
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective visible light-induced Ti–O bond splitting in an ansa-titanocene dihydroxido complex†‡
Christian
Godemann
a,
Laura
Dura
a,
Dirk
Hollmann
*a,
Kathleen
Grabow
a,
Ursula
Bentrup
a,
Haijun
Jiao
a,
Axel
Schulz
ab,
Angelika
Brückner
*a and
Torsten
Beweries
*a
aLeibniz-Institut für Katalyse e.V. der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: dirk.hollmann@catalysis.de; angelika.brueckner@catalysis.de; torsten.beweries@catalysis.de
bInstitut für Chemie, Universität Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany
First published on 8th January 2015
Abstract
Irradiation of a substituted ansa-titanocene(IV) dihydroxido complex with visible light induces Ti–O bond dissociation. In contrast to previous studies on structurally similar unbridged complexes, no side reactions are observed and formation of the Ti(III) species is highly selective. The formation of OH radicals was proved using a biradicaloid species.
The splitting of water into hydrogen and oxygen (2 H2O → 2 H2 + O2) is nowadays generally regarded as one of the key steps towards a sustainable and carbon neutral energy supply.1 Currently, several approaches are under investigation for this reaction, including electro- and photocatalysis as well as well-established techniques such as electrolysis.2 Since the first reports of photocatalytic water splitting at a TiO2/Pt array by Fujishima and Honda,3 chemists have strived for a fundamental understanding of the processes at the molecular level. A detailed mechanistic insight is highly desirable in order to optimise both, heterogeneous and homogeneous catalyst systems. However, to date only very few examples for defined reactions of water with early transition metal centres are known. Especially oxidative additions are almost unprecedented for complexes of group 3–5 metals.4 Models for overall water splitting at defined transition metal compounds were described by Milstein and co-workers for a ruthenium pincer complex5 as well as by Kunkely and Vogler for an osmocene species.6
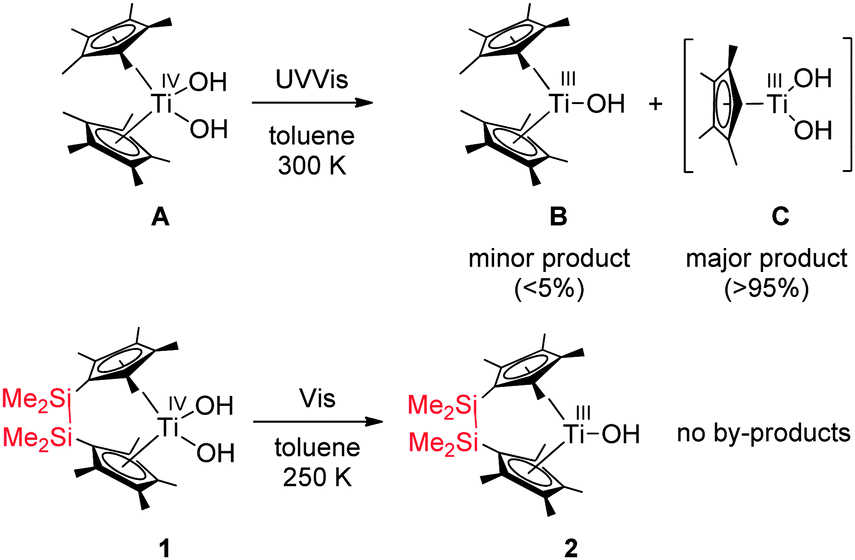
Inspired by the work of Fujishima and Honda, investigations in our group are focused on titanium complexes for modelling the elementary steps of overall water splitting. Here hydrogen formation from water (i.e. water reduction) can be performed in the absence of light.7 A model for the significantly more challenging water oxidation via Ti–O elimination under light irradiation was described as well; however, the main light-induced process was identified as Cp* elimination (∼95%).8 Herein we describe the application of ansa-titanocene complexes to realise selective Ti–O bond splitting even under visible light (λ > 420 nm) irradiation (Scheme 1) as well as a novel method for the identification of produced OH radicals, which serve as an equivalent for hydrogen peroxide and thus for molecular oxygen.
