) octahedra and CuS4 tetrahedra are stacked along the b-axis (see Fig. S7, ESI†).16,25 DFT calculations revealed that the valence band edge of LTC composed of Cu 3d and S 3p hybrid orbitals was localized around CuS4 tetrahedra and the conduction band edge composed of Ti 3d orbitals was localized around TiOxS6−x (x = 4 or 5) octahedra.16 Such a disassociated structure could be favorable for efficient charge separation. Furthermore, photoexcited electrons and holes could move through linear chains of TiOxS6−x and CuS4, respectively. Accordingly, charge carriers could migrate over a long distance, comparable to the length of the rod-like LTC particles.
In summary, Pt nanoparticles were loaded on Sc-LTC by photodeposition in the presence of H2PtCl6 and K2C2O4. The nanoparticles were selectively loaded on the edge of the rod-like Sc-LTC particles, taking advantage of the unique charge transport character of LTC. It was experimentally confirmed that the transfer of the photoexcited electrons was restricted one-dimensionally along the long axis of the rod-like Sc-LTC particles. This is because the crystal structure of LTC is made up of one-dimensionally stacked TiOxS6−x octahedra and CuS4 tetrahedra consisting of the conduction and valence band edges, respectively. Sc-LTC/Au photoelectrodes modified with Pt by the photodeposition method exhibited higher photocurrents and thus higher HC-STH than those modified by Pt sputtering. LTC is a prominent candidate for solar-driven water splitting owing to the excellent onset potential of the photocathodic current in addition to the narrow band gap and the abundance of the constituent elements.
This work was supported by the Artificial Photosynthesis Project of the Ministry of Economy, Trade and Industry (METI) of Japan, and Grants-in-Aids for Specially Promoted Research (no. 23000009) and for Young Scientists (B) (no. 25810112) of the Japan Society for the Promotion of Science (JSPS).
Notes and references
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- Y. Moriya, T. Takata and K. Domen, Coord. Chem. Rev., 2013, 257, 1957 CrossRef CAS PubMed.
- K. Sivula, F. L. Formal and M. Grätzel, ChemSusChem, 2011, 4, 432 CrossRef CAS PubMed.
- T. Hisatomi, H. Dotan, M. Stefik, K. Sivula, A. Rothschild, M. Grätzel and N. Mathews, Adv. Mater., 2012, 24, 2699 CrossRef CAS PubMed.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347 CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766 CrossRef CAS PubMed.
- M. Liu, L. Wang, G. Lu, X. Yao and L. Guo, Energy Environ. Sci., 2011, 4, 1372 CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165 CrossRef CAS PubMed.
- G. Liu, J. C. Yu, G. Q. Lu and H. M. Cheng, Chem. Commun., 2011, 47, 6763 RSC.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167 RSC.
- M. Katayama, D. Yokoyama, Y. Maeda, Y. Ozaki, M. Tabata, Y. Matsumoto, A. Ishikawa, J. Kubota and K. Domen, Mater. Sci. Eng., B, 2010, 173, 275 CrossRef CAS PubMed.
- T. Suzuki, T. Hisatomi, K. Teramura, Y. Shimodaira, H. Kobayashi and K. Domen, Phys. Chem. Chem. Phys., 2012, 14, 15475 RSC.
- J. Liu, T. Hisatomi, G. Ma, A. Iwanaga, T. Minegishi, Y. Moriya, M. Katayama, J. Kubota and K. Domen, Energy Environ. Sci., 2014, 7, 2239 CAS.
- T. Minegishi, N. Nishimura, J. Kubota and K. Domen, Chem. Sci., 2013, 4, 1120 RSC.
- G. Ma, T. Minegishi, D. Yokoyama, J. Kubota and K. Domen, Chem. Phys. Lett., 2011, 501, 619 CrossRef CAS PubMed.
- M. Moriya, T. Minegishi, H. Kumagai, M. Katayama, J. Kubota and K. Domen, J. Am. Chem. Soc., 2013, 135, 3733 CrossRef CAS PubMed.
- J. Zhao, T. Minegishi, L. Zhang, M. Zhong, G. Nakabayashi, G. Ma, T. Hisatomi, M. Katayama, S. Ikeda, N. Shibata, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2014, 53, 11808 CrossRef CAS PubMed.
- P. S. Surdhar, S. P. Mezyk and D. A. Armstrong, J. Phys. Chem., 1989, 93, 3360 CrossRef CAS.
- F. Forouzan, T. C. Richards and A. J. Bard, J. Phys. Chem., 1996, 100, 18123 CrossRef CAS.
- J. J. Testa, M. A. Grela and M. I. Litter, Environ. Sci. Technol., 2004, 38, 1589 CrossRef CAS.
- V. Meignen, L. Cario, A. Lafond, Y. Moelo, C. Guillot-Deudon and A. Meerschaut, J. Solid State Chem., 2004, 177, 2810 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation processes and SEM images of LTC, schematic of the PT method, PEC curves of Pt deposition, and other experimental procedures. See DOI: 10.1039/c4cc10297e |
| This journal is © The Royal Society of Chemistry 2015 |