
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbaborane-based alkynylphosphanes and phospholes†
Anika
Kreienbrink
a,
Menyhárt B.
Sárosi
ab,
Robert
Kuhnert
a,
Peter
Wonneberger
a,
Anna I.
Arkhypchuk
c,
Peter
Lönnecke
a,
Sascha
Ott
c and
Evamarie
Hey-Hawkins
*a
aInstitut für Anorganische Chemie, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Fax: (+49)-341-9739319
bFaculty of Chemistry and Chemical Engineering, Babeş-Bolyai University, Arany János 11, RO-400028, Cluj-Napoca, Romania. E-mail: msarosi@chem.ubbcluj.ro; Fax: (+40)264-590818
cDepartment of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: sascha.ott@kemi.uu.se; Fax: (+46)-018-4717340
First published on 12th November 2014
Abstract
The reaction of 1,2-bis(N,N-dimethylaminochloro-phosphanyl)-1,2-dicarba-closo-dodecaborane(12) and lithiated phenylacetylene gives a five-membered dihydrophosphole derivative with an exocyclic phosphanyl group. This unexpected reaction opens new possibilities for the synthesis of carbaborane-containing phosphorus heterocycles. P,P′-alkynylated 1,2-bis(phosphanyl)-1,2-dicarba-closo-dodecaborane(12)s are obtained from alkynylchlorophosphanes and dilithiated carbaborane. This new class of alkynes can be used for future applications in cyclizations and polymerizations.
Alkynylphosphanes [>P–C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–] have attracted considerable attention, not only due to their unique electronic properties that arise from interactions of phosphorus-based orbitals with the π system of the alkyne unit, but also because of their interesting spectroscopic and thermodynamic properties.1 Furthermore, owing to the well-established coupling chemistry of the acetylene moiety, they are appealing building blocks for more elaborate molecular architectures such as phosphapericyclynes and oligomers.2 In addition, the acetylenes can be engaged in subsequent chemistry, and allow, for example, the formation of a variety of hetero- and carbocycles, such as phospholes3a and diphosphinidenecyclobutenes.3b,c
C–] have attracted considerable attention, not only due to their unique electronic properties that arise from interactions of phosphorus-based orbitals with the π system of the alkyne unit, but also because of their interesting spectroscopic and thermodynamic properties.1 Furthermore, owing to the well-established coupling chemistry of the acetylene moiety, they are appealing building blocks for more elaborate molecular architectures such as phosphapericyclynes and oligomers.2 In addition, the acetylenes can be engaged in subsequent chemistry, and allow, for example, the formation of a variety of hetero- and carbocycles, such as phospholes3a and diphosphinidenecyclobutenes.3b,c
The combination of acetylenes with electron-deficient moieties offers the possibility to prepare monodisperse oligomers or polymers with high electron-acceptor capacities. Acetylenic scaffolds with fullerene units4 and cyanoethynylethenes5 are probably the best studied compounds in this context. The combination of alkynylphosphanes with ortho-carbaborane would be an interesting addition to this field, as such structures would merge the electron-withdrawing and -delocalizing properties of an ortho-carbaborane6 with the electronic properties and reactivity of the alkynylphosphane moiety. Such combinations of carbaboranes with alkynylphosphanes would make fascinating building blocks for new polymeric or heterocyclic systems.
