
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-aqueous semi-solid flow battery based on Na-ion chemistry. P2-type NaxNi0.22Co0.11Mn0.66O2–NaTi2(PO4)3†
Edgar
Ventosa
*a,
Daniel
Buchholz
bcd,
Stefan
Klink
e,
Cristina
Flox
a,
Luciana Gomes
Chagas
bcd,
Christoph
Vaalma
bc,
Wolfgang
Schuhmann
e,
Stefano
Passerini
bc and
Joan Ramon
Morante
af
aCatalonia Institute for Energy Research, Jardins de les Dones de Negre, e1, 08930 Sant Adria de Besos, Barcelona, Spain. E-mail: edgar.ventosa@rub.de
bHelmholtz Institute Ulm (HIU) Electrochemistry I, Helmholtzstrasse 11, 89081 Ulm, Germany
cKarlsruhe Institute of Technology (KIT), P.O. Box 3640, 76021 Karlsruhe, Germany
dInstitute of Physical Chemistry, University of Muenster, Corrensstrasse 28/30, Muenster, 48149, Germany
eAnalytical Chemistry – Center for Electrochemical Sciences (CES), Ruhr-University Bochum, Universitätsstr. 150, 44780 Bochum, Germany
fDepartament d'Electronica, Facultat de Fisica, Universitat de Barcelona, Martí i Franques 1, 08028 Barcelona, Spain
First published on 22nd December 2014
Abstract
We report the first proof of concept for a non-aqueous semi-solid flow battery (SSFB) based on Na-ion chemistry using P2-type NaxNi0.22Co0.11Mn0.66O2 and NaTi2(PO4)3 as positive and negative electrodes, respectively. This concept opens the door for developing a new low-cost type of non-aqueous semi-solid flow batteries based on the rich chemistry of Na-ion intercalating compounds.
Redox flow batteries (RFB) are promising technologies for energy storage due to the long life, low cost, high round-trip efficiency and independent scalability of energy and power capabilities.1–6 Semi-solid flow batteries (SSFBs) are a special class of RFB, in which anolyte and catholyte consist of flowable suspensions of solid active materials rather than dissolved redox species.7–11 Thus, the concentration of active redox centres in the anolyte and catholyte of the SSFB can be significantly increased. Using intercalation type active materials such as those typically used in Li-ion batteries (LIBs), e.g. Li4Ti5O12, LiCoO2 or LiNi0.5Mn1.5O4, the energy densities can reach up to 300–500 W h L−1, which is more than 10 times higher than that of all-vanadium RFBs (40 W h L−1).7 Compared to LIBs, SSFBs present several advantages: (I) power and energy can be scaled independently, (II) the amount of inactive materials such as current collectors or housing is decreased, and (III) the manufacturing processes become simpler and more cost-effective.
Sodium-ion batteries (SIBs) attracted increasing attention in the past few years since sodium is abundant, inexpensive, and does not alloy with aluminium which allows for cheaper current collectors.12–14 Energy densities of ca. 200 W h kg−1 have been proposed to be achievable.14 Even more importantly, while sodium intercalation compounds do not necessarily exhibit similar performance like their lithium counterparts, sodium does offers an even larger variety of crystal chemistries than lithium. As such, SIB technology is still considered to be in its infancy, and new active materials are developed.15,16
A key aspect in both LIB and SIB is the formation of the so called solid electrolyte interphase on negative (SEI) and positive (CEI) electrodes operated outside the electrochemical stability window due to reductive or oxidative decomposition of the carbonate based electrolyte. In the case of SSFBs, the formation of these passivating films has a specific detrimental effect since it hinders the electrical connection between the current collector and single particles dispersed in the electrolyte. In consequence, Duduta et al.7 employed Li4Ti5O12 as negative electrode material since it operates above the reduction potential of carbonate electrolytes to construct the first proof of principle of non-aqueous Li-ion SSFB. For SIBs, however, the search for “SEI-free” negative electrodes is more difficult, since the intercalation of sodium into the analogues of Li4Ti5O12 or TiO2 does not operate within the stability window of the electrolyte.17,18
The NASICON material NaTi2(PO4)3 (NaTP), however, does operate at a very flat potential plateau located at around ca. 2.1 V vs. Na/Na+,19–21 which is well above the stability limit of typical electrolytes.22 As it can be seen from Fig. 1a, a charge capacity of 125 mA h g−1 can be utilized when cycled as solid film electrode versus metallic sodium in a three-electrode Swagelok cell, which is very close to the theoretical value of 133 mA h g−1.
