
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
No photocatalyst required – versatile, visible light mediated transformations with polyhalomethanes†
Johannes F.
Franz
ab,
Wolfgang B.
Kraus
ab and
Kirsten
Zeitler
*ab
aInstitut für Organische Chemie, Universität Leipzig, D-04103 Leipzig, Germany. E-mail: kzeitler@uni-leipzig.de; Fax: +49-341-97-36599
bInstitut für Organische Chemie, Universität Regensburg, D-93053 Regensburg, Germany
First published on 2nd April 2015
Abstract
A visible light mediated, but photocatalyst-free method for the oxidative α-CH functionalization of tertiary amines with a broad scope of carbon- and heteroatom nucleophiles using polyhalomethanes has been developed. In addition, the pivotal visible light triggered activation of polyhalomethanes offers mild conditions for efficient Kharasch-type additions onto non-activated olefins. Preliminary mechanistic studies are reported.
Visible light photocatalysis has recently emerged as a versatile strategy for challenging C–H activation reactions of sp3-hybridized carbon atoms allowing for the direct and selective construction of C–X and C–C bonds without the need of substrate pre-activation.1 The use of photoredox catalysis for the facile generation of reactive intermediates, such as iminium ions2 or α-amino radicals3 from tertiary amines4 is highly attractive due to the mild conditions and hence its potential compatibility for multicatalytic processes.5 As an alternative to cross dehydrogenative coupling reactions (CDC),6 which are generally either metal-catalyzed or mediated by organic oxidants, a multitude of novel methods employing different photocatalysts have been established.7 Using the CDC activation of tetrahydroisoquinolines (THIQ) as a benchmark reaction typical photocatalysts range from organometallic complexes8 (mostly Ru2+ and Ir3+ based) and simple organic dyes9 to MOFs, graphene oxides as well as organic and inorganic semiconductors.10 Dual catalysis involving photocatalysis,11 which allows access to unprecedented transformations by a beneficial combination of the concurrent catalytic generation of both electrophile and nucleophile, has been developed for metal and organocatalytic activation modes. In the context of THIQ iminium derivatives as electrophiles the nucleophilic catalytic intermediates have been formed via metal catalysis12,13 as well as via covalent14–16 and non-covalent organocatalysis including highly enantioselective approaches to β-amino acid esters17 and α-acylated tertiary amines18 or propargylic amines.12b
