
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An IrIV-containing polyoxometalate†
Sergey A.
Adonin‡
ab,
Natalya V.
Izarova‡
c,
Claire
Besson
cd,
Pavel A.
Abramov
ab,
Beatrix
Santiago-Schübel
e,
Paul
Kögerler
 *cd,
Vladimir P.
Fedin
ab and
Maxim N.
Sokolov
*ab
*cd,
Vladimir P.
Fedin
ab and
Maxim N.
Sokolov
*ab
aNikolaev Institute of Inorganic Chemistry SB RAS, 630090 Novosibirsk, Russia. E-mail: caesar@niic.nsc.ru; Fax: +7 383-330-94-89; Tel: +7 383-316-58-45
bNovosibirsk State University, 630090 Novosibirsk, Russia
cPeter Grünberg Institute – PGI 6, Forschungszentrum Jülich, D-52425 Jülich, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de
dInstitute of Inorganic Chemistry, RWTH Aachen University, D-52074 Aachen, Germany
eCentral Institute for Engineering, Electronics and Analytics – ZEA 3, Forschungszentrum Jülich, D-52425 Jülich, Germany
First published on 28th November 2014
Abstract
[HIrIVW6O24]7−, representing the first Ir-containing Anderson–Evans-type polyanion and the first structurally characterized IrIV-based polyoxometalate, has been isolated as its hydrated sodium salt and characterized by single-crystal X-ray diffraction, mass spectrometry, and IR, UV-Vis and EPR spectroscopy. Cyclic voltammetry indicates that the IrIV ions in [HIrW6O24]7− can undergo reversible one-electron reduction and oxidation, resulting in IrIII and IrV derivatives.
The chemistry of polyoxometalates (POMs) incorporating noble metals represents a rapidly developing area, the interest in which is driven by various fundamental and applied aspects.1 Members of this class demonstrate a range of promising properties, in particular, catalytic activity in water splitting2,3 and oxidation of various organic substrates.4 We stress, though, that the number of publications on POM complexes of Ru and Pd by far exceeds those dedicated to all remaining noble metals (Pt, Au, Ir, Os and Rh) taken together. In particular, the chemistry of Ir-containing POMs for a long time has been limited to various POM-supported IrI- and IrIII-organometallic complexes (e.g. [IrI(1,5-COD)]+, [IrIII(Cp*)]2+etc.),5 some of which, e.g. [(1,5-COD)IrINb3P2W15O62]8−, are pre-catalysts for cycloalkene oxygenation with O2.5b–d Recently a number of Keggin and Wells–Dawson type POMs with covalently grafted heteroleptic cyclometalated IrIII-complexes exhibiting effective charge separation between the IrIII-based chromophore and the POM unit were reported.6 The first reliably characterized iridium-containing POM incorporating no organometallic fragments, [(IrIIICl4)(P2W20O72)]15−, was reported as late as in 2009 by Hill's group and identified as a water oxidation catalyst.3 Two years later, the synthesis, characterization and reactivity of Keggin-type complexes [PW11O39IrIIIL]n− (n = 4 or 5; L = H2O, OH, CH3, Cl, SCN) were reported.7 Yet, POMs incorporating IrIV centers, which are of interest as promoters or intermediates in oxidation reactions, remain unknown. For this oxidation state, we expect Ir-containing polyoxotungstates or molybdates to adopt the Anderson–Evans structure, in analogy to known RhIII,8 PtIV,9,10 and even PdIV derivatives.11
We report the preparation of [HIrIVW6VIO24]7− (IrW), the first polyoxometalate incorporating IrIV, and its characterization by single-crystal X-ray analysis, mass spectrometry, IR, UV-Vis and EPR spectroscopy, as well as cyclic voltammetry.