 | ||
| Fig. 1 Potential profile of (a) carbon coated (2 wt%) NaTi2(PO4) and (b) P2-type NaxNi0.22Co0.11Mn0.66O2 in the second cycle at 0.1C (12 mA g−1) as solid electrodes. Electrolyte solutions: (a) 1 M NaClO4 in PC, (b) 1 M NaPF6 in PC. Three-electrode Swagelok cell in which the counter and reference electrode was metallic sodium. Temperature: 22 °C. | ||
The positive electrode material should likewise operate at high potentials just below the potential of electrolyte oxidation. As shown in Fig. 1b, P2-type NaxNi0.22Co0.11Mn0.66O2 (NaNCM) has been demonstrated to store reversibly ca. 130 mA h g−1 in the range 4.3–2.1 V with excellent cyclibility.23–25 Detailed structural characterization of NaTP and NaNCM is given in the ESI.† On the base of the electrochemical performances of these materials, we selected NaTP and NaNCM as negative and positive electrode material for the construction of a non-aqueous sodium-based SSFB.
In the semi-solid flow cell configuration, NaTP and NaNCM were, together with 1.3 wt% conductive additive, dispersed in 0.5 M NaPF6 in ethyl carbonate/dimethyl carbonate (EC:DMC) as anolyte and catholyte. As already suggested before, SSFB are best evaluated under intermittent flow.7 Although NaTP was expected to be the charge limiting electrode due to the slightly smaller total charge capacity, it is apparent from Fig. 2 that the system was limited by the positive electrode, since the upper cut-off voltage of the battery was reached before reaching the end of the potential plateau of NaTP. Galvanostatic Intermittent Titration Technique (GITT) revealed that overpotentials during charge and discharge (ca. 0.5 V) at a current density of 0.5 mA cm−2 (ca. 0.35 C) mostly derive from the ohmic overpotential at the positive electrode, which prevents most of the suspension from accessing the charge capacity observed in solid electrode above 4.0 V.
 | ||
| Fig. 2 GITT potential curves of the SSFB consisting of suspensions of P2-type NaxNi0.22Co0.11Mn0.66O2–C/NaTi2(PO4)3 (positive and negative electrodes, respectively) at 0.5 mA cm−2 (ca. 0.35 C) in static. The upper part displays the potentials profiles of the positive and negative electrode versus a reference of Na/Na+, and the bottom part depicts the resulting voltage of the battery. | ||
Decreasing the current density to 0.17 mA cm−2 (ca. 0.1 C), indeed, resulted in a significantly improved specific charge capacity of the suspensions (Fig. 3). Fig. 3a shows the voltage profiles of three subsequent cycles for the first injection. A reversible specific charge capacity of ca. 80 mA h gcathode−1 was obtained for the first three cycles demonstrating the good reversibility of the electrochemical processes. The reversible charge capacity of the suspension (80 mA h gNaNCM−1) was below the value obtained using the solid electrode (130 mA h gNaNCM−1) which is likely due to the fact that the charge capacity of NaNCM above 4.0 V vs. Na/Na+ (Fig. 1) was not accessible due to high overpotentials.
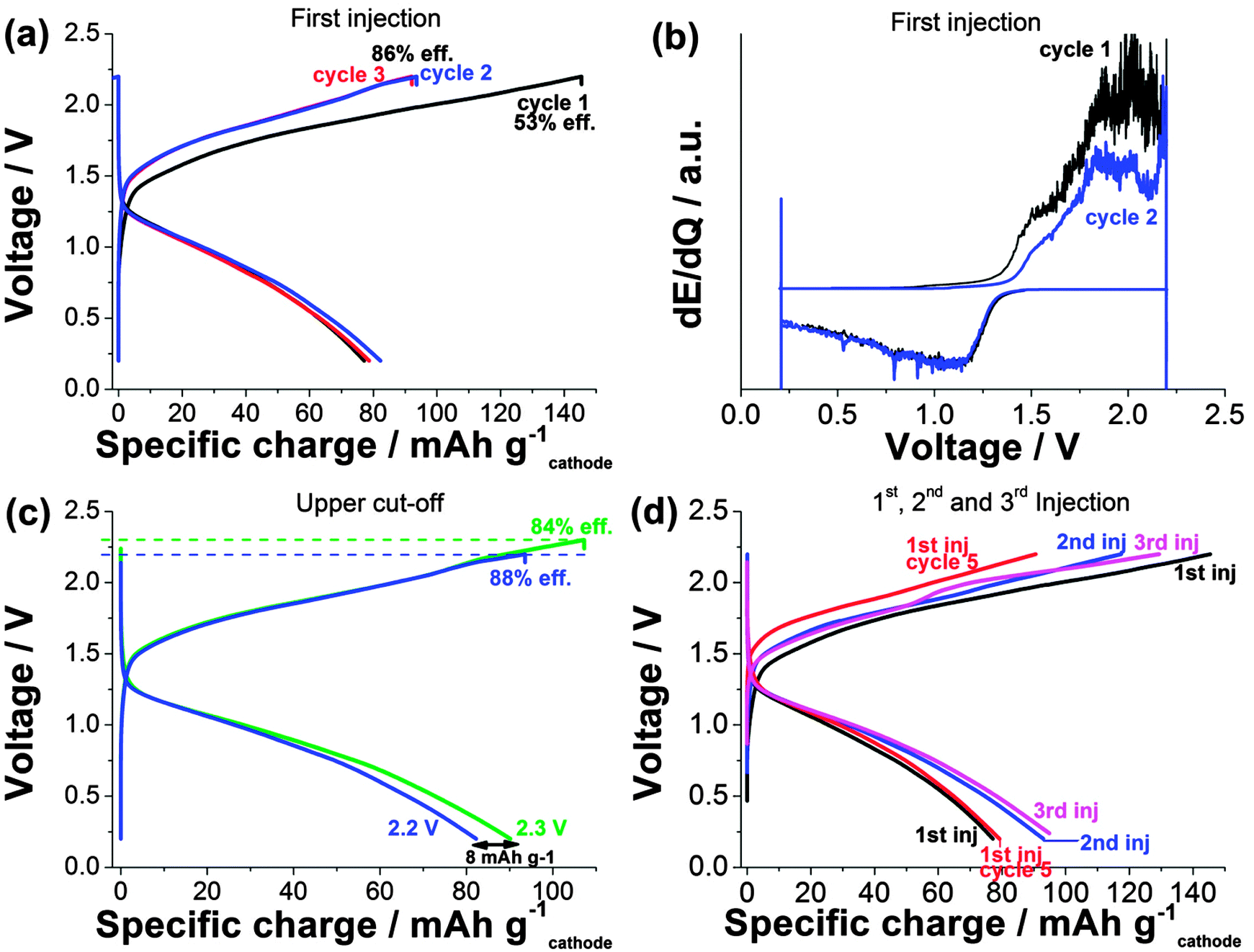 | ||
| Fig. 3 (a) Voltage profiles of the first three cycles of the first injection of SSFB consisting of suspensions of P2-type NaxNi0.22Co0.11Mn0.66O2–C/NaTi2(PO4)3 (positive and negative electrodes, respectively) at 0.17 mA cm−2 in static (ca. 65 mg of active material in the anolyte and catholyte compartments). (b) Differential voltage plot of the first injection. (c) Voltage profiles of the first injection with two different upper cut-off voltages (2.2 V and 2.3 V). (d) Voltage profiles of the first cycle of the three first injections as well as the fifth cycle of the first injection. | ||
The coulombic efficiency increased from 53% to 86–88% after the first cycle. The reduction of surface groups of carbon at the negative electrode as well as some electrolyte decomposition at the positive side occurring mainly during the first cycle were assumed to be the source of the observed modulation in efficiency. The differential voltage plot (Fig. 3b) indicates that the charge process occurred mainly in voltage regions located at around 1.9 V, whereas the discharge started at 1.35 V, reached the maximum at 1.1 V and then continues to the lower cut-off voltage. Increasing the upper cut-off voltage from 2.2 V to 2.3 V led to an increment in the reversible specific charge of 8 mA h gNaNCM−1. On the other hand, the coulombic efficiency decreased from 88% to 84%, which was probably related with electrolyte decomposition at the positive electrode. This suggests that upper cut-off voltages above 2.2 V are not suitable. Fig. 3d depicts the voltage profiles of three subsequent injections of suspensions. Between the injections, the suspensions were flown for at least 10 min (at 3 mL min−1 for a total volume of 6 mL of suspension) to homogenize the entire suspension. The fifth cycle of the first injection is included for comparison. An increase of 15 mA h gNaNCM−1 was observed from the first to the second injection and it stabilized from the second to the third one. The decrease of irreversible processes and an improved electrical percolation of the suspensions after several days of stirring were possibly responsible for the increase of reversible specific charge.