There are several synthetic approaches for alkynylphosphanes, the most widely applied of which is the nucleophilic attack of an acetylide at halophosphanes.7 Therefore, in a first attempt to obtain P,P′-alkynylated 1,2-bis(phosphanyl)-1,2-dicarba-closo-dodecaborane(12)s, 1,2-bis(N,N-dimethylaminochlorophosphanyl)-1,2-dicarba-closo-dodecacaborane (1)8 was treated with two equivalents of lithiated phenylacetylene (Scheme 1).9
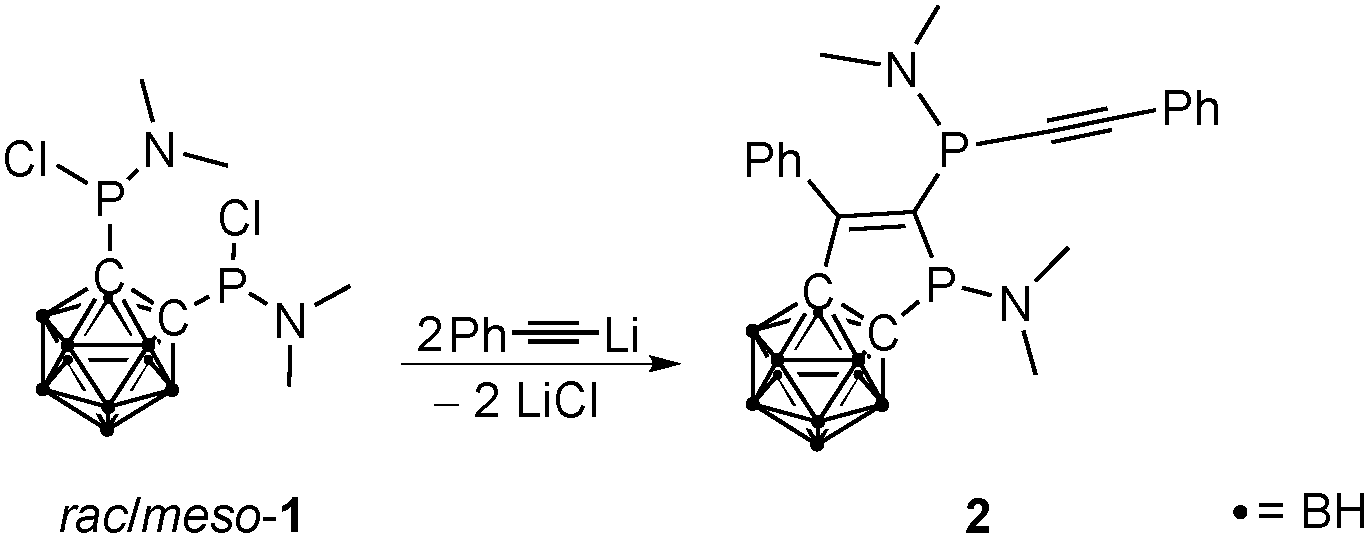 | ||
| Scheme 1 Synthesis of carbaborane-based alkynylphosphane-substituted phosphole 2. | ||
The 31P NMR spectrum of the reaction mixture showed the formation of the proposed product (6a, see Scheme 3) as well as many other products. Attempts to isolate 6a failed; however, yellow crystals of the highly substituted five-membered heterocycle 2 were isolated in 20% yield (Scheme 1). Compound 2 is a highly functionalized dihydrophosphole to which a carbaborane unit is fused via two carbon centers, with a dimethylamino group at the phosphole P atom as well as an exocyclic alkynylphosphanyl substituent.
The 31P{1H} NMR spectrum of 2 (in C6D6) exhibits two doublets at 35.3 (P2, see Fig. 1) and 87.6 ppm (P1, 2JPP = 20.7 Hz). The exocyclic alkynyl group is observed in the 13C{1H} NMR spectrum as two doublets at 78.1 (1JCP = 90.0 Hz) and 108.3 ppm (3JCP = 4.2 Hz), and the signals for C3 and C4 (Fig. 1) of the endocyclic double bond are observed as two doublets of doublets at 149.2 (1JCP = 43.1, 43.3 Hz) and 150.0 ppm (2JCP = 5.1, 5.6 Hz). Furthermore two doublets are observed for the carbon cluster atoms of the heterocycle at 84.6 (1JCP = 20.5 Hz) and 88.0 ppm (2JCP = 6.8 Hz). The 11B{1H} NMR spectrum shows five signals in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:2:4.
:1:2:2:4.