In the context of our own interest in novel synergistic and cooperative catalytic activation modes combining organocatalysis with photoredox catalysis,19 we also investigated the photocatalytic approach to iminium ion precursors from tertiary amines, namely THIQs. Apart from the great number of metal and organocatalyzed oxidative processes, two major groups of photocatalytic methods have been described: aerobic techniques in the presence of air or O2 and procedures which rely on the addition of external oxidants, such as alkyl halogenides (bromomalonate, BrCCl3etc.)20 or nitro compounds2,18 to regenerate the photocatalyst (Scheme 1). With respect to the above mentioned required compatibility to potentially unstable nucleophilic catalytic intermediates we decided to focus on methods with external oxidants to avoid detrimental interactions of reactive oxygen intermediates such as superoxide radical anions or peroxides.21
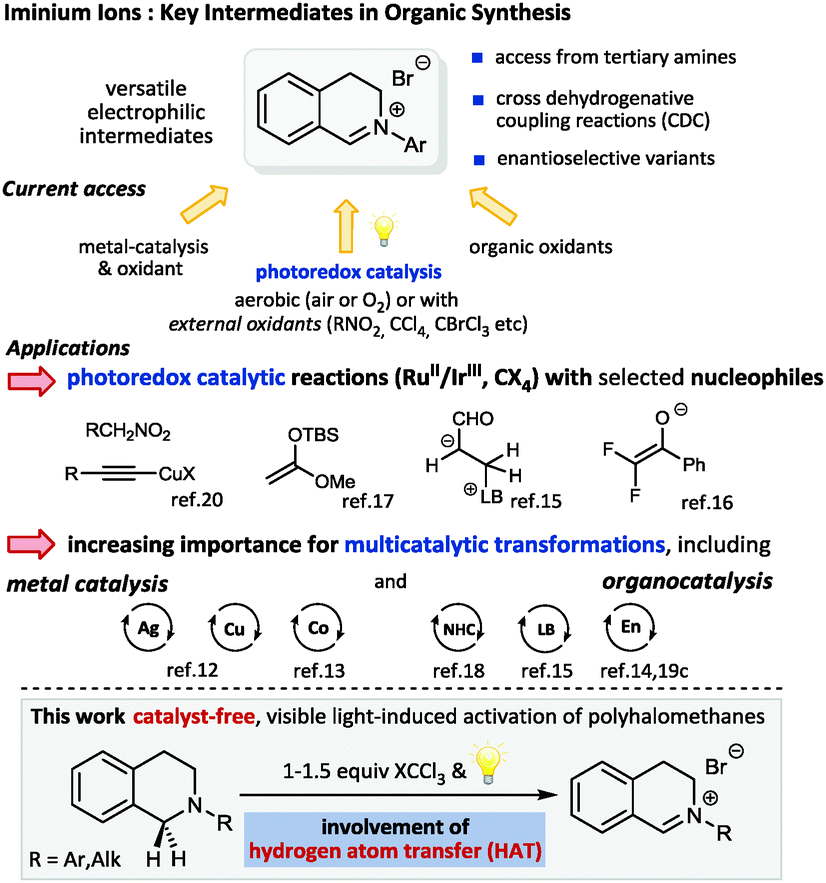 | ||
| Scheme 1 Iminium ions: access strategies and applications. | ||
Based on the well-documented weak C–Hal bonds in polyhalomethanes (low bond dissociation energies (BDEs), e.g. Br–CCl3: 55.3 kcal mol−1),22 being in the energy range accessible by visible light together with the established ability of CCl3˙ radicals to serve as acceptors in hydrogen atom transfer reactions (HAT), we questioned whether these prerequisites would allow a direct, photocatalyst-free, visible light mediated access to iminium intermediates from tertiary amines.23 With respect to BDEs the trichloromethyl radical (H–CCl3 BDE ≈ 95 kcal mol−1) should readily abstract a hydrogen atom from amine substrates (α-C–H BDE ≈ 84–90 kcal mol−1)22 to generate α-amino radicals and subsequently the crucial iminium ions upon either an electron or atom transfer pathway. We hoped to provide an alternative to the oxidation-potential-gated methods and hence to introduce a highly practical general pathway to a C–H functionalization using visible light as sole driving force for substrate partners with weak C–H bonds. Moreover, we anticipated that such a strategy would be ideally suited for multicatalytic transformations and the implementation of substrates that might form detrimental by-products via undesired photocatalytic processes.24 Herein we describe the successful realization of this proposal and present preliminary studies providing insights to the mechanism.
