
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-surface reductive coupling of aldehydes on Au(111)†
Oscar Díaz
Arado
ab,
Harry
Mönig
*ab,
Jörn-Holger
Franke
c,
Alexander
Timmer
ab,
Philipp Alexander
Held
d,
Armido
Studer
*d and
Harald
Fuchs
*ab
aPhysikalisches Institut, Westfälische Wilhelms-Universität Münster, Wilhelm-Klemm-Strasse 10, 48149 Münster, Germany. E-mail: harry.moenig@uni-muenster.de; fuchsh@uni-muenster.de
bCenter for Nanotechnology (CeNTech), Heisenbergstrasse 11, 48149 Münster, Germany
cDepartment of Physics, Université Libre de Bruxelles, Campus Plaine-CP 231, 1050 Brussels, Belgium
dOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 16th February 2015
Abstract
On-surface synthesis of a polyphenylene vinylene oligomer via reductive coupling of a terephthalaldehyde derivative on Au(111) is reported. Scanning tunneling microscopy and photoelectron spectroscopy experiments confirmed oxygen dissociation and its desorption from the surface. Density functional theory calculations provided a reasonable reaction mechanism involving reactive sites on the substrate.
On-surface synthesis of covalently interlinked organic nanostructures under ultrahigh vacuum (UHV) conditions has obtained extensive attention in recent years.1 Several reactions, such as the Ullmann coupling,2 formation of Schiff bases,3 condensation of boronic acids,4 acylation reactions,5 homocoupling of alkanes6 and alkynes7 and more recently, azide-alkyne cycloadditions8 have been successfully conducted on surfaces. Each approach aims to the same objective: to develop well-ordered functional nanostructures on surfaces, which sometimes are difficult or even impossible to synthesize via classical solution phase chemistry.6 Despite the versatility of these on-surface chemical processes for such a purpose, the investigation of new reaction types that could allow the preparation of more complex nanostructures with tunable properties remains of great interest in this field.
One suitable candidate for on-surface synthesis could be the equivalent of the reductive coupling of carbonyls in solution (McMurry reaction),9 which is known to produce materials with attractive electronic properties like polyphenylene vinylene (PPV). In particular, aldehydes have been shown to form covalent bonds after reductive coupling, which is catalyzed by Ti and V.9,10 Resembling the McMurry-type chemistry, in the present study we use a combination of scanning tunneling microscopy (STM), X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) to investigate the on-surface coupling of 2,5-dihexylterephthalaldehyde (DTA 1) monomers on the Au(111) surface under UHV conditions (Fig. 1). We will show that successful C–C coupling of DTA proceeds directly on Au(111) by temperature induced C–H activation of the aldehyde moiety, resulting in a para-polyphenylene vinylene derivative (p-PPV 2) adsorbed on the substrate. Given their great potential for the design of electronic devices and particularly organic solar cells research,11 the bottom-up growth of PPV-based polymers directly on substrates opens a new possibility for the development and fabrication of functional organic nanomaterials.
 | ||
| Fig. 1 Investigated on-surface reaction. | ||
The design of the DTA molecules allows them to be thermally deposited on the surface at a moderate sublimation temperature. Moreover, the reactants are charged with two aldehyde moieties, which allow this particular compound to undergo oligomerization. DTA 1 was next sublimated onto the Au(111) surface kept at room temperature. STM experiments were performed to study their adsorption and self-assembly behavior, revealing a single densely packed monolayer of monomers 1 on the substrate (Fig. 2a). Within the self-assembled monolayer (DTA-SAM), the aldehyde moieties are oriented along the ![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) direction (inset in Fig. 2a), following a periodicity of a = (0.81 ± 0.04) nm [0.92 nm, calculated value for the gas phase]. When comparing the calculated center-to-center distance with a, a reasonable quantitative agreement is found, where the small difference can be ascribed to the absence of the Au(111) surface in the gas phase calculations, which restrains out-of-plane movements of the molecular components.
direction (inset in Fig. 2a), following a periodicity of a = (0.81 ± 0.04) nm [0.92 nm, calculated value for the gas phase]. When comparing the calculated center-to-center distance with a, a reasonable quantitative agreement is found, where the small difference can be ascribed to the absence of the Au(111) surface in the gas phase calculations, which restrains out-of-plane movements of the molecular components.
