
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipase active site covalent anchoring of Rh(NHC) catalysts: towards chemoselective artificial metalloenzymes†
M.
Basauri-Molina
,
C. F.
Riemersma
,
M. A.
Würdemann
,
H.
Kleijn
and
R. J. M.
Klein Gebbink
*
Organic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584CG Utrecht, The Netherlands. E-mail: r.j.m.kleingebbink@uu.nl
First published on 11th March 2015
Abstract
A Rh(NHC) phosphonate complex reacts with the lipases cutinase and Candida antarctica lipase B resulting in the first (soluble) artificial metalloenzymes formed by covalent active site-directed hybridization. When compared to unsupported complexes, these new robust hybrids show enhanced chemoselectivity in the (competitive) hydrogenation of olefins over ketones.
The embedding of synthetic metallocatalysts in protein scaffolds allows for the development of a so-called 2nd coordination sphere around the metallic center, which can result in catalytic selectivity due to the intrinsically chiral and bulky character of the protein macromolecules, blocking specific sterically demanding transition states in a catalytic reaction.1 This way, proteins as naturally abundant supports represent an alternative to the development of chiral, bulky or enlarged ligands. Moreover, the solubility of organometallic species, normally restricted to organic media, is expanded by the nature of the protein scaffold, whose macromolecular nature additionally allows for a facilitated separation of the artificial enzymes.
Pioneering work from Whitesides,2 optimized and extended by Ward,3 on the supramolecular (strept)avidin–biotin system has provided early examples of the development of artificial metalloenzymes. Other hybridization strategies include alkylation of amino acid residues,4 the use of apomyoglobin with metallated artificial cofactors5 and metal–DNA hybrids.6 Enantio- and regioselective catalysis can be achieved or further optimized by mutagenic treatment of the proteomic scaffold.7
Recently, we have studied the active site-directed (ASD) hybridization strategy using organometallic phosphonate esters that covalently and irreversibly bind to the active serine residue of lipases. In this manner, protein–ECE metallopincer hybrids8 and supported Ru(Cp)–protein bifunctional catalytic hybrids9 for lipase labeling and dynamic kinetic resolution, respectively, were developed. ASD hybrids promise robustness of the covalent hybrid for ease of manipulation with reduced leaching, characterization by mass spectrometry, and anchoring of the metallocatalyst in a naturally selective environment (i.e. the lipase active site). Furthermore, phosphonate esters are not restricted to a particular lipase thus potentially allow for the screening of different protein environments.10
Our next goal was to further develop the catalytic applications of the ASD method by the use of a versatile ligand (see Scheme 1). To this end, N-heterocyclic carbenes (NHCs) are attractive water- and oxygen-tolerant spectator ligands that present robust σ-donating monodentate coordination towards a myriad of metallic centers without restriction of the remaining coordination sites for catalytic performance.11 Functionalization of the N-substituents has allowed for their immobilization12 and they have been applied recently in the construction of Grubbs-catalyst/protein artificial enzymes,13 however, to the best of our knowledge, the plausible selective behavior of such hybrids has not yet been documented.
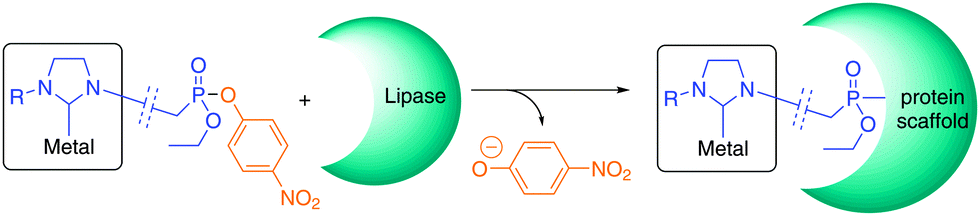 | ||
| Scheme 1 Covalent inhibition of lipases with P-functionalized phosphonate esters towards metal(NHC)–protein hybrids. | ||
Rhodium species were our first target given their relatively facile synthesis. The absence of this metal in biological systems makes it an attractive moiety to extend enzyme reactivity. The hydrogenation of acetophenone in water with Rh(NHC)-based catalysts has been studied by Herrmann and Kühn and proves the reactivity of such catalytic centers in aqueous media.14 With other types of ligands, e.g. arenes or bidentate phosphines, the reduction of ketones as well as olefins via transfer hydrogenation with artificial enzymes has been addressed by Ward15 and Reetz.16 When monodentate phosphites were addressed by de Vries,17 bulky protecting groups were needed to avoid their oxidation; yet no selectivity was found in spite of satisfactory catalytic activity. Without the use of protein scaffolds, Noyori's ruthenium catalysts are among the current state-of-the-art catalysts for hydrogenations, which in function of their phosphine ligands achieve excellent enantioselectivities in organic media.18
After our successful experience with cutinase from fusarium solani pisi, a lipase that reacts with hydrophobic esters without an initial activation step,19 as host in the formation of semi-synthetic enzymes,8,9 we addressed the preparation of Rh(NHC)–protein semi-synthetic hybrids to form (the first) catalytically active cutinase metallohybrid.20 and studied its behavior in hydrogenations to explore the (enantio)selectivity of the active site-embedded Rh(NHC) fragment.
Accordingly, a phosphonate cofactor was designed for an orthogonal metal-NHC–protein orientation. Allylphosphonamidate 1 and brominated imidazolinium salt 2 were synthesized and cross-coupled to form N-tethered NHC ligand 3. Deprotonation with the non-nucleophilic base potassium hexamethyldisilazide (KHMDS), followed by treatment with [Rh(μ-Cl)(cod)]2 (cod = cycloocta-1,5-diene) led to rhodium compound 4 as a mixture of isomers. Next, the dimethylamide group was substituted with p-nitrophenolate (pNP) towards complex Rh-pNP (Scheme 2). The mono p-nitrophenyl ethyl P-propylphosphonate motif used here has proven an effective inhibitor of cutinase and CalB,8,9,21 in contrast to dinitrophenyl phosphonates, which can lead to slow hydrolysis of the phosphorous–serine bond.16b Distinctive chemical shifts of the P-nucleus in NMR for 1 through Rh-pNP, facilitate its characterization in combination with mass spectrometry (for details on synthesis and characterization, see the ESI†).
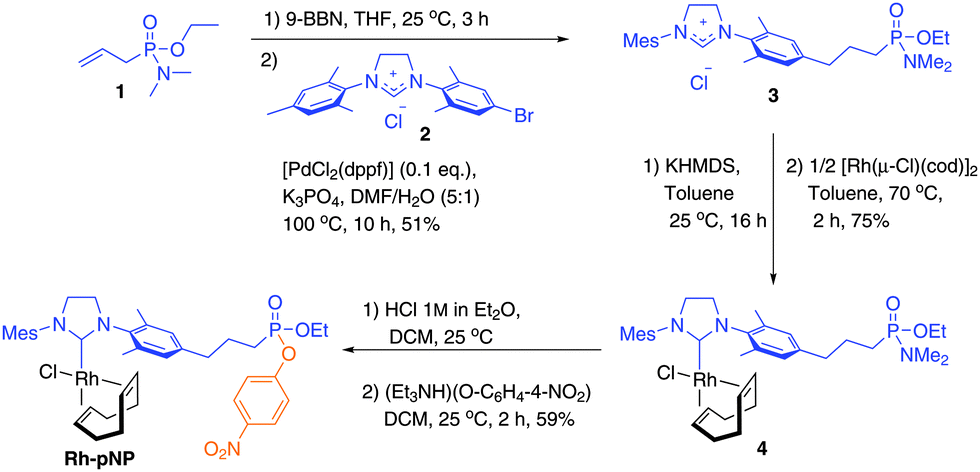 | ||
| Scheme 2 Synthesis of lipase inhibitor Rh-pNP. | ||
Next, cutinase was treated with an excess of Rh-pNP for the formation of the hybrid. A dialyzed and denatured aliquot (10% formic acid) was analyzed by ESI-MS, showing complete conversion of cutinase and formation of the desired Rh-cut hybrid (Fig. 1, top). Secondary peaks with considerably lower intensity were also observed. These originate from minor impurities in Rh-pNP, which due to their smaller size have a high inhibitory competence (see ESI†), and do not arise from decomposition of the hybrid. Also, a single hybridization stoichiometry of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was found, which discarded the association of Rh-pNP with the four cysteine (all in cystine form) and six lysine residues at the exterior of the enzyme. In addition, no residual hydrolytic cutinase activity was found when p-nitrophenyl butyrate (pNPB) was treated with Rh-cut, leading to a hydrolysis of pNPB only by the buffer, again confirming full inhibition of cutinase (Fig. 1, bottom). These observations demonstrate the covalent character and single site hybridization in forming Rh-cut.
:1 was found, which discarded the association of Rh-pNP with the four cysteine (all in cystine form) and six lysine residues at the exterior of the enzyme. In addition, no residual hydrolytic cutinase activity was found when p-nitrophenyl butyrate (pNPB) was treated with Rh-cut, leading to a hydrolysis of pNPB only by the buffer, again confirming full inhibition of cutinase (Fig. 1, bottom). These observations demonstrate the covalent character and single site hybridization in forming Rh-cut.
 | ||
| Fig. 1 ESI-MS analysis of Rh-cut hybrid (top, [M]10+ calc.: 2127.43, found: 2126.59) and the determination of residual hydrolytic activity of cutinase (bottom). | ||
The catalytic performance of Rh-cut was evaluated in the hydrogenation of acetophenone, a prochiral ketone, under a H2 atmosphere (40 bar) at room temperature for 20 h in aqueous biphasic conditions with CH2Cl2 (5%, v/v) in TrisHCl buffer at pH 8.5. The organic solvent was chosen to promote the interaction of the protein with organic substrates; CH2Cl2 also has low acidity and low coordination behavior with organometallics. At these conditions no product was observed. The same was observed when the non-supported catalyst analog [RhCl(SIMes)(cod)] 5 (where SIMes = 1,3-dimesityl-4,5-dihydroimidazolin-2-ilydene) was used (Table 1, entries 1 and 2). When the reaction with 5 was not buffered, 1-phenylethanol was produced in 12% yield and this significantly improved to a yield of 90% using a NaH2PO4/Na2HPO4 buffer at pH 8.5 (Table 1, entries 3 and 4). The difference in conversion using different buffers is attributed to the interference of the high concentration of chlorides in TrisHCl buffer with the Rh center. Accordingly, using phosphate buffer, Rh-cut proved to be an active artificial enzyme for the hydrogenation of acetophenone, albeit at a lower yield of 27% (Table 1, entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Substrate | Buffer | Product formationb (%) |
| a Reactions carried out with H2 (40 bar) at room temperature for 20 h (see ESI; single measurements). b Determined by chiral GC and GC-MS. c Yield for 1-phenylethanol and methyl acetylalaninate, respectively. d Reaction stopped at 40 min. | ||||
| 1 | Rh-cut | 6 | TrisHCl | 0 |
| 2 | 5 | 6 | TrisHCl | 0 |
| 3 | 5 | 6 | No buffer | 12 |
| 4 | 5 | 6 | Phosphate | 90 |
| 5 | Rh-cut | 6 | Phosphate | 27 |
| 6 | 5 | 7 | Phosphate | >99 |
| 7 | Rh-cut | 7 | Phosphate | >99 |
| 8 | Rh-calb | 7 | Phosphate | >99 |
| 9 | Rh-calb | 6 | Phosphate | 0 |
| 10 | 5 | 6 + 7 | Phosphate | 71, 93c |
| 11 | Rh-cut | 6 + 7 | Phosphate | 15, 78c |
| 12d | 5 | 6 + 7 | Phosphate | 15, 70c |
| 13d | Rh-cut | 6 + 7 | Phosphate | 0, 63c |

Next, we investigated the hydrogenation of a prochiral olefin, methyl 2-acetamidoacrylate, using 5 and Rh-cut under equivalent conditions as the previous reaction and found excellent yields with both catalysts towards the desired methyl acetylalaninate product (Table 1, entries 6 and 7).
Although a clear decrease of activity of the Rh(NHC) motif by its anchoring in the protein was found for the hydrogenation of the ketone, hydrogenation of the olefin did not suffer from a decrease in product formation. This difference in the influence of the protein could originate from a limited approach of acetophenone to the metallic center brought forward by the proteomic surroundings, or by dative interactions towards the metal center, e.g. from N and O donors reducing the oxophilicity of the catalyst; in these scenarios promoting the formation of the alkane from the olefin but hampering the formation of the alcohol from the ketone.
Rhodium catalysts are known to show higher catalytic rates in the hydrogenation of olefins than in the hydrogenation of ketones.22 Hence, the difference in yield between 1-phenylethanol and methyl acetalaninate by Rh-cut does not show by itself a selectivity gain derived from the hybridization, but the comparison of the yields achieved in both transformations between the unsupported catalyst 5 and the Ru-cut hybrid does show a larger influence of the protein over the ketone hydrogenation. In none of the reactions any enantioselectivity in product formation was observed.
The latter observations encouraged us to investigate the anchoring of the rhodium catalyst in a different protein environment. Candida antarctica lipase B (CalB), a lipase with similar exposure of the active site to solvent but at a deeper location,23 was chosen given our experience on its successful inhibition with metal-functionalized phosphonate esters.9 A similar methodology as for the previous inhibition was followed resulting in complete inhibition of this enzyme by a ten-fold excess of Rh-pNP towards the 1:1 metalloprotein Rh-calb. Treatment of pNPB with this hybrid showed no residual hydrolytic activity of CalB according to the release profile of pNP (see the ESI†). Successful use of a single inhibitor with different enzymes shows the versatility of the ASD method in protein scope applicability.
Studying Rh-calb in catalytic hydrogenations again showed no product enantioselectivity. On the other hand, the difference in reactivity between acetophenone and methyl 2-acetamidoacrylate was larger than with the Rh-cut analog. The effect of CalB over the Rh(NHC) motif seemed more pronounced allowing for complete blocking of the ketone in contrast to quantitative hydrogenation of the olefin (Table 1, entries 8 and 9), suggesting full chemoselectivity by this artificial enzyme.
In order to obtain more insight in the apparent chemoselectivity gained with the hybrids, we performed a series of competition experiments using both substrates in the reaction mixture with Rh-cut and 5 (Table 1, entries 10 and 11). The product yields were slightly lower than in the separate reactions. This decrease is attributed to the distribution of the catalyst's turnover over the two substrates. The olefin was hydrogenated in 93% and 78% by 5 and Rh-cut, respectively; in the same manner, the reduction of the ketone yielded 71% and 15%. These results again show an accentuated discrimination of the ketone when the metalloenzyme is used (Fig. 2), supporting that the protein backbone increases the difference between the reaction rates of the susbtrates. At shorter reaction times (40 min, Table 1, entries 12 and 13) the ketone was reduced in some 15% by 5, whereas Rh-cut afforded no ketone reduction at all (at 63% yield of olefin reduction product), which further supports the observed protein-induced chemoselectivity. The group of Lu has previously reported chemoselectivity when Mn–protein artificial enzymes prevented consecutive overoxidation of thioanisole, as opposed to the unsupported catalyst.24 Our current study shows a change in reactivity of different chemical functionalities in different substrates.
 | ||
| Fig. 2 Reaction outcome of the competitive hydrogenation of acetophenone vs. methyl 2-acetamidoacrylate (right) and the corresponding chiral GC chromatograms (center) comparing catalysts 5 [Rh(cod)(SIMes)Cl] and Rh-cut (left). | ||
In conclusion, we have demonstrated for the first time catalytic activity for a soluble artificial metallo-enzyme based on the ASD inhibition of lipases. The lipase hybrids reported here catalyze the hydrogenation of the olefin methyl 2-acetamidoacrylate in excellent yields and ambient temperature but show a protein-induced discrimination in the hydrogenation of the ketone acetophenone. The more sterically demanding active site of CalB as anchoring site resulted in exclusive hydrogenation of the olefin by the corresponding hybrid. This complete chemoselectivity was achieved as well by the cutinase derived hybrid in shorter reaction times.
While excellent chemoselective hydrogenation catalysts, preferring either ketone or olefin reduction, have been developed for application in organic synthesis,18 our current results represent, to the best of our knowledge, the first example of chemoselectivity in reactions catalyzed by artificial metalloenzymes, thereby extending the selectivity repertoire of this specific class of catalysts. These findings may lead to the development of more advanced catalytic tools for the selective conversion of a target substrate in a complex mixture of substrates, which is of interest to the fields of systems catalysis and synthetic biology.
This work was supported financially by the Dutch National Research School Combination-Catalysis (NRSCC). The authors thank Dr Arjan Barendregt from the Biomolecular Mass Spectrometry and Proteomics group at Utrecht University for his help in the MS analysis of the hybrids.
Notes and references
- (a) P. J. Deuss, R. den Heeten, W. Laan and P. C. J. Kamer, Chem. – Eur. J., 2011, 17, 4680 CrossRef CAS PubMed; (b) F. Rosati and G. Roelfes, ChemCatChem, 2010, 2, 916 CrossRef CAS; (c) M. R. Ringenberg and T. R. Ward, Chem. Commun., 2011, 47, 8470 RSC; (d) J. C. Lewis, ACS Catal., 2013, 3, 2954 CrossRef CAS.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306 CrossRef CAS.
- (a) T. R. Ward, Acc. Chem. Res., 2011, 44, 47 CrossRef CAS PubMed; (b) M. Creus and T. R. Ward, Prog. Inorg. Chem., 2012, 57, 203 CAS.
- (a) W. Laan, B. K. Munoz, R. den Heeten and P. C. J. Kamer, ChemBioChem, 2010, 11, 1236 CrossRef CAS PubMed; (b) R. den Heeten, B. K. Munoz, G. Popa, W. Laan and P. C. J. Kamer, Dalton Trans., 2010, 39, 8477 RSC.
- M. Allard, C. Dupont, V. Munoz Robles, N. Doucet, A. Lledos, J.-D. Marechal, A. Urvoas, J.-P. Mahy and R. Ricoux, ChemBioChem, 2012, 13, 240 CrossRef CAS PubMed.
- (a) A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083 RSC; (b) A. J. Boersma, B. de Bruin, B. L. Feringa and G. Roelfes, Chem. Commun., 2012, 48, 2394 RSC.
- (a) T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5716–5722 CrossRef PubMed; (b) M. T. Reetz, Angew. Chem., Int. Ed., 2001, 40, 284 CrossRef CAS; (c) F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495 CrossRef CAS PubMed.
- (a) C. A. Kruithof, M. A. Casado, G. Guillena, M. R. Egmond, A. van der Kerk-van Hoof, A. J. R. Heck, R. J. M. Klein Gebbink and G. van Koten, Chem. – Eur. J., 2005, 11, 6869 CrossRef CAS PubMed; (b) L. Rutten, B. Wieczorek, J.-P. B. A. Mannie, C. A. Kruithof, H. P. Dijkstra, M. R. Egmond, M. Lutz, A. L. Spek, P. Gros, R. J. M. Klein Gebbink and G. van Koten, Chem. – Eur. J., 2009, 15, 4270 CrossRef CAS PubMed; (c) B. Wieczorek, D. J. M. Snelders, H. P. Dijkstra, V. Kees Versluis, M. Lutz, A. L. Spek, M. R. Egmond, R. J. M. Klein Gebbink and G. van Koten, Organometallics, 2012, 31, 2810 CrossRef CAS.
- B. Wieczorek, A. Träff, P. Krumlinde, H. P. Dijkstra, M. R. Egmond, G. van Koten, J.-E. Bäckvall and R. J. M. Klein Gebbink, Tetrahedron Lett., 2011, 52, 1601 CrossRef CAS PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383 CrossRef CAS PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemine Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed.
- For a review on Chiral N-heterocyclic carbenes as stereo directing ligands in asymmetric catalysis see: (a) V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (b) L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270 CrossRef CAS PubMed , and references therein.
- (a) C. Mayer, D. G. Gillingham, T. R. Ward and D. Hilvert, Chem. Commun., 2011, 47, 12068 RSC; (b) C. Lo, M. R. Ringenberg, D. Gnandt, Y. Wilson and T. R. Ward, Chem. Commun., 2011, 47, 12065 RSC; (c) T. Matsuo, C. Imai, T. Yoshida, T. Saito, T. Hayashi and S. Hirota, Chem. Commun., 2012, 48, 1662 RSC; (d) F. Philippart, M. Arlt, S. Gotzen, S.-J. Tenne, M. Bocola, H.-H. Chen, L. Zhu, U. Schwaneberg and J. Okuda, Chem. – Eur. J., 2013, 19, 13865 CrossRef CAS PubMed; (e) A. Kajetanowicz, A. Chatterjee, R. Reuter and T. R. Ward, Catal. Lett., 2014, 144, 373 CrossRef CAS.
- H. Syska, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2012, 703, 56 CrossRef CAS PubMed.
- (a) J. Collot, J. Gradinaru, N. Humbert, M. Skander, A. Zocchi and T. R. Ward, J. Am. Chem. Soc., 2003, 125, 9030 CrossRef CAS PubMed; (b) M. Skander, N. Humbert, J. Collot, J. Gradinaru, G. Klein, A. Loosli, J. Sauser, A. Zocchi, F. Gilardoni and T. R. Ward, J. Am. Chem. Soc., 2004, 126, 14411 CrossRef CAS PubMed; (c) G. Klein, N. Humbert, J. Gradinaru, A. Ivanova, F. Gilardoni, U. E. Rusbandi and T. R. Ward, Angew. Chem., Int. Ed., 2005, 44, 7764 CrossRef CAS PubMed; (d) M. Skander, C. Malan, A. Ivanova and T. R. Ward, Chem. Commun., 2005, 4815 RSC; (e) C. Letondor, N. Humbert and T. R. Ward, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4683 CrossRef CAS PubMed; (f) T. R. Ward, Chem. – Eur. J., 2005, 11, 3798 CrossRef CAS PubMed; (g) C. Letondor, A. Pordea, N. Humbert, A. Ivanova, S. Mazurek, M. Novica and T. R. Ward, J. Am. Chem. Soc., 2006, 128, 8320 CrossRef CAS PubMed.
- (a) M. T. Reetz, Tetrahedron, 2002, 58, 6595 CrossRef CAS; (b) M. T. Reetz, M. Rentzsch, A. Pletsch and M. Maywald, Chimia, 2002, 56, 721 CrossRef CAS.
- L. Panella, J. Broos, J. Jin, M. W. Fraaije, D. B. Janssen, M. Jeronimus-Stratingh, B. L. Feringa, A. J. Minnaard and J. G. de Vries, Chem. Commun., 2005, 5656 RSC.
- (a) R. Noyori, Angew. Chem., Int. Ed., 2013, 52, 79 CrossRef CAS PubMed; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed.
- C. Martinez, P. Geus, M. Lauwereys, M. Matthyssens and C. Cambillau, Nature, 1992, 356, 615 CrossRef CAS PubMed.
- An initial report on the catalytic activity of a cutinase-based hybrid is reported in: B. Wieczorek, Semisynthetic Pincer Metalloenzymes, PhD thesis, Utrecht University, 2009 Search PubMed.
- H. P. Dijkstra, H. Sprong, B. N. H. Aerts, C. A. Kruithof, M. R. Egmond and R. J. M. Klein Gebbink, Org. Biomol. Chem., 2008, 6, 523 CAS.
- G. Shang, W. Li and X. Zhang, Catalytic Asymmetric Synthesis, Transition Metal-catalyzed Homogeneous Asymmetric Hydrogenation, John Wiley & Sons, Inc., 2010, p. 343 Search PubMed.
- According to comparison of the reported crystal structures of the lipases. These data was downloaded from the RSCB Protein Data Base: http://www.rcsb.org/pdb/home/home.do.
- J.-L. Zhang, D. K. Garner, L. Liang, Q. Chen and Y. Lu, Chem. Commun., 2008, 1665 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopical characterization of new compounds. See DOI: 10.1039/c4cc09700a |
| This journal is © The Royal Society of Chemistry 2015 |