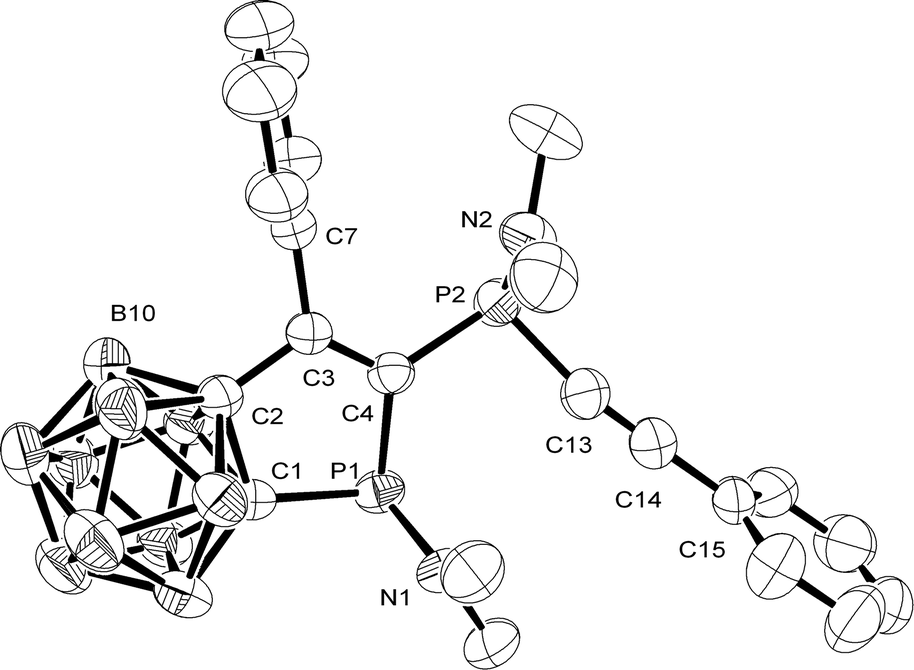 | ||
| Fig. 1 Molecular structure of 2. Selected bond lengths (Å) and angles (°): C1–C2 1.624(3), C2–C3 1.506(3), C3–C4 1.342(2), C1–P1 1.878(2), P1–C4 1.842(2), C4–P2 1.836(2), P2–C13 1.776(3), C13–C14 1.198(3), B–B 1.749–1.785, B–C 1.706–1.733, C1–C2–C3 118.1(2), C2–C3–C4 116.1(2), C3–C4–P1 116.8(2), C4–P1–C1 89.58(9), P1–C1–C2 108.9(1), P1–C4–P2 118.3(1) (hydrogen atoms have been omitted for clarity; ellipsoids are shown at 50% probability). Only the SP, SP isomer is shown. | ||
An X-ray structure analysis was carried out for 2 (Fig. 1).10 Yellow crystals of 2 were obtained at −20 °C from n-hexane. The endocyclic C2–C3, C3–C4, C1–P1, and P1–C4 bond lengths (1.506(3), 1.342(2), 1.878(2), and 1.842(2) Å) in 1-diethylamido-2,3-dihydro-1H-phosphole 2 compare well with the corresponding bonds found in 1H-phosphindole-1-sulfides11 or pentaphenyl-2,3-dihydro-1H-phosphole.12 The C1–C2 bond (1.624(3) Å) is longer than in these heterocycles (1.391(4), 1.523(3) Å),9,10 but similar to those of carbaborane-substituted 1,2-diphosphetanes (1.645(4), 1.631(5) Å).13 The P2–C13 and C13–C14 bond lengths (1.776(3), 1.198(3) Å) are in the same range as found in [8-(dimethylamino-κN)-1-naphthalenyl-κC]-diphenylethynylphosphorus (1.786(9), 1.184(9) Å).14 The phosphorus atoms P1 and P2 in the isolated rac isomer of compound 2 have a pyramidal, and the nitrogen atoms N1 and N2 a trigonal-planar environment.
A mechanism for the formation of 2 was proposed on the basis of computational and 31P NMR spectroscopic studies. The first step of the sequence is the cleavage of one P–C bond in 1 by lithiated phenylacetylene, resulting in intermediates A and 3 (Scheme 2 and ESI†). Such exo-polyhedral C–E bond (E = P, C) cleavage in mono- and disubstituted dicarba-closo-dodecaborane(12)s has been reported earlier.15 The second equivalent of lithiated phenylacetylene reacts with 3 to give the dialkynyl(amido)phosphane 4 (Scheme 2). Subsequently, the Li–Ccluster bond of A inserts into one of the carbon–carbon triple bonds of 4, and 2 is formed after LiCl elimination. The addition of organolithium compounds to carbon–carbon triple bonds has been described in the literature.16 For the investigation of the proposed mechanism (Scheme 2), alkynylphosphanes 3 and 4 were synthesized independently and fully characterized. On the basis of their 31P NMR shifts, both species could be identified in the reaction solution, and the formation of 4 by reaction of 1 and lithiated phenylacetylene was thus verified. Compound 4 polymerizes very fast above 0 °C and seems to be responsible for the dark color of the reaction solution. Polymerization of 4 is presumably also responsible for the moderate yield of 2.