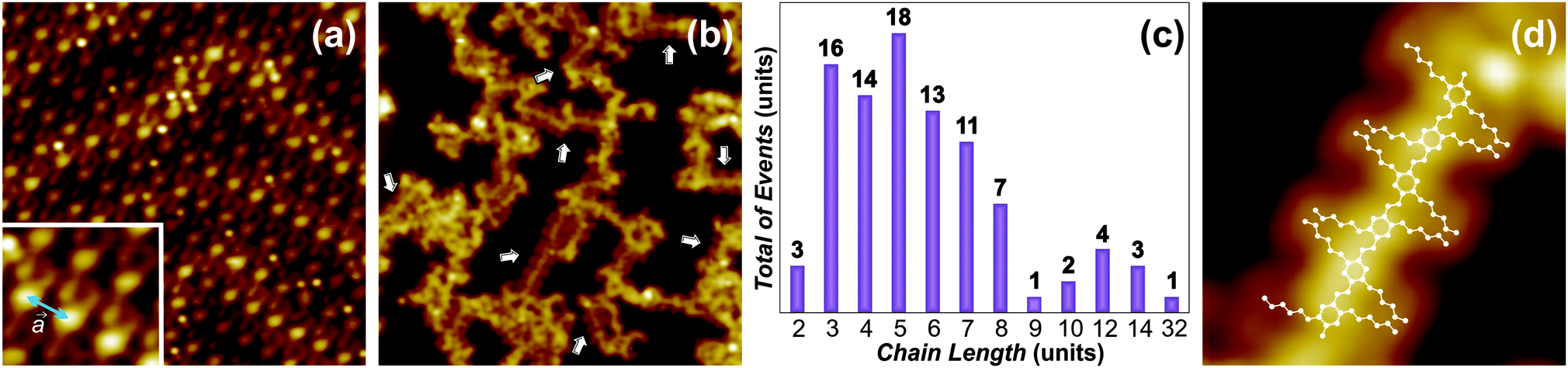 | ||
| Fig. 2 Deposition and on-surface reaction of DTA monomers on Au(111). (a) STM image (12 × 12 nm2; U = −1.50 V; I = 400 pA) after the deposition onto the surface kept at room temperature. The inset shows the intermolecular distance between adjacent monomers along the aldehyde moiety direction. (b) STM image (30 × 30 nm2; U = −0.65 V; I = 7 pA) after the deposition onto the surface kept at 250 °C. The targeted coupling reaction is achieved, producing p-PPV oligomers (white arrows) on the surface. (c) Oligomers 2 chain length distribution. (d) High resolution STM image (5.7 × 8.5 nm2; U = 0.65 V; I = 7 pA) of the p-PPV products 2. The inset schematically represents the chemical structure of the reacted specie. | ||
As a consecutive step, the DTA-SAM was annealed at temperatures ranging from 100 °C to 250 °C. STM experiments after the annealing at T = 250 °C (activation temperature) revealed disordered oligomers on the Au(111) substrate (see ESI†), indicating that a possible activation of the aldehyde moiety and subsequent C–C coupling could have occurred. Given the large dissociation energy for the oxygen of the aldehyde moiety,12 the requirement of such high activation temperature is not surprising. However, we found that the pronounced desorption the reactants undergo at such temperature is a limiting factor for the on-surface coupling. To circumvent this limitation, DTA monomers were deposited onto the Au(111) surface kept at 250 °C. STM imaging after the deposition revealed that oligomerization was accomplished (white arrows in Fig. 2b), with about 6% of the reactants forming the targeted p-PPV products (details in the ESI†). Moreover, the products 2 are surrounded, in a ratio ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, by a disordered phase which is difficult to characterize with STM imaging. An improvement of these factors was not observed in experiments on other Au substrates (see ESI†). However, XPS experiments on Au(111) allowed to exclude the presence of oxygen on the surface after the on-surface reaction (see below). Therefore, the disordered phase could consist of entangled oligomers 2 and/or oxygen-free side-products.