As outlined in Table 1 we started our search for the best conditions with the direct transformation of N-phenyl THIQ 1 to its corresponding iminium salt 2. An initial evaluation of the reagent CBrCl3 revealed that stochiometric amounts seemed to be sufficient, however a small excess (Table 1, entry 4) ensured a cleaner reaction within a shorter time frame. Similar results could be obtained by irradiation with a household compact fluorescent light bulb or sunlight (entries 5, 6). For the successful transformation to the iminium salt the presence of light proved to be essential; lower energy light sources, such as green light were not sufficient (entries 7, 8). The thermal reaction25 in acetonitrile at 70 °C or 100 °C using toluene as solvent (decomposition) was not competitive.26 Employment of less activated CCl4 (Cl–CCl3 BDE = 70.9 kcal mol−1;22λcorr ≈ 404 nm) still allows completion of the reaction using our standard conditions, however with a strongly increased reaction time.26
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. of CBrCl3 | Light source | Temperature | Reaction timeb | Yieldc (%) |
| a 0.25 mmol of THIQ in 0.5 mL acetonitrile, x mmol of CBrCl3 as noted. b In min. c Determined by 1H NMR spectroscopy using bromoform (1 equiv.) as internal standard. d Large scale experiment using 2.5 mmol of THIQ. | |||||
| 1 | 3.0 | Blue LEDs | rt | 30 | 91 |
| 2 | 1.0 | Blue LEDs | rt | 30 | 97 |
| 3 | 1.0 | Blue LEDs | rt | 60 | 99 |
| 4 | 1.5 | Blue LEDs | rt | 30 | Quant. |
| 5 | 1.5 | 22 W CWF bulb | rt | 30 | Quant. |
| 6 | 1.5 | Sunlight | rt | 30 | 91 |
| 7 | 1.5 | No light | 70 °C | 30 | 23 |
| 8 | 1.5 | Green LEDs | rt | 30 | 14 |
| 9d | 1.5 | Blue LEDs | rt | 30 | 95 |
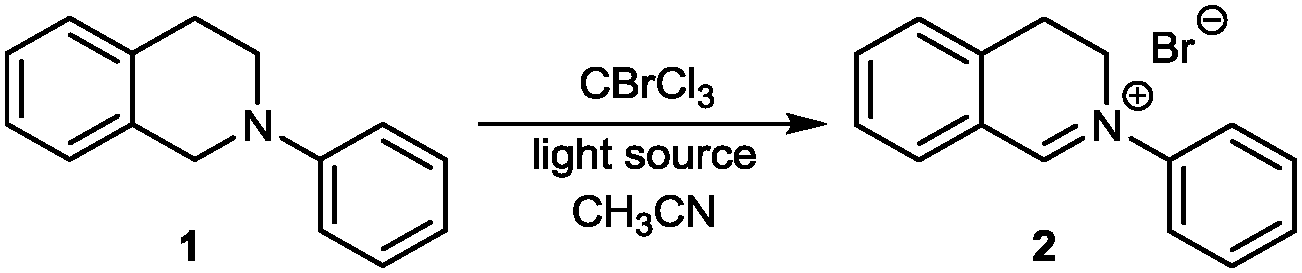
An additional comprehensive survey on solvent effects26 demonstrated the robustness of this transformation. The reaction could be successfully conducted in organic solvents ranging from less polar ethers, halogenated solvents and alkanes to more polar alcohols, but also in water. Having established our best conditions we examined the scope of different THIQs in this transformation: both electron-poor and electron-rich substrates allowed for an efficient generation of their corresponding iminium ions (Fig. 1).27 Notably, our protocol also allows transformations of N-alkyl THIQs to their corresponding iminium salts 6 and 7, which have only scarcely been applied28 in visible light photoredox catalyses due to their higher oxidation potential.29 Employment of N-acyl THIQ substrates with further increased oxidation potential29c leads to highly reactive N-acyliminium salts30 which cannot be isolated. However, C–H activation of these substrates (for a further example, see transformation of dimethylformamide (DMF) in Fig. 3) was proven by isolation of their corresponding by-products, such as a cyclized derivative stemming from the corresponding Boc-protected THIQ.26