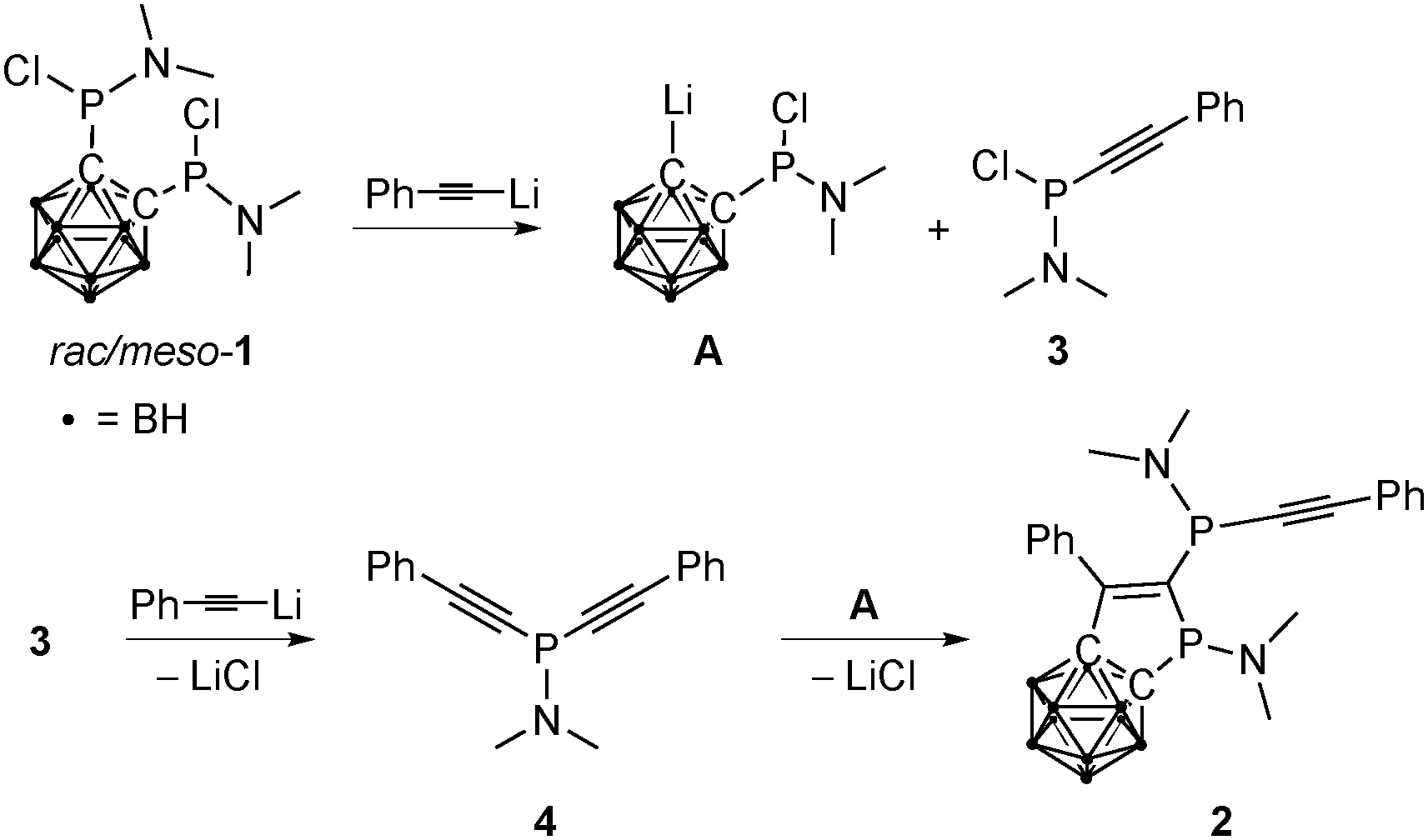 | ||
| Scheme 2 Proposed mechanism for the formation of 2 and the observed byproduct 4. | ||
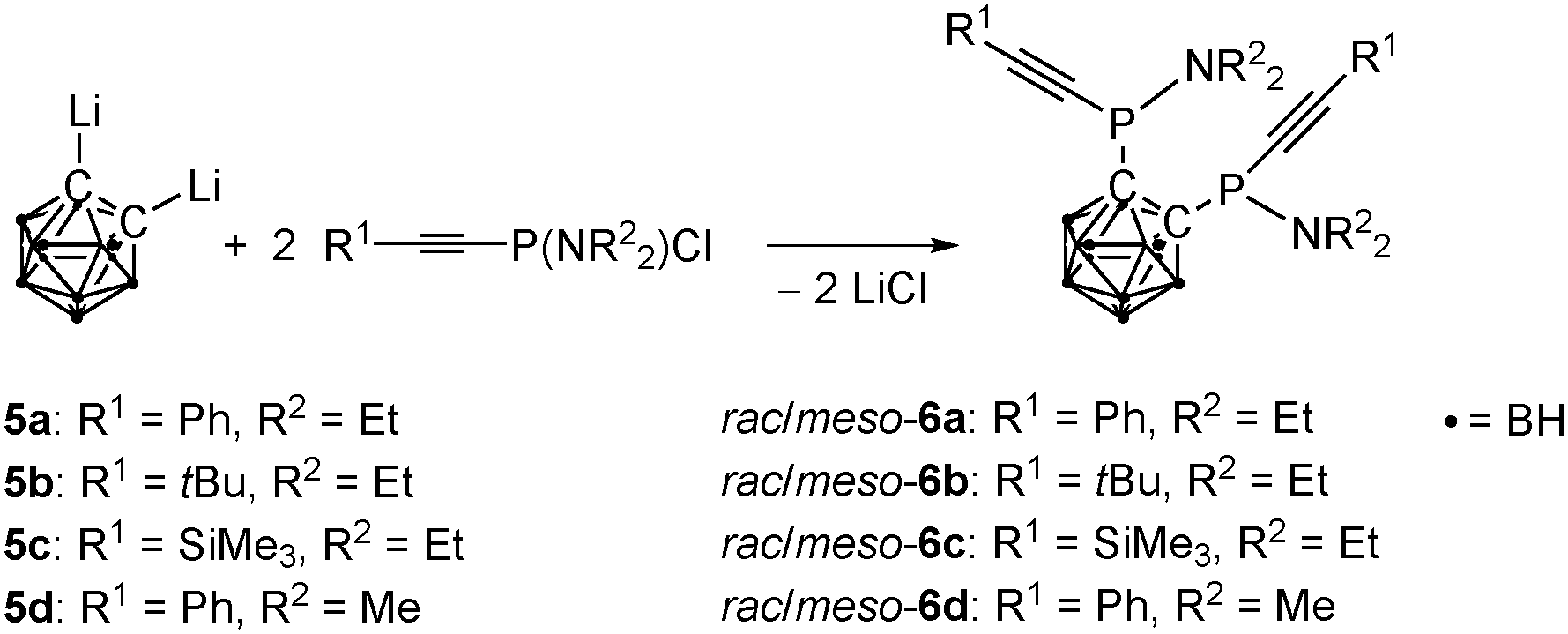 | ||
| Scheme 3 Synthesis of P,P′-alkynylated 1,2-bis(N,N-dialkylaminophosphanyl)-1,2-dicarba-closo-dodecaborane(12)s 6a–6d. | ||
While the unexpected reactivity of 1 towards lithium acetylides gives rise to the unusual phosphole 2, the initially desired bis(alkynylphosphanyl)carbaboranes remain elusive by this synthetic strategy. They can, however, be prepared in a different procedure from alkynylchlorophosphanes 5a–5d and lithiated ortho-carbaborane (Scheme 3) in a twofold nucleophilic substitution reaction. The bis(alkynylphosphanyl)carbaboranes 6a–6d are stable under ambient conditions and were obtained in moderate to good yields.