IrW assembles in condensation reactions of in situ-formed IrIV hydroxo complexes with tungstate ions in aqueous acetate media. In a first step, IrCl3·3H2O was dissolved in a CH3COONa solution and the pH of the reaction mixture was adjusted to 12 with 6 M NaOH. Heating of the solution led to a gradual color change from brown-green to blue and then to purple. Previous studies on alkaline solutions of hexachloroiridate(III) have shown that the processes in such reaction systems include substitution of Cl− ligands in the inner coordination sphere of the IrIII ions by OH− groups and the subsequent oxidation of thus formed hydroxo–oxo complexes of IrIII in air.12 During the reaction, the color change of the IrIV-containing solutions from blue to purple is apparently caused by (not well-defined) oligomerization processes. Another hypothesis postulates transformation of the blue superoxo dimeric [(OH)5IrIV(μ-O2−)IrIV(OH)5]3− species into the purple [(OH)4IrIV(μ-O22−)(μ-OH)IrIV(OH)5]3− complex, based on UV-Vis and EPR studies of 3 M KOH solutions of [IrCl6]3−.12f Next, the solution of the in situ-formed IrIV hydroxo complexes was added dropwise to a solution containing the lacunary [B-α-PW9O34]9− polyanion under careful control of pH kept strictly between 6 and 7 by addition of diluted nitric acid. [B-α-PW9O34]9− is unstable in solution and acts as a source of free WO42− species, which effectively assemble and condense around IrIV, resulting in IrW. This is conceptually similar to the controlled formation of MnIII species by slow hydrolysis of [Mn12(CH3COO)16(H2O)4O12] in the synthesis of Mn-substituted polyoxotungstates.13 It should be noted that both monoprotonated and non-protonated forms of [IrIVWVIO24]8− (IrW′) can be isolated as the hydrated sodium salts Na6H[HIrIVWVIO24]·26H2O (Na–IrW) and Na8[IrIVWVIO24]·26H2O (Na8–IrW′), respectively. The exact protonation state depends on the final pH of the reaction mixture.
Thus, pure Na–IrW crystals form when the final pH of the reaction mixture before crystallization is set within the range of 6.1–6.5. At higher final pH (7.0–7.5) crystals of Na8–IrW′ form as a byproduct to Na–IrW. No crystals could be isolated at pH > 7.6 where one could expect formation of pure Na8–IrW′. We herein focus on the monoprotonated species and provide structural data for Na8–IrW′ in the ESI.†
Our attempts to rationalize the synthetic procedure by reacting IrIV solutions directly with Na2WO4 at various ratios were not successful and resulted in formation of the paratungstate salt Na10[H2W12O42]·25H2O as the main product, as established via single-crystal X-ray measurements and IR spectroscopy. We conjecture that an initial excess of IrIV ions coupled with the gradual release of WO42− species into the reaction medium are key to successful preparation of IrW. Alternatively, IrW can be prepared by prolonged heating of K2[IrF6] with Na2WO4 in water at neutral pH, however, this reaction is not very well reproducible and the IrW salts could be isolated only in a low yield (≤10%).
The compound Na6H[HIrIVW6VIO24]·26H2O (Na–IrW) crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The IrW polyanions display an Anderson–Evans-type ring structure comprising a central IrIVO6 octahedron surrounded by six distorted edge-sharing WVIO6 octahedra (Fig. 1). All metal centers in the IrW6 core define a single plane and form a virtually regular Ir-centered W6 hexagon (W⋯W: 3.223–3.345 Å; Ir⋯W: 3.234–3.316 Å). IrW has idealized D3d symmetry and thus belongs to the A-type family of polyanions with Anderson–Evans structure which is typical for [M'W6O24]n− heteropolyoxotungstates. In contrast, some of the polyoxomolybdates [M'Mo6O24]n− may also adopt a bent conformation with C2v symmetry (usually referred as a B-type Anderson–Evans structure), which was e.g. found in [H4PtMo6O24]4−,9a,c–e [H2SbMo6O24]5−