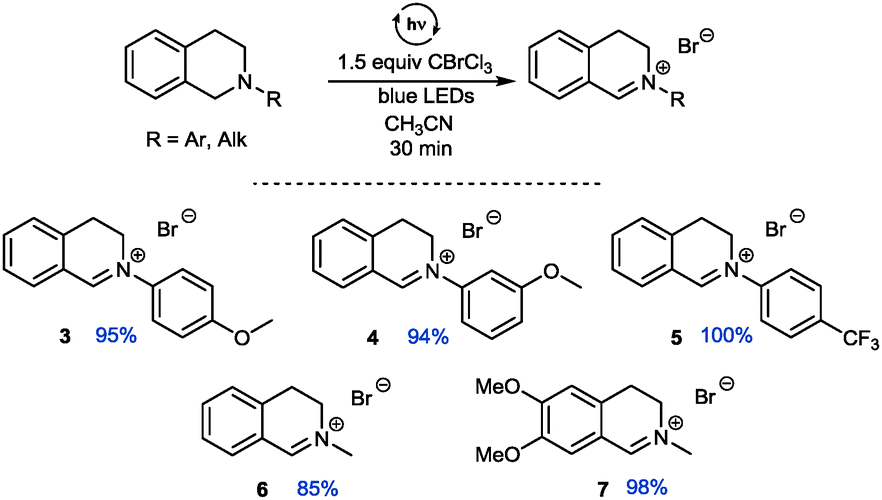 | ||
| Fig. 1 Visible light assisted generation of iminium ions from N-aryl and N-alkyl THIQs (yields determined by NMR using CH2Br2 or CHBr3 as internal standard). | ||
Next we checked the applicability of our conditions for subsequent C–C and C–X cross coupling reactions. We examined literature known nucleophiles which were directly added to the reaction mixture after irradiation with blue light.9a,12,15,20 Both phosphorous and carbon nucleophiles reacted smoothly with the pregenerated iminium salt in a sequential one-pot fashion and without further manipulation of the reaction conditions required (Fig. 2A). Apart from C–H-acidic nitromethane (aza-Henry reaction) and malonate providing excellent yields, electron-rich indole proved to be a competent substrate for the corresponding arylation reaction. Next we focused on expanding the method to multicatalytic processes. As outlined in Fig. 2B our method is applicable to both metal and organocatalytic in situ generation of nucleophiles. Cu-catalyzed alkynylation12,20 proceeds in excellent yield and Lewis-base catalyzed Morita–Baylis–Hillman15 reaction affords the acroleinated THIQ 12 in good yield. All reactions are easily handled and provide similar yields to the related transformations using an additional photocatalyst.
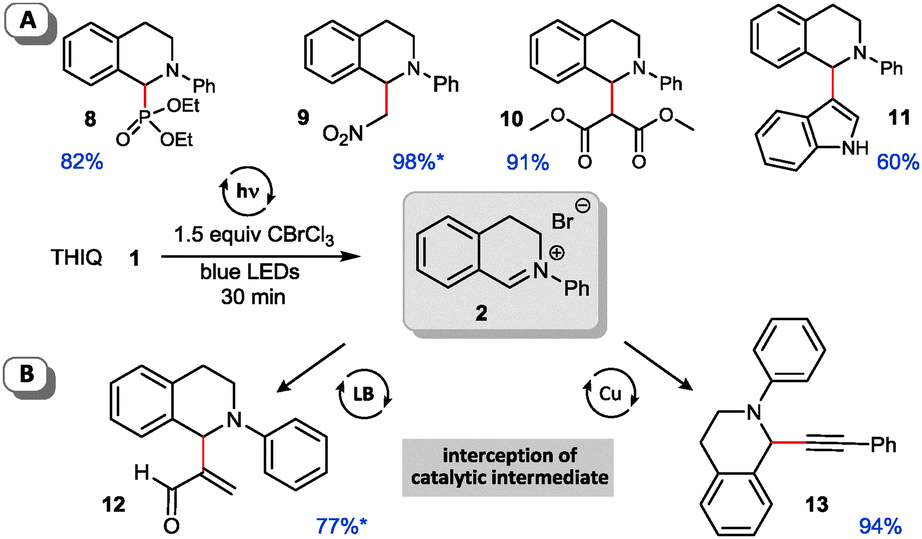 | ||
| Fig. 2 Cross coupling reactions of iminium bromide salts with carbon and heteroatom nucleophiles (yields determined by NMR using CH2Br2 as internal standard, isolated yields*). | ||
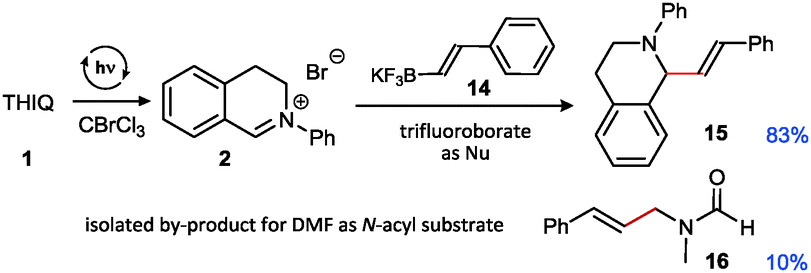 | ||
| Fig. 3 Cross coupling with potassium styryl trifluoroborate (yields determined by NMR using CH2Br2 as internal standard). | ||
In addition, we could extend the scope of C-nucleophiles to trifluoroborate salts which are also competent reaction partners. This is perhaps most remarkable as photocatalytic pathways either using aerobic conditions (e.g. with Ru or xanthene dyes as photocatalysts where occurring oxygen species21 may contribute to the decomposition of trifluoroborate 14)31 or with external oxidants were not equally successful for providing the corresponding vinylation product in a sequential one-pot fashion (best yield: 58%). This is most probably connected with a detrimental interaction of the substrate with remaining catalyst-derived species. Interestingly, as noticed in a control reaction to prove potential side reactions, DMF can also undergo styrenylation under similar conditions, albeit in low yield only (not optimized26).