Compounds 1, 2, and 6d were investigated by cyclic voltammetry (CV) to elucidate the electronic communication between the different P-substituents in the three compounds and the carbaborane unit. Phosphole 2 features one irreversible oxidation at Ep,a = 0.85 V vs. Fc+/0, and thus at a potential that is very similar to that found for 6d (Ep,a = 1.00 V). It is thus tempting to assign this process to an oxidation of mainly the P(NMe2)(CCR) substituent, which is present in both compounds. Supporting this assignment is the notion that the oxidation potential of the P(NMe2)(CCR) group in 2 can be expected to be somewhat lower than that of 6d due to the presence of the conjugated endocyclic double bond in the former. Neither CV of the two compounds shows a reductive feature within the solvent window down to −2.3 V vs. Fc+/0, in analogy to the reported reduction potential of the unsubstituted, parent dicarba-closo-dodecaborane(12).17 The only cathodic process that is observed for the three compounds is an irreversible reduction of rac/meso-1 at Ep,c = 2.20 V (see ESI†) which is assigned to the electron-deficient phosphane units, rather than to processes that are localized on the carbaborane core.17
In conclusion, the reaction of 1,2-bis(N,N-dimethyl-aminochlorophosphanyl)-1,2-dicarba-closo-dodecaborane(12) (1) and lithiated phenylacetylene gave an unusual five-membered heterocycle 2 containing an exocyclic alkynylphosphane unit and a dimethylamino group at the phosphole P atom. The high degree of functionalization should offer multiple possibilities for the synthesis of future carbaborane-containing phosphorus heterocycles. 1,2-Bis(phosphanyl)-1,2-dicarba-closo-dodecaborane(12)s 6a–6d are accessible from the reaction of alkynylchlorophosphanes and dilithiated ortho-carbaborane. Comparative cyclic voltammetric studies show that the HOMO of 2 is localized mainly on the P(NMe2)(CCR) group. The dihydrophosphole unit is thus rather inert to oxidation, an effect that could point towards strong electronic coupling to the electron-deficient carbaborane that is fused onto the phosphole ring. Further studies on the reactivity of this new class of alkynylphosphanes are underway.
Financial support of the Konrad-Adenauer Stiftung (doctoral grant for A.K.), the Graduate School Leipzig School of Natural Sciences – Building with Molecules and Nano-objects (BuildMoNa), COST Action CM1302 SIPs and COST Action CM0802 PhoSciNet (STSM for A.K. in Uppsala), the German Academic Exchange Service (DAAD) (short-term research stay at Universität Leipzig for M.B.S.), and the Swedish Research Council is gratefully acknowledged. We thank Chemetall GmbH for a generous donation of butyllithium.
Notes and references
- (a) L. T. Scott and M. Unno, J. Am. Chem. Soc., 1990, 112, 7823–7825 CrossRef CAS; (b) D. E. Cabelli, A. H. Cowley and M. J. S. Dewar, J. Am. Chem. Soc., 1981, 103, 3290–3292 CrossRef CAS; (c) I. K. Ahmad, H. Ozeki and S. Sato, J. Chem. Phys., 1997, 107, 1301–1307 CrossRef CAS; (d) P. Bharathi and M. Periasamy, Organometallics, 2000, 19, 5511–5513 CrossRef CAS.
- (a) S. G. A. van Assema, P. B. Kraikivskii, S. N. Zelinskii, V. V. Saraev, G. Bas de Jong, F. J. J. de Kanter, M. Schakel, J. C. Slootweg and K. Lammertsma, J. Organomet. Chem., 2007, 692, 2314–2323 CrossRef CAS; (b) R. Shiozawa and K. Sakamoto, Chem. Lett., 2003, 32, 1024–1025 CrossRef CAS; (c) G. Märkl, T. Zollitsch, P. Kreitmeier, M. Prinzhorn, S. Reitinger and E. Eibler, Chem. – Eur. J., 2000, 6, 3806–3820 CrossRef; (d) S. G. A. van Assema, G. B. de Jong, A. W. Ehlers, F. J. J. de Kanter, M. Schakel, A. L. Spek, M. Lutz and K. Lammertsma, Eur. J. Org. Chem., 2007, 2405–2412 CrossRef CAS; (e) L. T. Scott and M. J. Cooney, in Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, Wiley-VCH, Weinheim, 2007, pp. 321–351 Search PubMed.
- (a) M. Yoshifuji, Y. Ichikawa, N. Yamada and K. Toyota, Chem. Commun., 1998, 27–28 RSC; (b) M. Yoshifuji, Y. Ichikawa, K. Toyota, E. Kasashima and Y. Okamoto, Chem. Lett., 1997, 87–88 CrossRef; (c) J. Möbus, Q. Bonnin, K. Ueda, R. Fröhlich, K. Itami, G. Kehr and G. Erker, Angew. Chem., 2012, 124, 1990–1993 ( Angew. Chem., Int. Ed. , 2012 , 51 , 1954–1957 ) CrossRef.
- L. Isaacs, P. Seiler and F. Diederich, Angew. Chem., 1995, 107, 1636–1639 ( Angew. Chem., Int. Ed. Engl. , 1995 , 34 , 1466–1469 ) CrossRef.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed.