. The IrW polyanions display an Anderson–Evans-type ring structure comprising a central IrIVO6 octahedron surrounded by six distorted edge-sharing WVIO6 octahedra (Fig. 1). All metal centers in the IrW6 core define a single plane and form a virtually regular Ir-centered W6 hexagon (W⋯W: 3.223–3.345 Å; Ir⋯W: 3.234–3.316 Å). IrW has idealized D3d symmetry and thus belongs to the A-type family of polyanions with Anderson–Evans structure which is typical for [M'W6O24]n− heteropolyoxotungstates. In contrast, some of the polyoxomolybdates [M'Mo6O24]n− may also adopt a bent conformation with C2v symmetry (usually referred as a B-type Anderson–Evans structure), which was e.g. found in [H4PtMo6O24]4−,9a,c–e [H2SbMo6O24]5−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (ref. 14) and heptamolybdate [Mo7O24]6− ions.15
(ref. 14) and heptamolybdate [Mo7O24]6− ions.15
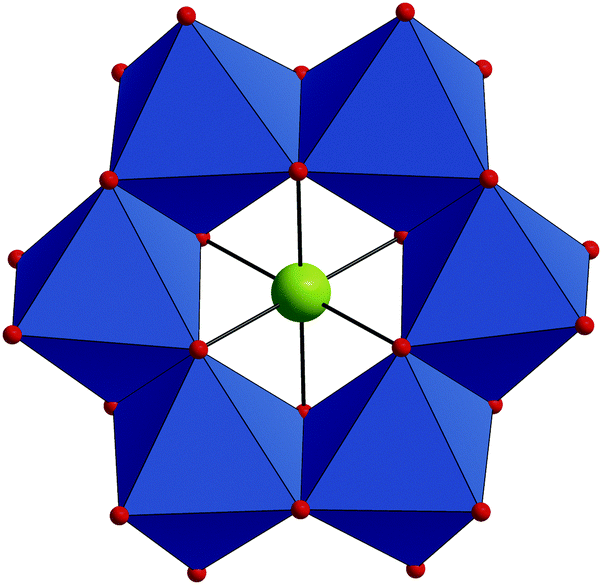 | ||
| Fig. 1 Structure of IrW. Color code: WO6, blue octahedra; Ir, green; O, red. | ||
There are three types of oxygen atoms in IrW: μ3-O linking IrIV and two WVI centers (Ir–O: 1.994(5)–2.022(5) Å; W–O: 2.142(5)–2.293(5) Å), μ2-O bridging two WVI ions (W–O: 1.923(5)–1.987(5) Å) and terminal O atoms (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 1.735(5)–1.747(5) Å). The average Ir–O bond length in IrW of 2.004 Å is comparable with that of 2.013 Å observed in the only structurally characterized IrIII-containing all-inorganic polyoxotungstate [(IrIIICl4)P2W20O72]15−,3 and with that in IrIV oxides, e.g. CaIrO3 (2.057 Å).16 The W–O distances in IrW are similar to the W–O bond lengths in other [XW6O24]n− species (X = PtIV, TeIV, MnIV, n = 8; X = SbV, n = 7).10,17 Bond valence sum calculations (Table S2, ESI†) suggest a disorder of a proton over two inversion center related μ3-O atoms (O123) in IrW. The second proton, which should be present in the structure of Na–IrW from the charge balance considerations (only six Na+ countercations were found both in X-ray diffraction and elemental analyses of Na–IrW), is either highly disordered over the terminal oxygens of the IrW polyanions (BVS 1.60–1.67) or, more likely, is associated with an oxygen of a co-crystallized water molecule, i.e. as a H3O+ cation. In the crystal lattice of Na–IrW the Na+ countercations and the crystal water molecules assemble in the parallel infinite zig-zag {Na6(μ2-H2O)10(H2O)t14}n6n+ chains. The IrW polyanions are packed between the chains (see ESI† for details) and form an extended net of hydrogen bonds with the H atoms of the H2O molecules (Fig. S1 and Table S4, ESI†).
O: 1.735(5)–1.747(5) Å). The average Ir–O bond length in IrW of 2.004 Å is comparable with that of 2.013 Å observed in the only structurally characterized IrIII-containing all-inorganic polyoxotungstate [(IrIIICl4)P2W20O72]15−,3 and with that in IrIV oxides, e.g. CaIrO3 (2.057 Å).16 The W–O distances in IrW are similar to the W–O bond lengths in other [XW6O24]n− species (X = PtIV, TeIV, MnIV, n = 8; X = SbV, n = 7).10,17 Bond valence sum calculations (Table S2, ESI†) suggest a disorder of a proton over two inversion center related μ3-O atoms (O123) in IrW. The second proton, which should be present in the structure of Na–IrW from the charge balance considerations (only six Na+ countercations were found both in X-ray diffraction and elemental analyses of Na–IrW), is either highly disordered over the terminal oxygens of the IrW polyanions (BVS 1.60–1.67) or, more likely, is associated with an oxygen of a co-crystallized water molecule, i.e. as a H3O+ cation. In the crystal lattice of Na–IrW the Na+ countercations and the crystal water molecules assemble in the parallel infinite zig-zag {Na6(μ2-H2O)10(H2O)t14}n6n+ chains. The IrW polyanions are packed between the chains (see ESI† for details) and form an extended net of hydrogen bonds with the H atoms of the H2O molecules (Fig. S1 and Table S4, ESI†).