At this stage we undertook an in situ IR tracing of the standard THIQ reaction to gain further mechanistic insights (Fig. 4). This experiment clearly reveals the fast and clean transformation of the THIQ starting material 1 to its corresponding iminium salt 2 in approx. 8 min according to the indicative vibrational bands at 1604 cm−1 (THIQ 1) and 1640 cm−1 for developing iminium salt 2.
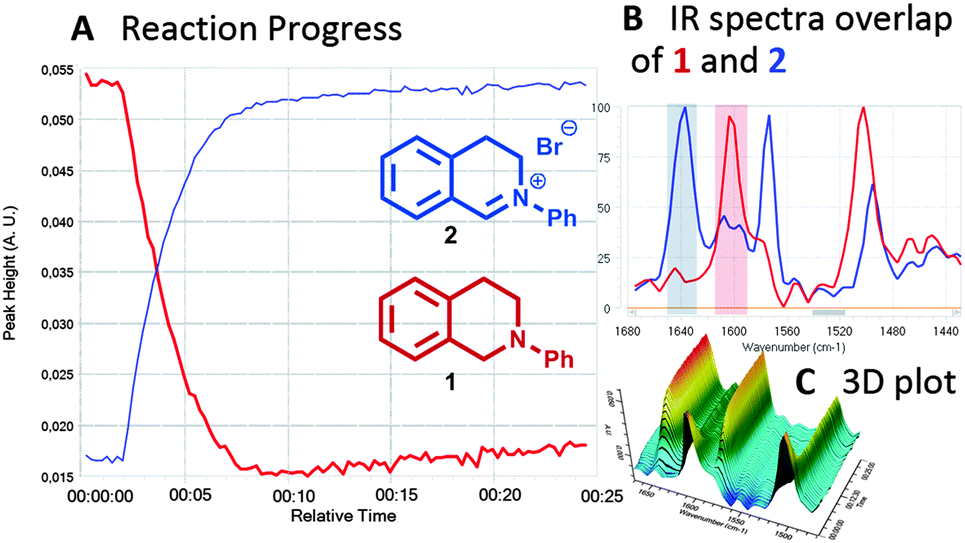 | ||
| Fig. 4 In situ IR-reaction monitoring of visible light-assisted generation of THIQ iminium ion 2 from THIQ 1 with CBrCl3. | ||
Further control experiments with short periods of irradiation and subsequent dark periods resulted in interruption of the reaction progress in the absence of light and recovery of the former reactivity upon further illumination, hence demonstrating the light dependence of the reactions. This suggests that, if a radical chain mechanism is operative, the corresponding chain might only be very short as only during irradiation periods significant reaction progress is observed.26 To additionally exclude a possible involvement of UV light we irradiated the sample through a UV filter (75% w/v NaNO2 solution, cut-off: λ = 400 nm).26 No differences in yield and performance for the formation of the iminium bromide 2 were observed. Based on the observation of a short-lived, intense blue coloration of the THIQ solution containing CBrCl3 upon irradiation,26 the participation of a redox pathway via the known formation of amine polyhalomethane electron donor acceptor (EDA) complexes32 might be operative. However, due to the aforementioned low BDE of C–Br bonds and the observed positive results in transforming of substrates with higher oxidation potentials, we questioned whether this reaction is solely promoted by the formation of an EDA complex. Based on both cyclovoltammetric measurements and a series of KI/starch