:8, by a disordered phase which is difficult to characterize with STM imaging. An improvement of these factors was not observed in experiments on other Au substrates (see ESI†). However, XPS experiments on Au(111) allowed to exclude the presence of oxygen on the surface after the on-surface reaction (see below). Therefore, the disordered phase could consist of entangled oligomers 2 and/or oxygen-free side-products.
A representative sample of clearly identifiable 562 reacted monomers from different deposition experiments allowed the estimation of the oligomers 2 chain length distribution (Fig. 2c). Oligomers with a length of n = 3–8 units (reacted monomers) are obtained more frequently, whereas the maximum length observed corresponded to n = 32 units. It is interesting to note that the oligomers 2 do not follow the Au(111) herringbone reconstruction, as it has been observed for other on-surface reactions on this substrate.2 Their relative disorder could be due to the substrate reconstruction providing insufficient confinement during the reaction. The enhanced diffusion of the reactants 1 and products 2 on the substrate at the activation temperature could additionally contribute to such disorder. Furthermore, the absence of monomers 1 in the STM images indicates that in this case reactants also desorb, similar as with the DTA-SAM annealing at 250 °C. Moreover, STM tip manipulations were carried out to investigate the intra-molecular and molecule–substrate interaction for the oligomers 2 on Au(111) (see ESI†). These experiments showed the p-PPV products to weakly (non covalently) interact with the metal substrate, while the intra-molecular units are strongly (covalently) interlinked.
High resolution STM images (Fig. 2d) allowed the structural characterization of the newly formed oligomers. As it can be observed, the aliphatic chains follow a distinct zig-zag arrangement, likely induced by van der Waals interactions. More importantly, the products 2 are on a trans configuration, given that the cis alternative would not follow the linear arrangement observed on the surface. The existence of cis isomers in the disordered phase, however, cannot be excluded. We determined the center-to-center distance between adjacent phenyl rings experimentally (0.5 ± 0.2 nm) and by DFT calculations for the gas phase (0.67 nm). When compared with the unit cell direction a, corresponding to the aldehyde moiety orientation in the DTA-SAM (Fig. 2a), a significant reduction of the intermolecular distance (∼0.3 nm) is observed. The reduced center-to-center distance between the phenyl rings clearly indicates that oligomers 2 (schematically represented in Fig. 2d) were formed as a result of a reductive coupling process on the Au(111) surface.
To provide detailed chemical information about the observed on-surface coupling of the DTA monomers, XPS experiments were carried out. After depositing the monomers onto the Au(111) surface kept at room temperature, C1s and O1s spectra were recorded (Fig. 3a and b). In this case, the C1s region shows two distinct peaks which correspond well with contributions from three carbon species at binding energies of 284.7 eV (aromatic), 285.0 eV (hydrocarbon) and 287.6 eV (carbonyl). In addition, a single O1s peak is detected at a binding energy of 531.9 eV, corresponding well with physisorbed aldehyde oxygen. Quantitative analysis reproduced the composition of the DTA reactants (carbon = 91.8%; oxygen = 8.2%). Spectra collected after depositing DTA monomers onto the Au(111) surface kept at 250 °C show an absence of the oxygen-related signals. In this case, the previously observed O1s peak at 531.9 eV (Fig. 3c) and C1s peak at 287.6 eV (Fig. 3d) are not detected. Motivated by the configuration proposed for the p-PPV oligomers (Fig. 2d), the peak fitting of the C1s region in this case includes a new carbon specie consistent with the vinylene moiety of the products. This strongly evidences a complete desorption of oxygen from the surface, in agreement with previously reported studies for Au(111).13 Furthermore, this fact suggests the oxygen scission from the aldehyde as a prerequisite for double bond formation. Moreover, the formation of oxygenated side-products after the on-surface reaction can hereby be excluded.