 | ||
| Scheme 1 Light-induced conversion of non-bridged and Si bridged titanocene complexes (A and 1). | ||
To overcome the problem of Cp* elimination, we envisioned connecting the Cp* rings which could result in a strong chelating effect and thus possibly in more strongly bound cyclopentadienyl ligands. Therefore ansa-titanocene dihydroxido complexes were synthesised via a new “oxido” route, a method, which prevents the production of metallocene alkenyl-silanolate side products.9,10
Through extensive explorative testing of various conditions including different light filters and temperatures we discovered that selective Ti–O bond splitting occurred in toluene at 250 K. Irradiation of a solution of complex Me4Si2(C5Me4)2Ti(OH)2 (1) resulted in formation of a single Ti(III) species (2) detected by EPR spectroscopy (Fig. 1, g = 1.978, ATi = 9.7 G, line width ΔB = 3.9 G), accompanied by a colour change from yellow to dark green. Note that this g value is close to the non-bridged species B (g = 1.977, toluene, 23 °C), which was reported before.11a At 100 K the EPR spectrum showed a single species of rhombic symmetry, which had nearly the same g values as determined for Cp*2Ti(OH)11 (Fig. 1).
 | ||
| Fig. 1 EPR spectra of complex 2 generated from 1 by irradiation with visible light (λ > 420 nm) in toluene at 250 K (liquid, toluene) and 100 K (toluene, matrix) (black-experimental and red-simulated). Simulated EPR parameter:12T = 250 K: g = 1.978, ATi = 9.7 G, line width ΔB = 3.9 G; T = 100 K: g1 = 2.002, g2 = 1.984, g3 = 1.951, giso = 1.979, line width ΔB1 = 6.0 G, ΔB2 = 5.1 G, ΔB3 = 11.4 G. | ||
No other by-products e.g. cyclopentadienyl radicals were detected. 1H NMR spectra recorded after irradiation showed very broad signals confirming the presence of a significant amount of paramagnetic Ti(III). Furthermore, wavelength dependent measurements (band pass filter 700 to 350 nm, 50 nm steps, 10 nm band width) showed that evolution of this Ti(III) resonance even occurred with visible light-irradiation in the absence of high energetic UV light (Fig. 2). Prolonged irradiation with visible light (λ > 420 nm) at 250 K resulted in a continuous formation of the new Ti(III) complex without degradation (Fig. S1, ESI‡). This is well in line with the observed low absorbance of 1 in the visible region (Fig. S4, ESI‡).
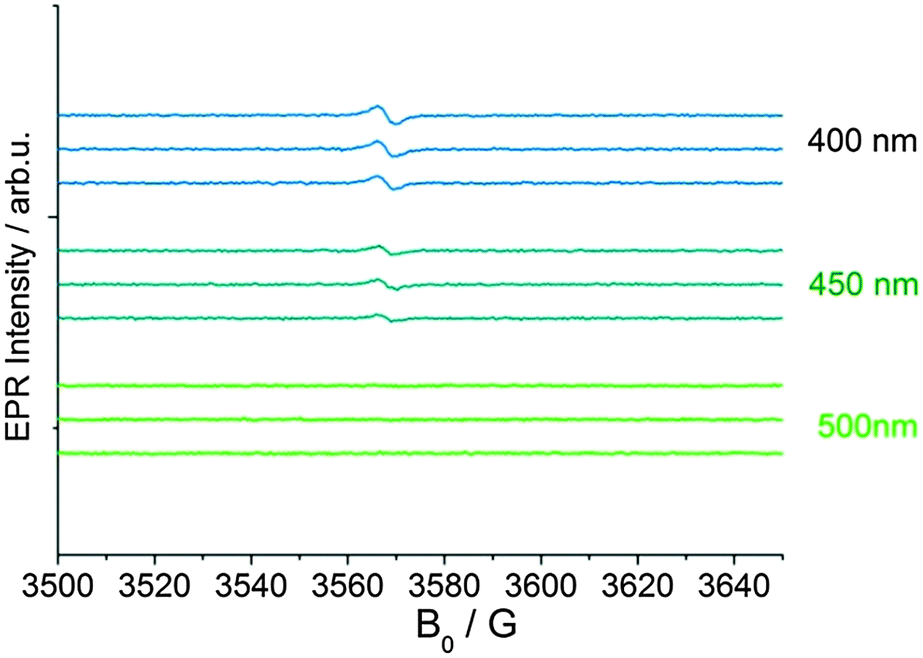 | ||
| Fig. 2 EPR spectra recorded during irradiation of complex 1 in toluene at 298 K using different optical band pass filters (10 nm band width). | ||
On a macroscopic level, the photo-reduction process can even be observed by slow degradation of a solution of the yellow complex 1 and formation of a dark green Ti(III) species in the presence of ambient daylight!