tests26 we can exclude a direct oxidation or the presence of an oxidant before irradiation; in addition, blue light irradition of CBrCl3 alone showed a positive KI/starch test. Hence, we additionally suggest an initial homolytic bond fission to yield a bromine and a trichloromethyl radical (see Scheme 2A) as a second, alternative pathway. To validate our preliminary mechanistic picture, we performed a Kharasch reaction (Fig. 5), i.e. the ATRA reaction of a polyhalomethane to non-activated alkenes.33,34 Typical ATRA initiations such as radical starters, Lewis acids or by high temperature or UV light have recently been supplemented by a number of photoredox catalytic transformations (using Ru, Ir and Cu based catalysts).35,36 Using a similar set-up as optimized for the THIQ transformations we could access the addition product 18 of the ω-hydroxy alkene 17 in excellent yield and selectivity comparable to catalyst mediated processes, however under remarkably mild conditions.
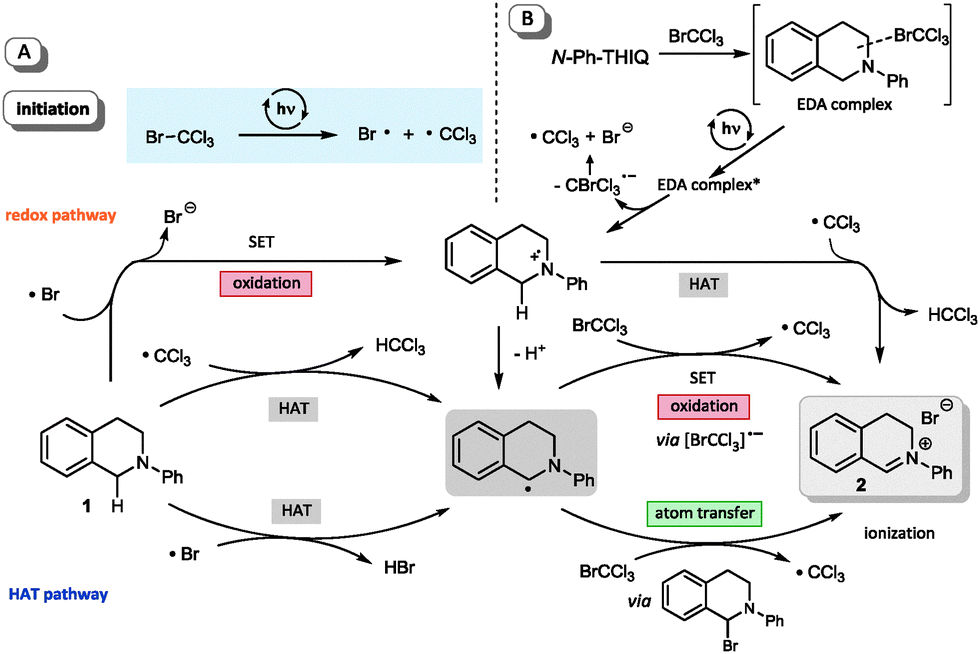 | ||
| Scheme 2 Mechanistic considerations. | ||
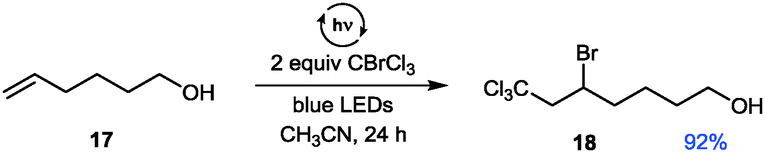 | ||
| Fig. 5 Kharasch addition of CBrCl3 with visible light. | ||
Scheme 2 summarizes our preliminary mechanistic picture. Besides an involvement of EDA complexes in terms of amine substrates (Scheme 2B), whose excitation facilitates charge transfer to generate both the amino radical cation and an instable CBrCl3 radical anion, a number of different mechanistic pathways are conceivable upon light-mediated fission of CBrCl3, namely both a redox pathway where the bromine radical generates the common amino radical cation, while the other routes builds on hydrogen atom transfers as crucial steps. The fact that we could also observe the formation of chloroform,26 also points to the important involvement of CCl3˙ radicals in HAT steps as hydrogen acceptor.