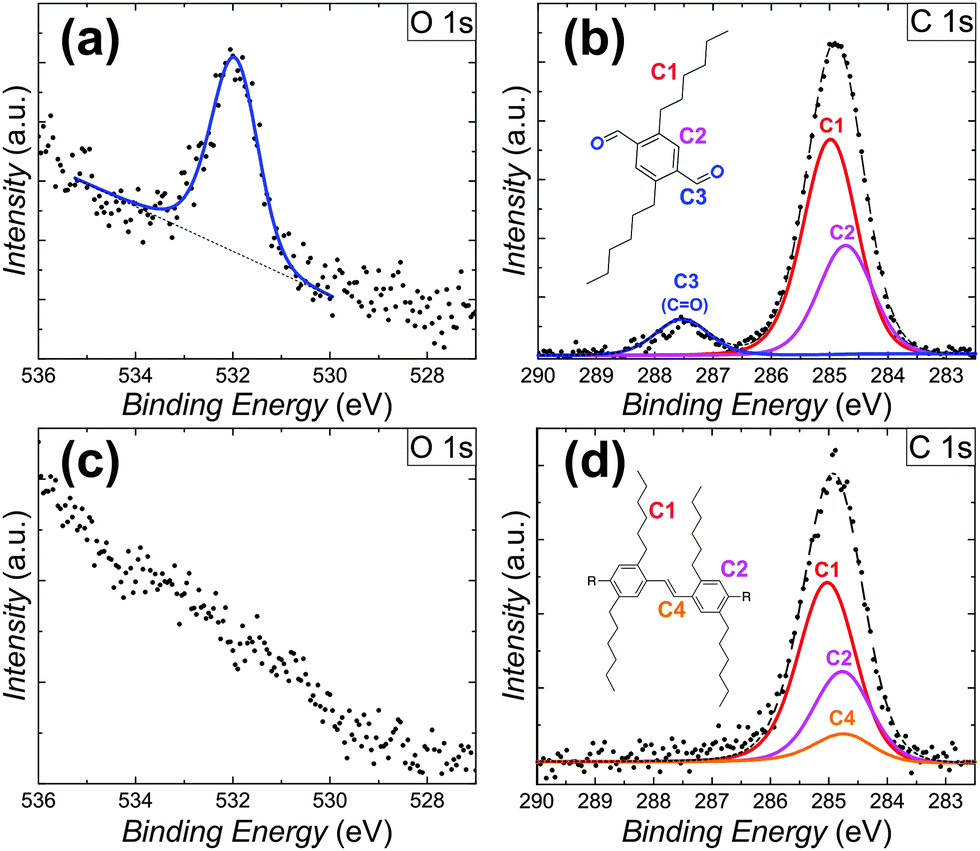 | ||
| Fig. 3 XPS analysis of the on-surface reaction. (a and b) XPS data of the C1s and O1s regions after deposition onto the substrate kept at room temperature. Peak fitting analysis reproduced the stoichiometry of intact monomers. (c and d) XPS data collected after the deposition onto the substrate kept at 250 °C show a complete absence of oxygen species. The C1s signal is consistent with the formation of p-PPV 2, but does not exclude contributions from oxygen-free side products in the disordered phase. | ||
In previous studies of the McMurry-type chemistry, the coupling mechanism is explained by the formation of vicinal diolate intermediates between a metal atom from the catalyst and the oxygen atom from the aldehyde moiety at room temperature. The diolates were shown to induce the dissociation of the oxygen atom and the C–C coupling after subsequent thermal treatment.10 However, our XPS experiments demonstrate that on Au(111) at room temperature, the O1s binding energy for the DTA-SAM (Fig. 3b) corresponds to an aldehyde specie.14 This is also in agreement with our DFT calculations (see ESI†), which are consistent with a weak physisorption of monomers 1 on the substrate, strongly suggesting that a covalently bound diolate is not formed. Furthermore, if diolates were formed directly after the deposition onto the Au(111) surface kept at 250 °C, chemisorbed DTA monomers and/or coadsorbed oxygen from the aldehyde moiety should be expected to remain on the metal substrate.10 In contrast, our XPS data show a complete absence of oxygen related signals, evidencing the desorption of unreacted monomers 1 and the dissociation of oxygen during the on-surface reaction forming the p-PPV products. Therefore, taking also the absence of well-established catalysts (titanium, zinc, vanadium or copper) in our experiments into consideration,9 an alternative reaction mechanism must be considered.