Attempts to isolate single crystalline material of the paramagnetic irradiation product suitable for an X-ray analysis failed because no analytically pure samples of the desired Ti(III) monohydroxido complex 2 could be isolated from the reaction mixture. Thus, in order to further corroborate the photo-reduction of complex 1, we prepared complex 2 by an alternative method starting from the aforementioned oxido species Me4Si2(C5Me4)2Ti(py)O (py = pyridine).10 Reduction of the latter with dihydrogen at elevated temperatures in toluene resulted in elimination of pyridine and selective reduction of the terminal oxido group and the Ti(IV) centre to yield the trivalent titanocene(III) monohydroxido complex 2. The EPR signal of 2 exactly corresponds to the Ti(III) complex obtained during irradiation (Fig. S2, ESI‡). Thus, photo-reduction of 1 exclusively yielded the Ti(III) complex 2. This reactivity is in contrast to that of the previously reported, unbridged complex Cp*2Ti(OH)2 (Scheme 1, A).8
In order to understand these observed differences between complexes A and 1 we have computed the thermodynamic parameters of their homolytic dissociation using BP86 DFT methods. All computational details are given in the Supporting Information. For the OH radical dissociation (Scheme 2), the dissociation enthalpies (84.31 vs. 84.96 kcal mol−1) and Gibbs energies (73.16 vs. 73.42 kcal mol−1) for complexes 1 and A are very close, indicating their similar Ti–OH bonding strengths.
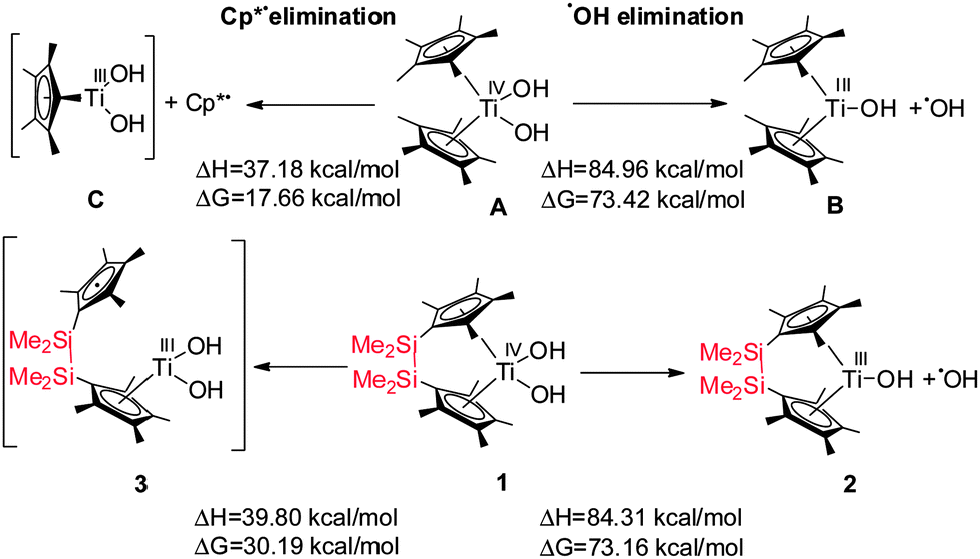 | ||
| Scheme 2 Dissociation enthalpies (ΔH) and Gibbs energies (ΔG) of selected photolytic bond splitting. | ||
For the dissociation of one of the cyclopentadienyl ligands (Cp* secession) as radical, the dissociation enthalpy for complex A is slightly lower than that for complex 1 (A: 37.18 vs.1: 39.80 kcal mol−1), indicating that the bond of the cyclopentadienyl ligand to Ti(III) in complex A is weaker than in 1 (Scheme 2, Schemes S3 and S4, ESI‡).