With respect to the BDEs the trichloromethyl radical (H–CCl3 BDE ≈ 95 kcal mol−1) should readily abstract a hydrogen atom from amine substrates (α-C–H BDE ≈ 84–90 kcal mol−1),22 but similarly a bromine radical (H–Br BDE ≈ 88 kcal mol−1) could still be able to trigger the formation of the central α-amino radical which might undergo numerous pathways towards the iminium ion 2.
In conclusion, we have demonstrated the applicability of visible (blue) light for the activation of polyhalomethanes in a broad range of different reactions. CDC coupling of THIQs as well as Kharasch-type addition reactions to olefins were achieved in good to excellent yields. As the light-assisted α-C–H activation of amines does not only rely on redox properties, but rather on suitably low BDEs of the corresponding C–H bonds, we expect this mild, metal-free and operational simple method to not only provide a valuable alternative to established catalyst-promoted procedures, especially in the context of dual or multicatalytic reactions, but also to be broader applicable for currently unattainable transformations. More detailed mechanistic studies are currently in progress.
Financial support from the DBU (fellowship to J. F. Franz) and the DFG (GRK 1626) is gratefully acknowledged. We also thank J. A. Allen and O. Jaurich (Mettler TOLEDO) for their support.
Notes and references
- For recent reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (c) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed.
- For a seminal example, see: A. G. Condie, J.-C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed.
- For seminal examples, see: (a) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672 CrossRef CAS PubMed; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed.
- For a recent review on photocatalytic α-functionalization of amines, see: L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC.
- For recent reviews, see: (a) R. C. Wende and P. R. Schreiner, Green Chem., 2012, 14, 1821 RSC; (b) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC.
- For a recent update, see: S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed.
- For an instructive review, see: L. Furst and C. R. J. Stephenson, in Carbon–carbon Bond Formations via Cross-Dehydrogenative-Coupling of C–H Bonds, ed. C.-J. Li, Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- (a) G. Zhao, C. Yang, L. Guo, H. Sun, C. Chen and W. Xia, Chem. Commun., 2012, 48, 2337 RSC; (b) M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 12709 RSC; (c) J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2013, 19, 6443 CrossRef CAS PubMed.
- (a) D. P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed; (b) Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682 RSC.
- (a) M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem. – Eur. J., 2012, 18, 3478 CrossRef CAS PubMed; (b) L. Möhlmann and S. Blechert, Adv. Synth. Catal., 2014, 356, 2825 CrossRef PubMed.
- (a) M. Neumann and K. Zeitler, in Chemical Photocatalysis, ed. B. König, de Gruyter, 2013 Search PubMed; for two recent examples, see: (b) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (c) Z. Zuo, D. Ahneman, L. Chu, J. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- (a) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem. – Eur. J., 2012, 18, 5170 CrossRef CAS PubMed; (b) I. Perepichka, S. Kundu, Z. Hearne and C.-J. Li, Org. Biomol. Chem., 2015, 13, 447 RSC.
- J.-J. Zhong, Q.-Y. Meng, B. Liu, X.-B. Li, X.-W. Gao, T. Lei, C.-J. Wu, Z.-J. Li, C.-H. Tung and L.-Z. Wu, Org. Lett., 2014, 16, 1988 CrossRef CAS PubMed.