To elucidate the mechanism driving the coupling of aldehydes on the Au(111) surface, we performed systematic DFT calculations. Hereinafter, the most energetically favored mechanism is described in detail, while less favored alternatives are presented in the ESI.† Two simplified DTA monomers adsorbed on the substrate with the carbonyl groups facing each other (Fig. 4; config. 1) are defined as the state with 0 eV. Taking into consideration the large dissociation energy for the aldehyde oxygen,12 we investigated the C–H activation of the carbonyl group via H abstraction by the substrate. We found that a surface adsorbed α-benzoyloxy benzyl radical is generated while the H-atom remains bound to the surface (Fig. 4; config. 2), increasing the total energy to 0.84 eV. This radical formally derives from the acyl radical addition to the carbonyl O-atom of the second reactant. This process can occur at a relatively low transition state energy of 1.13 eV. At this point, a transfer of the hydrogen atom back from the surface to the O-atom of the ester carbonyl group of the α-benzoyloxy benzyl radical leads to a 1,3-biradical (1.27 eV transition state energy), increasing the total energy to 0.90 eV (Fig. 4; config. 3). Following from the 1,3-biradical configuration, the reaction can progress more energetically favored following a two-step process: (i) the 1,3-biradical first cyclizes to a hydroxyoxirane (Fig. 4; config. 4) that is fairly stable at 0.72 eV, with a transition state energy of 2.22 eV; (ii) subsequent oxygen scission from the oxirane ring results in the dissociated oxygen atom adsorbed on the Au(111) surface and an enol (Fig. 4; config. 5) now at a total energy of 0.88 eV, with a transition state energy barrier of 1.81 eV.