A large difference between both complexes has been found for their dissociation Gibbs energies (A: 17.66 vs.1: 30.19 kcal mol−1). This might be due to the fact that the Cp* radical is completely split off from complex A, while it remains attached to complex 1via the bridge (3). The formation of 3 was experimentally proven by addition of the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) (Fig. S3, ESI‡). In the absence of the spin trap, complex 3 is short-lived due to the chelate/entropy effect which results in a fast recombination of the cyclopentadienyl ligand with the Ti(III) centre. As a consequence cyclopentadienyl radical dissociation from complex A is thermodynamically much more favourable than from complex 1. Hence, in the unbridged complex A, the Cp* radicals – once dissociated from the Ti centre – diffuse into the reaction solution, while immediate recombination of the intermediately formed cyclopentadienyl radical with the Ti(III) centre is much more likely for the bridged complex 1, as both sites remain in close proximity. These thermodynamic differences rationalise our experimentally observed difference in photo-induced reactivity between the bridged and unbridged complexes.
At ambient temperature degradation of the new formed Ti(III) complex 2 occurred with time (t > 2 h). To identify the further transformation pathway of complex 2 at 298 K, its behaviour during irradiation with visible light in toluene was investigated by in situ ATR-IR spectroscopy. In the OH region (Fig. 3A) a band at 3650 cm−1 was observed for the starting complex 1, which can be assigned to ν(OH) because in the related complex [Cp*2Ti–OH] the OH band was observed at 3652 cm−1.11 The intensity of this ν(OH) band decreased with progressive irradiation time and is slightly shifted from 3650 cm−1 to 3648 cm−1. Simultaneously, a new band appeared at 773 cm−1 increasing in intensity with extended time of irradiation with visible light (t > 5 h, Fig. 3B). This frequency is typical for ν(Ti–O–Ti) modes13 and, thus, suggests further transformation of 2 into polynuclear clusters containing Ti–O–Ti bridges, which have to be regarded as decomposition products of complex 1.
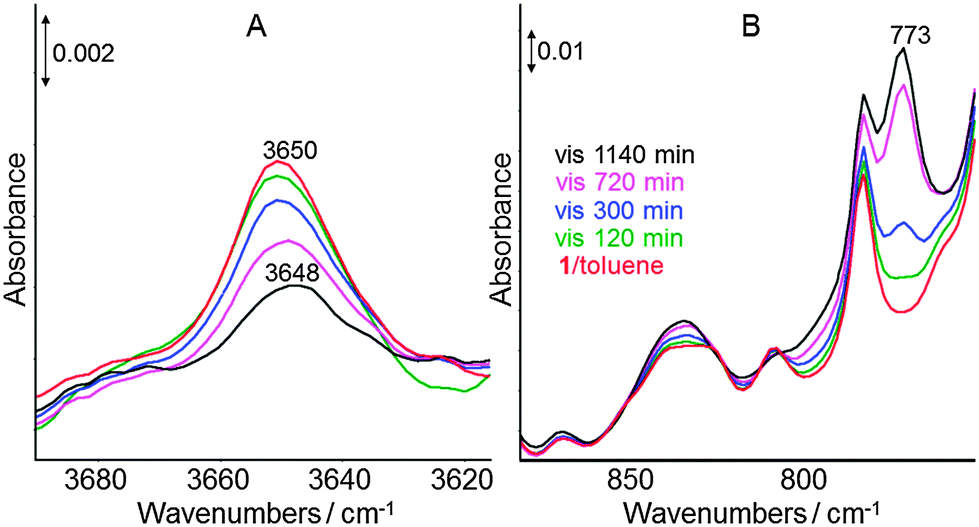 | ||
| Fig. 3 In situ ATR-IR spectra obtained during irradiation of complex 1 in toluene at 298 K using a 420 nm cut-off filter: characteristic OH region (A) and spectral region 875–750 cm−1 (B). | ||
However, both, DFT calculations (Scheme S5, ESI‡) as well as EPR experiments do not point to the formation of defined dimeric condensation products of 2.11b
To verify the production of OH radicals, different approaches were attempted. Note that in the previously described photo-induced decomposition of the unbridged complex A, the OH radicals recombined with the Cp* fragments to give the alcohol C5Me5OH.8 Application of spin trapping reagents such as DMPO provided no clear evidence. Here preferential trapping of the bridged cyclopentadienyl ligand occurred (vide supra).