The oxidation state of the iridium center (IrIV, 5d5, S = 1/2) was further confirmed by the characteristic EPR signal around g = 2 (Fig. 2). The complexity of the powder spectrum indicates the strong contribution of quadrupole as well as hyperfine coupling (I = 3/2 for 191Ir [natural abundance 37%] and 193Ir [63%]), similar to what has been observed for iridium(IV) impurities displaying a tetragonally distorted octahedral environment in MgO and CaO matrices.18 Unfortunately, low distortion of the iridium coordination sphere in IrW leads to a small split between g values, and the resulting overlap of the various components in our powder X-band spectrum precludes direct determination of the hyperfine and quadrupole coupling constants, while the large number of variables renders an exhaustive search of the parameter space impracticable.
 | ||
| Fig. 2 Powder X-band EPR spectrum of Na–IrW at 110 K. | ||
The ESI mass spectrum obtained by ionization of Na–IrW solution in H2O/MeOH is very complex and exhibits signals which could be assigned for the doubly negatively charged ion pairs {Na4H2[IrIVW6VIO24]}2− and {Na3H3[IrIVW6VIO24]}2− as well as sets of signals corresponding to various decomposition products, e.g. [HWO4]−, [HW2O7]−, NaxH5−x[IrW5O20]2−etc. (see ESI†). While it confirms the existence of the intact polyanions in aqueous solutions the obtained data do not answer the question if the partial decomposition starts to occur already in the solution or only happens during the ionization process. The stability of the polyanions in aqueous solution as well as aqueous acetate media with different pH has been further checked by UV-vis spectroscopy (see ESI† for details). Electrochemistry studies of IrW in aqueous CH3COONa solution at pH 6.1 (see ESI†) produce typical voltammograms (Fig. 3) that show two quasi-reversible redox couples with the corresponding peak potentials E1/2 located at 0.245 V (ΔE = 0.190 V at 100 mV s−1) and 0.700 V (ΔE = 0.200 V) vs. Ag/AgCl reference electrode (0.441 V and 0.896 V vs. SHE, respectively). These waves are attributed to the IrIII/IrIV and IrIV/IrV redox transformations within the polyanion [HIrW6O24]n−. The observed IrIII/IrIV potential value is similar to 0.18 V found for the IrIII/IrIV transition in [PW11O39IrIII(H2O)]4−, and the IrIV/IrV couple is close to the potential of irreversible oxidation of the Ir centers in the same POM 0.75 V (vs. Ag/AgCl).7a The significant shift to lower potentials as compared with the IrIII/IrIV couple observed in the hexachloroiridates (0.669 V vs. Ag/AgCl19) is consistent with the stabilization of higher oxidation states of transition metals generally observed in the oxygen environment provided by polyoxometalates. The relative broadness of the waves could be due to uncompensated solution resistance but this can be ruled out since no change was observed