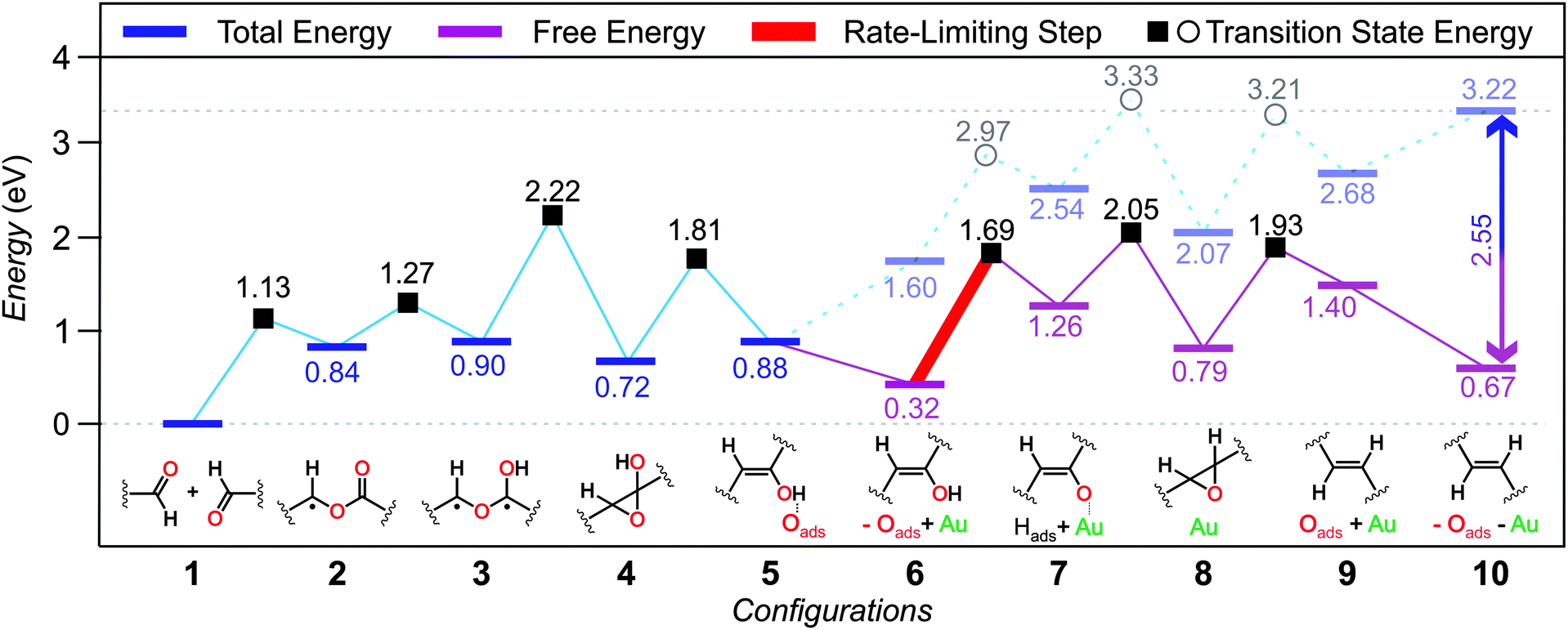 | ||
| Fig. 4 Energetics of the proposed reaction mechanism. Purple lines indicate free energy estimates after oxygen desorption at the experimental conditions. The rate limiting step appears highlighted in red. | ||
Since the XPS data showed that a complete desorption of all oxygen species occurred after the on-surface reaction, we can assume that the dissociated oxygen atom desorbed from the substrate at this point, probably as a molecular specie. It is important to note that the large entropy increase upon desorption of a molecular specie decreases the free energy of the system by 2.55 eV per O2 molecule at the experimental conditions, which provide the thermodynamic driving force for this desorption process.15 Another important aspect to be considered is the existence of many reactive sites on the Au(111) substrate due to surface reconstruction and/or surface diffusion of single Au atoms at elevated temperatures. Therefore, the next step of the reaction is calculated in a system where the oxygen atom is already removed from the surface and an extra Au atom mimicking a reactive site (see ESI†) is introduced (Fig. 4; config. 6), increasing the total energy to 1.60 eV and decreasing the free energy at the experimental conditions to 0.32 eV accordingly. From that point on, H donation to the substrate can occur after overcoming a transition state energy of 1.69 eV forming a Au-enolate (Fig. 4; config. 7) and leaving the system at a free energy of 1.26 eV. Hydrogen recombination with the Au-enolate results in the formation of an epoxide (Fig. 4; config. 8) now at a free energy of 0.79 eV after a transition state energy of 2.05 eV. Subsequent deoxygenation results in the dissociated oxygen adsorbed on the substrate (not bound to the extra Au atom) and the formation of the final product (Fig. 4; config. 9) at a free energy of 1.40 eV after a transition state energy of 1.93 eV. Given the poor interaction of the dissociated oxygen with the extra Au atom and that our XPS results demonstrate the absence of oxygenated species on the surface after the reaction, in the final configuration the oxygen and the extra Au atoms are removed from the system simultaneously (Fig. 4; config. 10). This results in an increase of the total energy of the system up to 3.22 eV and a reduction of its free energy down to 0.67 eV at the experimental conditions. The rate-limiting step of the proposed reaction mechanism is the 1.37 eV that the system must overcome for the formation of the Au-enolate in config. 7. It is important to note that such a barrier can be overcome at our experimental conditions.
In conclusion, we have shown the successful reductive coupling of an aldehyde on a Au(111) surface leading to the formation of a PPV derivative. The obtained oligomers 2 were found to be covalently interlinked and to remain weakly adsorbed on the substrate while oxygen completely desorbs during the reaction. Based on our results, the so far accepted reaction pathway for reductive aldehyde coupling involving a vicinal diolate intermediate is not dominating the observed on-surface reaction. Different alternatives were explored, and an energetically favored C–C coupling mechanism proceeding via initial C–H activation of the aldehydes followed by a deoxygenation process favored by gold adatoms on the surface is proposed. With this on-surface synthesis approach, nanostructures with tunable optoelectronic properties on substrates can be developed, thus increasing the existing pool of suitable reactions for bottom-up growth of organic nanostructures on surfaces.
Financial support by the Deutsche Forschungsgemeinschaft (DFG) through the SFB 858 (project B2) and the transregional collaborative research center TRR 061 (projects B3 and B7) is gratefully acknowledged.
References
-
(a) A. Gourdon, Angew. Chem., Int. Ed., 2008, 47, 6950–6953 CrossRef CAS PubMed
; (b) D. F. Perepichka and F. Rosei, Science, 2009, 323, 216–217 CrossRef CAS PubMed
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193–1197 CrossRef CAS PubMed
- S. Weigelt, C. Busse, C. Bombis, M. M. Knudsen, K. V. Gothelf, T. Strunskus, C. Wöll, M. Dahlbom, B. Hammer, E. Lgsgaard, F. Besenbacher and T. R. Linderoth, Angew. Chem., Int. Ed., 2007, 46, 9227–9230 CrossRef CAS PubMed
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed
- M. Treier, N. V. Richardson and R. Fasel, J. Am. Chem. Soc., 2008, 130, 14054–14055 CrossRef CAS PubMed
- D. Y. Zhong, J. H. Franke, S. K. Podiyanachari, T. Blömker, H. M. Zhang, G. Kehr, G. Erker, H. Fuchs and L. F. Chi, Science, 2011, 334, 213–216 CrossRef CAS PubMed
- H. Y. Gao, H. Wagner, D. Zhong, J. H. Franke, A. Studer and H. Fuchs, Angew. Chem., Int. Ed., 2013, 52, 4024–4028 CrossRef CAS PubMed
- O. Díaz Arado, H. Mönig, H. Wagner, J. H. Franke, G. Langewisch, P. A. Held, A. Studer and H. Fuchs, ACS Nano, 2013, 7, 8509–8515 CrossRef PubMed
-
(a) J. E. McMurry and M. P. Fleming, J. Am. Chem. Soc., 1974, 96, 4708–4709 CrossRef CAS
-
(a) L. Benz, J. Haubrich, R. G. Quiller, S. C. Jensen and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 15026–15031 CrossRef CAS PubMed
-
(a) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed
- X. Deng, B. K. Min, A. Guloy and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 9267–9270 CrossRef CAS PubMed
-
G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: the Scienta ESCA300 Database, Wiley, New York, 1992 Search PubMed
- F. Bebensee, K. Svane, C. Bombis, F. Masini, S. Klyatskaya, F. Besenbacher, M. Ruben, B. Hammer and T. Linderoth, Chem. Commun., 2013, 49, 9308–9310 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc09634g |
| This journal is © The Royal Society of Chemistry 2015 |