Moreover, attempts to trap the eliminated OH radicals using the hydroxylation reaction of terephthalic acid (TA) to give the fluorescent adduct 2-hydroxyterephthalic acid (TAOH)14 were not successful due to overlap of the fluorescence signals of the titanocene species with the expected fluorescence of the TAOH at 426 nm. Additionally, we observed interaction and even a condensation reaction between the titanocene complex 1 and TA to give a dinuclear carboxylate complex (6) (Scheme S2 and Fig. S5, ESI‡), thus indicating that the described method is not suited for the identification of OH radicals in this organometallic system.
However, direct confirmation of OH radicals was observed with the biradicaloid [PN(Ter)]215 which features a planar diphosphadiazane ring with two unpaired electrons located at the P atoms.16 This sterically demanding compound is capable of reacting with OH radicals but does not react with the metallocene unit. Irradiation of a mixture of complex 1 and 0.5 equivalents of the biradicaloid 4 in toluene gave a dark green solution due to formation of complex 2 (Scheme 3).
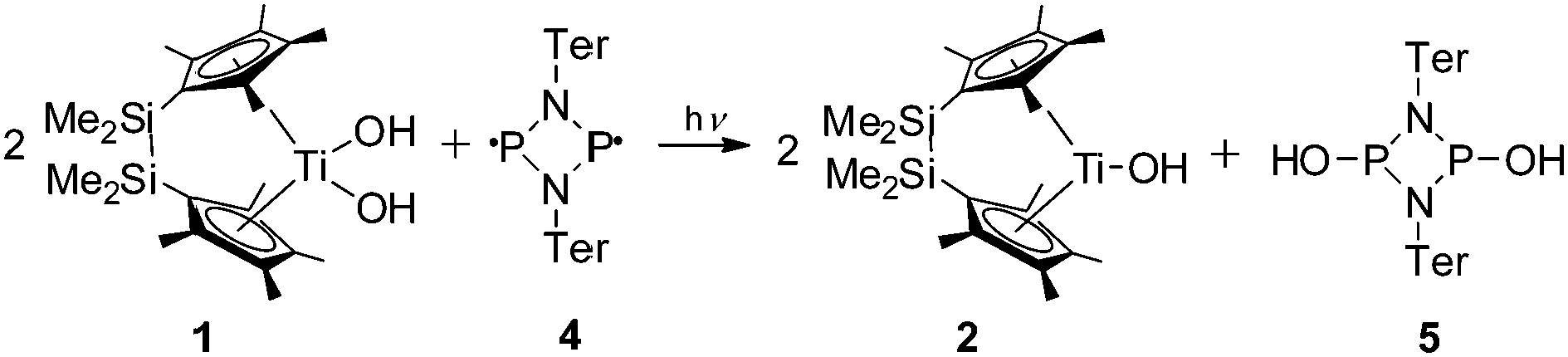 | ||
| Scheme 3 NMR scale reaction of complex 1 with the biradicaloid 4 (toluene-d8, room temperature, λ > 420 nm). | ||
31P NMR analysis of the reaction solution showed a single intense resonance at 211.2 ppm for [HO–P(μ-NTer)]2 (5), a value which is well in line with other cyclo-diphosphadiazanes such as [(MeO)P(μ-Nt-Bu)]2 (cis/trans: 133.7/202.4 ppm)17 or [ClP(μ-NTer)]2 (cis/trans: 227.4/264.1 ppm),18 thus indicating the formation of HO-addition product 5 in the reaction of biradicaloid 4 and OH radicals (Fig. S6, ESI‡).