upon increase of the electrolyte concentration. A deviation from reversibility is to be also excluded by the fact that the peak currents for the both processes remain proportional to the square root of the scan rate (Fig. S11, ESI†), indicating diffusion-controlled electrode reactions for scan rates up to 1 V s−1.20 The diffusion coefficient D = 2.62 × 10−6 cm2 s−1 calculated for [HIrIVW6O24]7− according to Randles–Sevcik equation21 compares well to the values reported for other polyoxotungstates.22 The broadness of the redox waves reflects the co-existence of multiple protonation states at pH = 6.1. This is supported by significant narrowing of the waves when the pH decreases to 4.3 (Fig. S12, ESI†), as well as by isolation of the non-protonated form, IrW′, at pH > 6.5 (vide supra). Detailed studies of the protonation phenomena are underway. The redox couples associated with reduction of tungsten(VI) centers in IrW are not accessible, which is typical for WVI centers in POMs possessing two terminal cis-oxo ligands.23
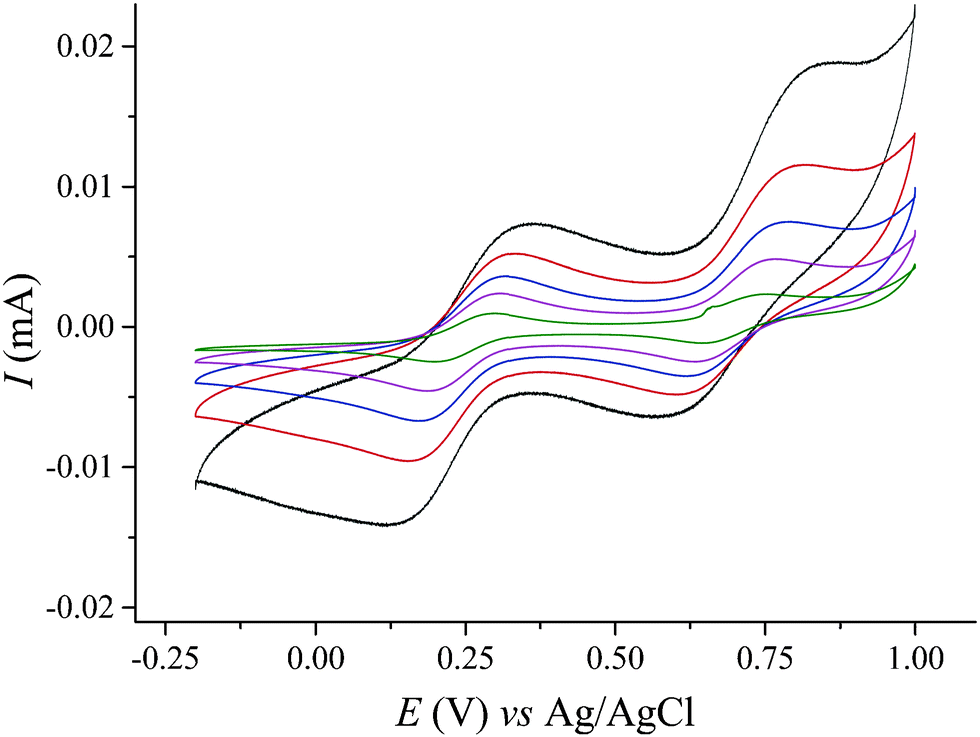 | ||
| Fig. 3 CV of 1.3 mM Na–IrW solution in 2 M NaOAc medium (pH 6.1) at different scan rates: 10 mV s−1 (green), 25 mV s−1 (pink), 50 mV s−1 (blue), 100 mV s−1 (red), 250 mV s−1 (black) vs. Ag/AgCl. | ||
In summary, the identified synthetic path leading to new IrIV-based polyoxotungstate with Anderson–Evans structure, [HIrIVW6O24]7−, comprises the use of in situ-formed IrIV oxo–hydroxo complexes and tungstate ions gradually released in solution. The IrW polyanion is the first example of a structurally characterized IrIV-containing POM as well as the first Ir-based POM with Anderson–Evans structure. The IrIV oxidation state in IrW has been directly confirmed by EPR spectroscopy and the cyclic voltammetry measurements showed the possibility of reversible oxidation and reduction of the IrIV ions in IrW resulting in IrV and IrIII species. Our results suggest existence of a rich IrIV-POM and possibly even IrV-POM chemistry that we are planning to elucidate.
PK appreciates EU financial support (ERC Starting Grant MOLSPINTRON, no. 308051). The work has been supported by RFBR grant No. 13-03-00012 (to MNS). Support by President fellowship to SAA is gratefully acknowledged.
Notes and references
- (a) P. Putaj and F. Lefebvre, Coord. Chem. Rev., 2011, 255, 1642 CrossRef CAS PubMed; (b) N. V. Izarova, M. T. Pope and U. Kortz, Angew. Chem., Int. Ed., 2012, 51, 9492 CrossRef CAS PubMed.
- See for example: (a) M. N. Sokolov, S. A. Adonin, P. A. Abramov, D. A. Mainichev, N. F. Zakharchuk and V. P. Fedin, Chem. Commun., 2012, 48, 6666 RSC; (b) H. J. Lv, Y. V. Geletii, C. C. Zhao, J. W. Vickers, G. B. Zhu, Z. Luo, J. Song, T. Q. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC; (c) A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262 RSC and references therein.