- (a) R. N. Grimes, Carboranes, Academic Press, New York, 1970 Search PubMed; (b) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (c) H. Beal, in Boron Hydride Chemistry, ed. E. Muetterties, Academic Press, New York, 1975 Search PubMed.
- E. Bernoud, R. Veillard, C. Alayrac and A.-C. Gaumont, Molecules, 2012, 17, 14573–14587 CrossRef CAS PubMed.
- S. Stadlbauer, R. Frank, I. Maulana, P. Lönnecke, B. Kirchner and E. Hey-Hawkins, Inorg. Chem., 2009, 48, 6072–6082 CrossRef CAS PubMed.
- Y. Wang, W.-X. Zhang, Z. Wang and Z. Xi, Angew. Chem., Int. Ed., 2011, 50, 8122–8126 CrossRef CAS PubMed.
- Structural data of 2: C22H32B10P2N2, M = 494.54, monoclinic, space group P21/c, a = 14.5505(3) Å, b = 15.3833(3) Å, c = 12.6324(3) Å, β = 92.549(2)°, V = 2824.8(1) Å3, Z = 4, ρcalcd = 1.163 Mg m−3, μ = 0.170 mm−1, θmax. = 26.37°, R(all data) = 0.0830, wR(all data) = 0.1411, T = 293(2) K, 5771 independent reflections, 409 parameters, no restraints, maximal residual electron density 0.400 e Å−3. The structure determination was carried out at room temperature because the sample was twinned at lower temperatures, e.g. 130 K, structure solution with SIR92 ( A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed ). Anisotropic refinement of all non-hydrogen atoms with SHELXL-97 ( G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed ). With the exception of the methyl groups, all H atoms were located on difference Fourier maps calculated at the final stage of the structure refinement. CCDC 976541 (2).
- Y. Miquel, A. Igau, B. Donnadieu, J.-P. Majoral, L. Dupuis, N. Pirio and P. Meunier, Chem. Commun., 1997, 279–280 RSC.
- K. Issberner, E. Niecke, E. Wittchow, K. H. Dötz and M. Nieger, Organometallics, 1997, 16, 2370–2376 CrossRef CAS.
- A. Kreienbrink, M. B. Sárosi, E. G. Rys, P. Lönnecke and E. Hey-Hawkins, Angew. Chem., 2011, 123, 4798–4800 ( Angew. Chem., Int. Ed. , 2011 , 50 , 4701–4703 ) CrossRef.
- S. B. Bushuk, F. H. Carré, D. M. H. Guy, W. E. Douglas, Y. A. Kalvinkovskya, L. G. Klapshina, A. N. Rubinov, A. P. Stupak and B. A. Bushuk, Polyhedron, 2004, 23, 2615–2623 CrossRef CAS.
- (a) A. V. Kasantsev, M. N. Zhubekova and L. I. Zakharkin, Zh. Obshch. Khim., 1971, 41, 2027–2033 Search PubMed; (b) A. Kreienbrink, P. Lönnecke, M. Findeisen and E. Hey-Hawkins, Chem. Commun., 2012, 48, 9385–9387 RSC; (c) L. I. Zakharkin and A. I. L'vov, J. Organomet. Chem., 1966, 5, 313–319 CrossRef CAS; (d) W. Neumann, M. Hiller, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2014, 43, 4935–4937 RSC.
- (a) S. C. Cohen, A. J. Tomlinson, M. R. Wiles and A. G. Massey, J. Organomet. Chem., 1968, 11, 385–392 CrossRef CAS; (b) A. Krebs, W. Born, B. Kaletta, W.-U. Nickel and W. Rueger, Tetrahedron Lett., 1983, 24, 4821–4824 CrossRef CAS; (c) A. I. Meyers and P. D. Pansegrau, Tetrahedron Lett., 1983, 24, 4935–4938 CrossRef CAS; (d) W. Bauer, M. Feigel, G. Mueller and P. von Rague Schleyer, J. Am. Chem. Soc., 1988, 110, 6033–6046 CrossRef CAS PubMed.
- K. Hosoi, S. Inagi, T. Kubo and T. Fuchigami, Chem. Commun., 2011, 47, 8632–8634 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures and data, details of computational studies. CCDC 976541. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08449g |
| This journal is © The Royal Society of Chemistry 2015 |