In summary, we have presented the selective visible light-induced photoreduction of a titanocene(IV) complex. Bridging of the ligand system resulted in significant stabilisation, thus preventing elimination of cyclopentadienyl ligands, which was previously observed for unbridged systems.8 Studies to close the model cycle of overall water splitting using bridged titanocene complexes are ongoing in our group; the results will be published in due course.
Financial support by the Federal Ministry for Education and Research (BMBF, Project Light2Hydrogen) is gratefully acknowledged. We thank Dr. Anke Spannenberg (LIKAT) for X-ray analysis of complex 6. T.B. would like to thank Prof. Uwe Rosenthal (LIKAT) for general support and fruitful discussions regarding titanocene chemistry.
Notes and references
- One widely discussed concept in this context is Power-To-Gas, see: G. Gahleitner, Int. J. Hydrogen Energy, 2013, 38, 2039 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- O. V. Ozerov, Chem. Soc. Rev., 2009, 38, 83 RSC.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstaninovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74 CrossRef CAS PubMed.
- H. Kunkely and A. Vogler, Angew. Chem., Int. Ed., 2009, 48, 1685 CrossRef CAS PubMed.
- (a) M. Kessler, S. Hansen, D. Hollmann, M. Klahn, T. Beweries, A. Spannenberg, A. Brückner and U. Rosenthal, Eur. J. Inorg. Chem., 2011, 627 CrossRef CAS; (b) D. Hollmann, K. Grabow, H. Jiao, M. Kessler, A. Spannenberg, T. Beweries, U. Bentrup and A. Brückner, Chem. – Eur. J., 2013, 19, 13705 CrossRef CAS PubMed.
- M. Kessler, S. Schüler, D. Hollmann, M. Klahn, T. Beweries, A. Spannenberg, A. Brückner and U. Rosenthal, Angew. Chem., Int. Ed., 2012, 51, 6272 CrossRef CAS PubMed.
- M. Kessler, S. Hansen, C. Godemann, A. Spannenberg and T. Beweries, Chem. – Eur. J., 2013, 19, 6350 CrossRef CAS PubMed.
- C. Godemann, E. Barsch, A. Spannenberg, R. Ludwig and T. Beweries, Eur. J. Inorg. Chem., 2014, 4068 CrossRef CAS.
- (a) M. Horacek, R. Gyepes, J. Kubista and K. Mach, Inorg. Chem. Commun., 2004, 7, 155 CrossRef CAS PubMed; (b) J. Pinkas, I. Cisarova, R. Gyepes, J. Kubista, M. Horacek and K. Mach, Organometallics, 2013, 32, 6306 CrossRef CAS.
- EPRSim32: T. Spalek, P. Pietrzyk and Z. Sojka, J. Chem. Inf. Model., 2005, 45, 18 CrossRef CAS PubMed . Subroutine diagonal + PT: F. E. Mabbs and D. Collison, Mol. Phys. Rep., 1999, 26, 39 Search PubMed.
- W. W. Lukens, Jr. and R. A. Andersen, Inorg. Chem., 1995, 34, 3440 CrossRef.
- L. Schwertmann, M. Wark and R. Marschall, RSC Adv., 2013, 3, 18908 RSC.
- Ter = 2,6-Mes2–C6H3 (Mes = 2,4,6-Me3C6H2).
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974 CrossRef CAS PubMed.
- R. Keat and D. G. Thompson, J. Chem. Soc., Dalton Trans., 1979, 1224 RSC.
- F. Reis, A. Schulz, A. Villinger and N. Weding, Dalton Trans., 2010, 39, 9962 RSC.
Footnotes |
| † This work is dedicated to Robin N. Perutz on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, EPR, UV-Vis and NMR spectra, computational and crystallographic details. CCDC 1011531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc09733e |
| This journal is © The Royal Society of Chemistry 2015 |