- R. Cao, H. Ma, Y. V. Geletii, K. I. Hardcastle and C. L. Hill, Inorg. Chem., 2009, 48, 5596 CrossRef CAS PubMed.
- See for example: (a) C. N. Kato, A. Shinohara, N. Moriya and K. No-miya, Catal. Commun., 2006, 7, 413 CrossRef CAS PubMed; (b) L. D. Dingwall, C. M. Corcoran, A. F. Lee, L. Olivi, L. M. Lynam and K. Wilson, Chem. Commun., 2008, 53 CAS; (c) R. Neumann and D. Barats, Adv. Synth. Catal., 2010, 352, 293 CrossRef; (d) M. N. Sokolov, S. A. Adonin, E. V. Peresypkina, P. A. Abramov, A. I. Smolentsev, D. I. Potemkin, P. V. Snytnikov and V. P. Fedin, Inorg. Chim. Acta, 2013, 394, 656 CrossRef CAS PubMed and references therein.
- (a) See ref. 1a the references therein and; (b) N. Mizuno, D. K. Lyon and R. G. Finke, J. Catal., 1991, 128, 84 CrossRef CAS; (c) N. Mizuno, H. Weiner and R. G. Finke, J. Mol. Catal. A: Chem., 1996, 114, 15 CrossRef CAS; (d) H. Weiner, A. Trovarelli and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 191, 253 CrossRef CAS; (e) G. Modugno, A. Monney, M. Bonchio, M. Albrecht and M. Carraro, Eur. J. Inorg. Chem., 2014, 2356 CrossRef CAS.
- (a) B. Matt, J. Moussa, L. M. Chamoreau, C. Afonso, A. Proust, H. Amouri and G. Izzet, Organometallics, 2012, 31, 35 CrossRef CAS; (b) B. Matt, X. Xiang, A. L. Kaledin, N. N. Han, J. Moussa, H. Amouri, S. Alves, C. L. Hill, T. Q. Lian, D. G. Musaev, G. Izzet and A. Proust, Chem. Sci., 2013, 4, 1737 RSC; (c) B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504 RSC.
- (a) M. N. Sokolov, S. A. Adonin, D. A. Mainichev, C. Vicent, N. F. Zakharchuk, A. M. Danilenko and V. P. Fedin, Chem. Commun., 2011, 47, 7833 RSC; (b) M. N. Sokolov, S. A. Adonin, P. L. Sinkevich, C. Vicent, D. A. Mainichev and V. P. Fedin, Dalton Trans., 2012, 41, 9889 RSC; (c) M. N. Sokolov, S. A. Adonin, P. L. Sinkevich, C. Vicent, D. A. Mainichev and V. P. Fedin, Z. Anorg. Allg. Chem., 2014, 640, 122 CrossRef CAS.
- (a) G. A. Barbieri, Atti Accad. Lincei, 1914, 23, 334 CAS; (b) Y. Ozawa, Y. Hayashi and K. Isobe, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 637 CrossRef; (c) G. Z. Kaziev, S. H. Quinones, V. K. Bel'skii, V. E. Zavodnik and I. V. Osminkina, Russ. J. Inorg. Chem., 2002, 47, 14 Search PubMed.
- For PtMo6 see for example: (a) U. Lee and Y. Sasaki, Chem. Lett., 1984, 1297 CrossRef CAS; (b) U. Lee, Bull. Korean Chem. Soc., 1988, 9, 256 CAS; (c) K.-M. Park and U. Lee, Chem. Lett., 1989, 1943 CrossRef CAS; (d) U. Lee and Y. Sasaki, Bull. Korean Chem. Soc., 1994, 15, 37 CrossRef CAS; (e) H.-C. Joo, K.-M. Park and U. Lee, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1659 CrossRef; (f) U. Lee, H.-C. Joo and K.-M. Park, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, i25 CAS.