The coulombic efficiency of the first cycle increased from the first to subsequent injections (53%, 79% and 75% for 1st, 2nd and 3rd injection, respectively). Nevertheless, the values of coulombic efficiency of the first cycle of the second and third injection were in between those of the first and the fifth cycle of the first injection (53% and 88%, respectively). In other words, (i) the amount of irreversible processes occurring in the first injection decreases for subsequent injections (2nd, 3rd, etc.) and (ii) the amount of irreversible processes occurring in the fifth cycle of the first injection is lower than that of the first cycle of subsequent fresh injections. This fact indicates that irreversible processes decrease after the first injection for each fresh injection and they continue decreasing until the entire volume of suspension has been cycled. On the other hand, the coulombic efficiency even after several cycles remained rather low (88%), which suggests that the voltage limit of 2.2 V to be slightly too high and electrolyte decomposition to occur, especially if the large active material-electrolyte contact area and the values obtained for Li-based SSFB (80% coulombic efficiency in the second cycle) are taken into account.7
In conclusion, we have successfully demonstrated that non-aqueous semi-solid flow batteries can operate on Na-ion based chemistry. This first proof of principle has been achieved by employing P2-type NaxNi0.22Co0.11Mn0.66O2 and NaTi2(PO4)3 as positive and negative electrode materials, respectively. The proposed battery stores 80 mA h gNaNCM−1 within the voltage range of 2.2–0.2 V. First results are encouraging but certainly indicate the need for a better understanding and control of irreversible charge losses in this type of battery. Although the energy density of this proof of concept was ca. 9 W h L−1 (6 W h kg−1), a proper selection and optimization of the electrolyte, active materials, especially the negative electrode, as well as cycling conditions will certainly result in a significantly improved electrochemical performance of sodium-based SSFBs, e.g. a 2.5 V battery with 30 vol% of active material in the suspensions would deliver ca. 150 W h L−1.
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 608621. European Regional Development Funds (ERDF-FEDER Programa Competitivitat de Catalunya 2007–2013) are also acknowledged.
Notes and references
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- C. P. de Leon, A. Frias-Ferrer, J. Gonzalez-Garcia, D. Szanto and F. C. Walsh, J. Power Sources, 2006, 160, 716–732 CrossRef PubMed.
- L. Joerissen, J. Garche and C. Fabjan, J. Power Sources, 2004, 127, 98–104 CrossRef CAS PubMed.
- M. Skyllas-Kazacos and F. Grossmith, J. Electrochem. Soc., 1987, 134, 2950–2953 CrossRef CAS PubMed.
- C. Flox, M. Skoumal, J. Rubio-Garcia, T. Andreu and J. R. Morante, Appl. Energy, 2013, 109, 344–351 CrossRef CAS PubMed.
- C. Flox, J. Rubio-Garcia, R. Nafria, R. Zamani, M. Skoumal, T. Andreu, J. Arbiol, A. Cabot and J. R. Morante, Carbon, 2012, 50, 2372–2374 CrossRef CAS PubMed.
- M. Duduta, B. Y. Ho, V. C. Wood, P. Limthongkul, V. E. Brunini, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2011, 1, 511–516 CrossRef CAS.
- S. Hamelet, T. Tzedakis, J.-B. Leriche, S. Sailler, D. Larcher, P.-L. Taberna, P. Simon and J.-M. Tarascon, J. Electrochem. Soc., 2012, 159, A1360–A1367 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Energy Environ. Sci., 2013, 6, 1552–1558 CAS.
- S. Hamelet, D. Larcher, L. Dupont and J.-M. Tarascon, J. Electrochem. Soc., 2013, 160, A516–A520 CrossRef CAS PubMed.
- F. Y. Fan, W. H. Woodford, Z. Li, N. Baram, K. C. Smith, A. Helal, G. H. McKinley, W. C. Carter and Y.-M. Chiang, Nano Lett., 2014, 14, 2210–2218 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 CAS.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007–15028 RSC.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y.-S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- L. Wu, D. Bresser, D. Buchholz, G. Giffin, C. Ramirez-Castro, A. Ochel and S. Passerini, Adv. Energy Mater. DOI:10.1002/aenm.201401142.
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS.
- C. Delmas, A. Nadiri and J. L. Soubeyroux, Solid State Ionics, 1988, 28–30, 419–423 CrossRef.
- S. Patoux and C. Masquelier, Chem. Mater., 2002, 14, 5057–5068 CrossRef CAS.
- A. Bhide, J. Hofmann, A. K. Dürr, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2014, 16, 1987–1998 RSC.
- D. Buchholz, A. Moretti, R. Kloepsch, S. Nowak, V. Siozios, M. Winter and S. Passerini, Chem. Mater., 2013, 25, 142–148 CrossRef CAS.
- D. Buchholz, L. Gomes Chagas, M. Winter and S. Passerini, Electrochim. Acta, 2013, 110, 208–213 CrossRef CAS PubMed.
- L. Gomes Chagas, D. Buchholz, L. Wu, B. Vortmann and S. Passerini, J. Power Sources, 2014, 247, 377–383 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD patterns, SEM images. See DOI: 10.1039/c4cc09597a |
| This journal is © The Royal Society of Chemistry 2015 |