- M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. Fabry, Chem. Commun., 2011, 47, 2360 RSC.
- Z.-J. Feng, J. Xuan, X.-D. Xia, W. Ding, W. Guo, J.-R. Chen, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Org. Biomol. Chem., 2014, 12, 2037 CAS.
- W. Li, X. Zhu, H. Mao, Z. Tang, Y. Chenga and C. Zhu, Chem. Commun., 2014, 50, 7521 RSC.
- G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112 RSC.
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed.
- (a) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed; (b) M. Neumann and K. Zeitler, Org. Lett., 2012, 14, 2658 CrossRef CAS PubMed; (c) M. Neumann and K. Zeitler, Chem. – Eur. J., 2013, 19, 6950 CrossRef CAS PubMed.
- D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed.
- Q. Liu, Y.-N. Li, H.-H. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2012, 18, 620 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- For a selection of recently published examples of catalyst-free transformations with visible light: (a) I. D. Arceo, A. Jurberg, A. Álvarez-Fernàndez and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef PubMed; (b) H. Jiang, J. R. Bak, F. J. López-Delgado and K. A. Jørgensen, Green Chem., 2013, 15, 3355 RSC; (c) E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438 RSC; (d) E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064 CrossRef CAS PubMed; (e) M. Tobisu, T. Furukawa and N. Chatani, Chem. Lett., 2013, 42, 1203 CrossRef CAS.
- The frequent use of various Ir catalysts for CDC reactions can e.g. trigger undesired dehalogenations of aromatic bromides: (a) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (b) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed ; for reported diminished yields, see ref. 12b. Carboxylic acid and trifluoroborate substrates might also suffer from the strong oxidation potential of Ir photocatalysts: see ref. 11b and c.
- (a) C. Dai, F. Meschini, J. M. R. Narayanam and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425 CrossRef CAS PubMed; (b) H. Ueda, K. Yoshida and H. Tokuyama, Org. Lett., 2014, 16, 4194 CrossRef CAS PubMed.
- See ESI† for details.
- For the X-ray structure of iminium salt 2, please refer to ESI.† CCDC 993641.
- W. Fu, W. Guo, G. Zou and C. Xu, J. Fluorine Chem., 2012, 140, 88 CrossRef CAS PubMed.
- For comparison: redox potential (all given vs. SCE) of (a) N-Ph-THIQ: 0.84 V, see ref. 25a; (b) N-Et-THIQ: 1.20 V, see: G. Pandey, K. S. Rani and G. Lakshmaiah, Tetrahedron Lett., 1992, 33, 5107 CrossRef CAS; (c) tertiary carbamates: 1.50–1.90 V, L. Haya, F. J. Sayago, A. M. Mainar, C. Cativiela and J. S. Urieta, Phys. Chem. Chem. Phys., 2011, 13, 17696 RSC.
- B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431 CrossRef CAS PubMed.
- For the intended photocatalytically induced oxidation of less stable boronic acids, see: (a) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed; (b) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286 CrossRef CAS PubMed.
- (a) L. Eberson and M. Ekström, Acta Chem. Scand., 1989, 43, 86 CrossRef CAS PubMed; (b) D. P. Stevenson and G. M. Coppinger, J. Am. Chem. Soc., 1986, 84, 149 CrossRef.
- (a) M. S. Kharasch, E. V. Jensen and W. H. Urry, Science, 1945, 102, 128 CAS; (b) M. S. Kharasch, E. V. Jensen and W. H. Urry, J. Am. Chem. Soc., 1946, 68, 154 CrossRef CAS.
- During the blue light irradiation of alkene 17 with CBrCl3 we did not observe any coloration of the reaction mixture; see ESI† for details.
- (a) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed; (b) M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 733 CrossRef PubMed.
- For a recent metal-free approach using aromatic aldehydes as energy transfer sensitizer, see: ref. 23d.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterization. CCDC 993641. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc10270c |
| This journal is © The Royal Society of Chemistry 2015 |