- For PtW6 see: (a) U. Lee, A. Kobayashi and Y. Sasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 817 CrossRef; (b) U. Lee, H. Ichida, A. Kobayashi and Y. Sasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 5 CrossRef; (c) U. Lee and Y. Sasaki, Taehan Hwahakhoe Chi, 1987, 31, 118 CAS; (d) R. E. Marsh and I. Bernal, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 300 Search PubMed; (e) U. Lee, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, 130 Search PubMed; (f) U. Lee, S.-J. Jang, H.-C. Joo and K.-M. Park, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, i116 Search PubMed; (g) U. Lee and H.-C. Joo, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, i33 CAS; (h) U. Lee, H.-C. Joo and K.-M. Park, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, i55 CAS; (i) U. Lee and H.-C. Joo, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, i86 CAS.
- S. Angus-Dunne, R. C. Burns, D. C. Craig and G. A. Lawrance, Z. Anorg. Allg. Chem., 2010, 636, 727 CrossRef CAS.
- (a) J. M. P. Cabral, J. Inorg. Nucl. Chem., 1964, 26, 1657 CrossRef CAS; (b) J. C. Chahg and C. S. Garner, Inorg. Chem., 1965, 4, 209 CrossRef; (c) A. A. El-Awady, E. J. Bounsall and C. S. Garner, Inorg. Chem., 1967, 6, 79 CrossRef CAS; (d) D. A. Fine, Inorg. Chem., 1969, 8, 1014 CrossRef CAS; (e) S. E. Castillo-Blum, D. T. Richens and A. G. Sykes, Inorg. Chem., 1989, 28, 954 CrossRef CAS; (f) D. A. Pankratov, P. N. Komozin and Y. M. Kiselev, Russ. J. Inorg. Chem., 2011, 56, 1794 CrossRef CAS.
- X. Fang, P. Kögerler, Y. Furukawa, M. Speldrich and M. Luban, Angew. Chem., Int. Ed., 2011, 50, 5212 CrossRef CAS PubMed.
- A. Ogawa, H. Yamato, U. Lee, H. Ichida, A. Kobayashi and Y. Sasaki, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1879 CrossRef.
- I. Lindqvist, Acta Crystallogr., 1950, 3, 159 CrossRef CAS.
- M. Sugahara, A. Yoshiasa, A. Yoneda, T. Hashimoto, S. Sakai, M. Okube, A. Nakatsuka and O. Ohtaka, Am. Mineral., 2008, 93, 1148 CrossRef CAS.
- SbW6: (a) U. Lee and Y. Sasaki, Bull. Korean Chem. Soc., 1987, 8, 1 CAS; (b) H. Naruke and T. Yamase, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 597 CrossRef; (c) H. Naruke, N. Kajitani and T. Konya, J. Solid State Chem., 2011, 184, 770 CrossRef CAS PubMed ; MnW6: ; (d) V. S. Sergienko, V. N. Molchanov, M. A. Porai Koshits and E. A. Torchenkova, Koord. Khim., 1979, 5, 936 CAS; (e) A. L. Nolan, R. C. Burns, G. A. Lawrance and D. C. Craig, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 729 Search PubMed; (f) A. V. Oreshkina, G. Z. Kaziev, S. H. Quinones, A. I. Stash and P. A. Shipilova, Russ. J. Coord. Chem., 2011, 37, 845 CrossRef CAS ; TeW6: ; (g) K. J. Schmidt, G. J. D. Schrobilgen and J. F. Sawyer, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1115 CrossRef.
- A. Raizman and J. T. Suss, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 1141 CrossRef CAS.
- P. George, I. H. Hanania and D. H. Irvine, J. Chem. Soc., 1957, 3048 RSC.
- A. M. Bond, Broadening Electrochemical Horizons, Oxford University Press, 2003 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, New York, 1980 Search PubMed.
- See for example: (a) C. Rong and F. C. Anson, Inorg. Chem., 1994, 33, 1064 CrossRef CAS; (b) G. Bernardini, A. G. Wedd, C. Zhao and A. M. Bond, Dalton Trans., 2012, 41, 9944 RSC.
- (a) D. H. Brown and J. A. Mair, J. Chem. Soc., 1958, 2597 RSC; (b) P. Souchay and R. Contant, Bull. Soc. Chim. Fr., 1973, 3287 CAS; (c) S. C. Termes and M. T. Pope, Transition Met. Chem., 1978, 3, 103 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF format; details on synthesis, crystallographic and electrochemical studies; crystal packing in Na–IrW, bond valence sum values; mass spectrometry; IR and UV-Vis spectra. See DOI: 10.1039/c4cc09271f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |