
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect reductive coupling of secondary amides: chemoselective formation of vicinal diamines and vicinal amino alcohols†‡
Pei-Qiang
Huang
*,
Qi-Wei
Lang
,
Ai-E
Wang
and
Jian-Feng
Zheng
Department of Chemistry and Fujian Provincial Key Laboratory for Chemical Biology, Collaborative Innovation Centre of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn; Tel: +86-592-2182240
First published on 25th November 2014
Abstract
We report the first one-pot reductive homocoupling reaction of secondary amides and cross-coupling reaction of secondary amides with ketones to give secondary vicinal diamines and amino alcohols. This method relies on the direct generation of α-amino carbon radicals from secondary amides by activation with trifluoromethanesulfonic anhydride, partial reduction with triethylsilane and samarium diiodide-mediated single-electron transfer. The reactions were run under mild conditions and tolerated several functional groups.
Amides are easily available1 and highly stable carbonyl compounds. These features make them excellent starting materials and synthetic intermediates for a number of useful transformations,2 including amide group-directed C–H functionalization.3 After these transformations, it is often necessary to convert the amide group, which is at a high level of oxidation state, to a functionality at a lower oxidation state. In this regard, chemoselective transformations of amides into amines and ketones via C–C bond formation, as a class of redox economical reactions,4 have attracted considerable attention.5–9 Although significant progress has been made recently after the discovery of the well-known Kulinkovich–de Meijere reaction,5 most of the methods involve the chemoselective or controlled generation of electrophilic intermediates such as iminoyl triflates, iminium ions, or nitrilium ions, followed by the capture of these reactive intermediates using π-nucleophiles7 or organometallic reagents.8,9 If reactive species other than electrophilic iminium ions were generated directly from amides, many other subsequent reactions other than nucleophilic addition could be anticipated. Along these lines, few examples involving the direct generation of α-amino carbenes10 or α-amino carbon radicals11 from tertiary amides have been reported. The direct generation of α-amino radicals from the more challenging secondary amides for C–C bond formation has not been reported probably because of the presence of a free N–H group in these amides.
As part of our goal of developing new C–C bond formation reactions that employ stable amides as substrates,8,9b,c we now report the generation of α-amino carbon radicals from secondary amides and the application of these reactive species in the development of the first one-pot synthesis of vicinal diamines and vicinal amino alcohols from secondary amides. Vicinal diamines and vicinal amino alcohols are privileged scaffolds widely present in synthetically useful chiral ligands, auxiliaries and bioactive compounds.12 Although many methods have been developed for the synthesis of these structural motifs,12b,13,14 it is still highly desirable to develop methods that use stable and easily available starting materials.
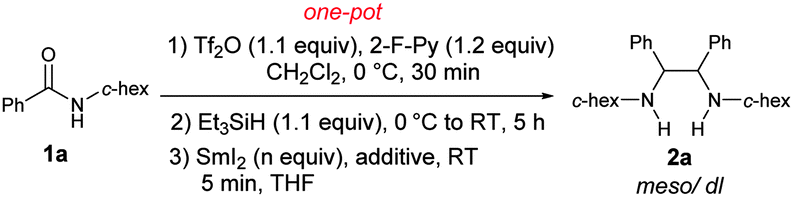
Our investigation was initiated by examining the reductive homocoupling of benzamide 1a (Table 1). 1a was treated sequentially with trifluoromethanesulfonic anhydride (Tf2O)15 (1.1 equiv.) and 2-F-Py9,16 (1.2 equiv.) at 0 °C for 30 min, Et3SiH17,18 at 0 °C to RT for 5 h, and SmI2 (ref. 19) (3.0 equiv.) for 5 min. To our delight, the desired diamine 2a was obtained in 86% yield with a meso/dl ratio of 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46 (Table 1, entry 1). No N-benzyl-cyclohexylamine as a result of unimolecular reduction was observed. In the presence of a catalytic amount of NiI219d (1% mol), a slightly improved yield of 88% was obtained. However increasing the amount of SmI2 to 3.5 equiv. produced no additional improvement in the yield (entry 3), lowering its quantity to 2.2 equiv. was shown to be detrimental (entry 4). On the other hand, replacing NiI2 with other additives, including t-BuOH, HMPA or Yb(OTf)3, failed to improve the diastereoselectivity (Table 1, entries 5–7). Hence, 3.0 equiv. of SmI2 and 1 mol% NiI2 were determined to be optimal for the reductive coupling reaction.20
:46 (Table 1, entry 1). No N-benzyl-cyclohexylamine as a result of unimolecular reduction was observed. In the presence of a catalytic amount of NiI219d (1% mol), a slightly improved yield of 88% was obtained. However increasing the amount of SmI2 to 3.5 equiv. produced no additional improvement in the yield (entry 3), lowering its quantity to 2.2 equiv. was shown to be detrimental (entry 4). On the other hand, replacing NiI2 with other additives, including t-BuOH, HMPA or Yb(OTf)3, failed to improve the diastereoselectivity (Table 1, entries 5–7). Hence, 3.0 equiv. of SmI2 and 1 mol% NiI2 were determined to be optimal for the reductive coupling reaction.20
|
|
|||
|---|---|---|---|
| Entry | Additive | SmI2 (equiv.) | % yielda (meso:dl)b |
| a Isolated yields. b Determined by 1H NMR analysis of the benzylic protons of the mixture obtained from a preliminary column chromatographic separation. | |||
| 1 | None | 3.0 | 86 (54:46) |
| 2 | NiI2 (1 mol%) | 3.0 |
88 (53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :47) :47)
|
| 3 | NiI2 (1 mol%) | 3.5 | 88 (53:47) |
| 4 | NiI2 (1 mol%) | 2.2 | 74 (53:47) |
| 5 | t BuOH (2 equiv.) | 3.0 | 86 (54:46) |
| 6 | HMPA (2 equiv.) | 3.0 | 89 (54:46) |
| 7 | Yb(OTf)3 (1 equiv.) | 3.0 | 88 (55:45) |
With the optimized reaction conditions in hand, the scope of the one-pot reductive homocoupling reaction was explored by varying the substituents on the phenyl ring and the amidyl nitrogen (Table 2). Electron-donating groups (entries 2 and 3), halogens (entries 4–6), and electron-withdrawing groups (entries 4–7) were shown to be well tolerated on the phenyl ring. A cyano group and an ester, often considered to be sensitive and labile under reductive conditions, were found to be compatible with the current process, furnishing the desired diamine products in moderate yields (entries 8–9). Meanwhile, amides bearing primary (entry 10) and secondary (entries 1–9 and 11–15) alkyl substituents were suitable substrates. The introduction of a sterically hindered t-Bu group (entry 16) or a phenyl ring (entry 17), however, completely abolished product formation. Lastly, non-benzamides such as thiophenyl amide 1r (entry 18) and cyclohexyl amide 1s (entry 19) also underwent reductive coupling to produce 2r in 65% yield and 2s in 54% yield, respectively. However, the homocoupling of other aliphatic amides gave low yields.
|
|
|||
|---|---|---|---|
| Entry | Substrate (R1, R2) | Producta (%) |
meso:dlb |
|
a Isolated yields.
b
meso:dl ratios, determined by 1H NMR analysis of the benzylic protons of the mixture obtained from a preliminary column chromatographic separation.
c Determined by 1H NMR analysis of the aromatic protons.
d The reaction was treated with Tf2O at −78 °C for 10 min and then at 0 °C for another 10 min.
e
N-benzyl-1-cyclohexylmethanamine was obtained in 20% yield.
|
|||
| 1 | 1a (Ph, c-hex) | 2a (88) | 53:47 |
| 2 | 1b (4-MeC6H4, c-hex) | 2b (90) | 55:45 |
| 3 | 1c (4-MeOC6H4, c-hex) | 2c (80) | 61:39c |
| 4 | 1d (4-FC6H4, c-hex) | 2d (86) | 56:44 |
| 5 | 1e (4-ClC6H4, c-hex) | 2e (88) | 57:43 |
| 6 | 1f (4-BrC6H4, c-hex) | 2f (79) | 54:46 |
| 7 | 1g (4-CF3C6H4, c-hex) | 2g (81) | 54:46 |
| 8 | 1h (4-NCC6H4, i-Pr) | 2h (58) | 55:45 |
| 9 | 1i (4-MeO2CC6H4, i-Pr) | 2i (41) | 54:46 |
| 10 | 1j (Ph, n-Bu) | 2j (71) | 70:30 |
| 11 | 1k (Ph, i-Bu) | 2k (83) | 60:40 |
| 12d | 1l (Ph, c-propyl) | 2l (66) | 78:22 |
| 13 | 1m (Ph, c-pentyl) | 2m (88) | 55:45 |
| 14 | 1n (Ph, i-Pr) | 2n (93) | 58:42 |
| 15 | 1o (4-MeC6H4, i-Pr) | 2o (94) | 59:41 |
| 16 | 1p (Ph, t-Bu) | 2p (0) | — |
| 17 | 1q (Ph, Ph) | 2q (0) | — |
| 18 | 1r (2-thienyl, c-hex) | 2r (65) | 60:40 |
| 19e | 1s (c-hex, Bn) | 2s (54) | 52:48 |
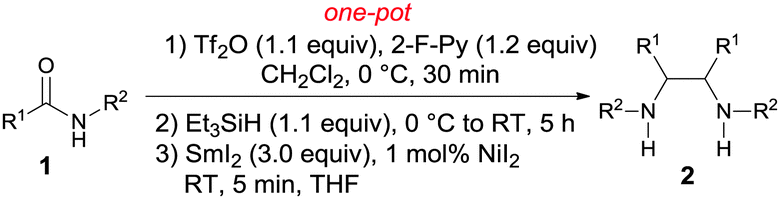
Our success with the reductive homocoupling prompted us to turn our attention to the more challenging cross-coupling reactions of secondary amides with ketones. Under the reaction conditions established for the homocoupling reaction, 1n was subjected to amide activation and controlled reduction before mixing with 2.0 equiv. of cyclopentanone (Table 3). Under these conditions, the desired cross-coupling product 3a was obtained in 27% yield, along with the homocoupling product 2n in 63% yield (entry 1). Attempts to increase the yield of 3a by varying the amount of SmI2 or ketone used, or by altering the reaction temperature, were unsuccessful.
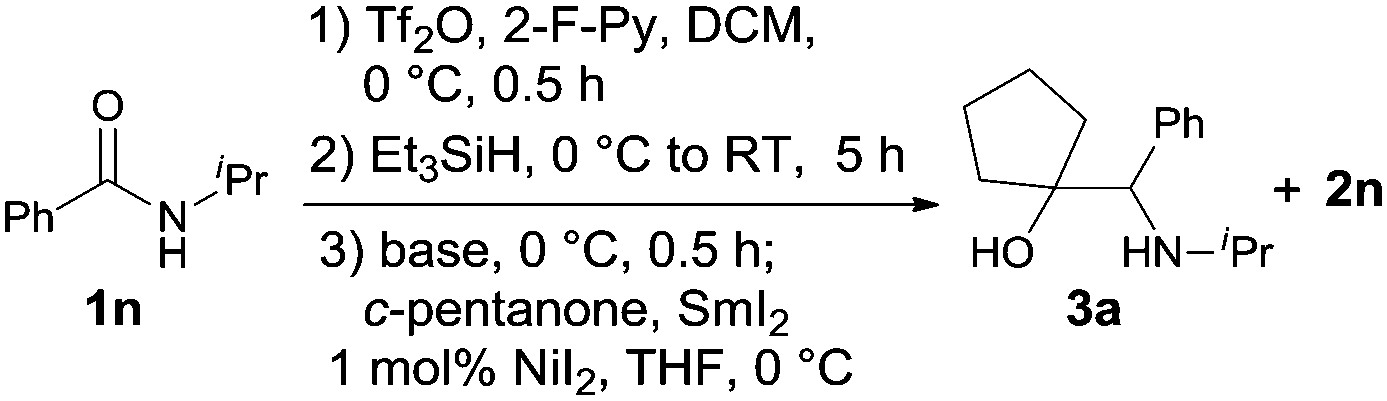
Gratifyingly, the addition of 1.5 equiv. of Et3N to the reaction before the introduction of cyclopentanone dramatically increased the yield of 3a to 66%, and concomitantly, limited the formation of 2n to 25% yield (Table 3, entry 2). Increasing the amount of ketone used to 3.0 equiv. (entry 3) further tilted the reaction toward cross coupling (76% of 3a and 8% of 2n). Using an even higher amount (6.0 equiv.), however, did not result in an additional increase in yield (entry 4). Conversely, changing the amount of SmI2 to above or below 2.5 equiv. invariably lowered the amino alcohol formation (entries 5–7). Hence, the best conditions for the cross-coupling reaction consisted of the use of 1.5 equiv. of Et3N, 3.0 equiv. of ketone, and 2.5 equiv. of SmI2.
The scope of the cross-coupling reaction was investigated (Table 4). Cyclic ketones with ring sizes ranging from 4 to 8 all reacted efficiently affording good yields (products 3a–3e, 60–76% yields). Acyclic ketones were also suitable coupling partners; however, 8.0 equiv. of the ketone were needed to ensure a good yield of the amino alcohol. For the amide part, both secondary and primary alkyl N-substituents were tolerated with the latter being inferior. The reaction of cyclopentanone with the benzamides bearing either electron-donating or electron-withdrawing groups on the benzene ring afforded the expected vic-amino alcohols 3m–3p in 53–79% yields.
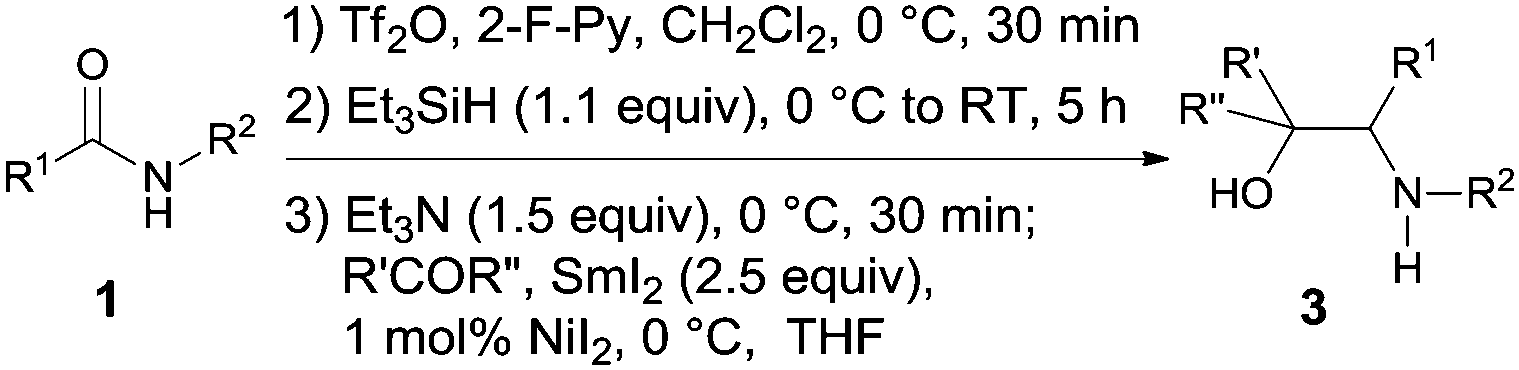
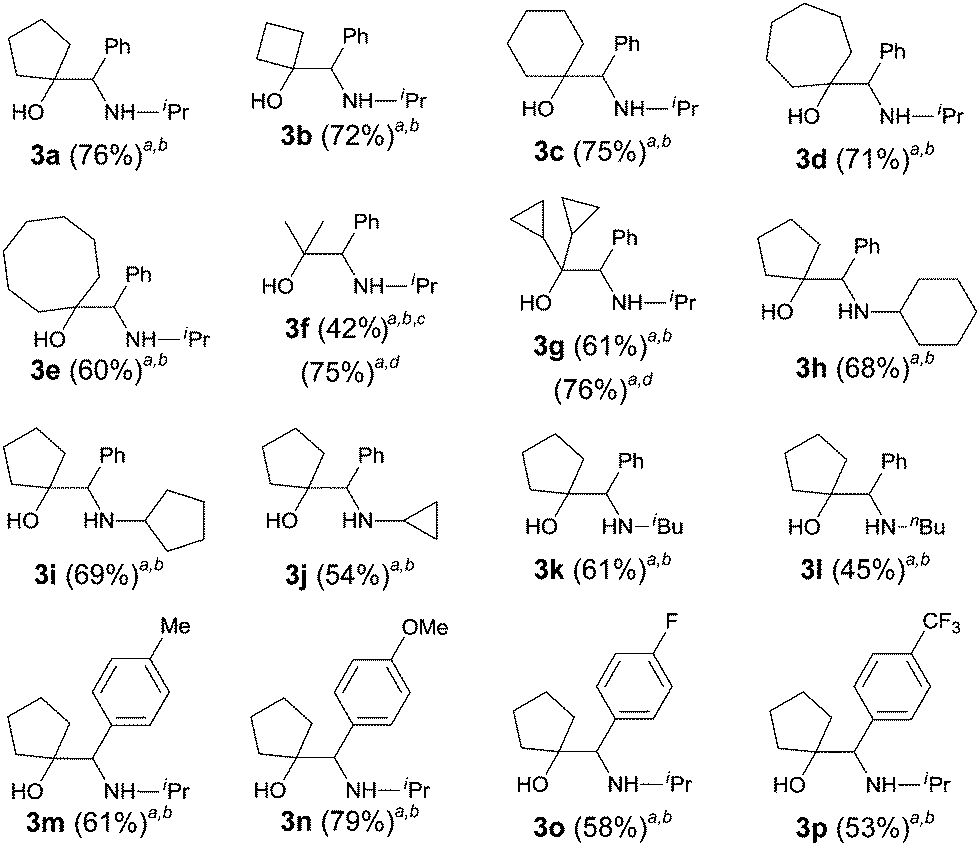
A plausible reaction mechanism for the coupling reactions is depicted in Scheme 1. The treatment of secondary amide 1 with Tf2O yielded reactive nitrilium ion A,9b–d,16 which is then partially reduced with triethylsilane17,18 to give the protonated imine B. The highly reactive protonated imine B is then subjected to the SmI2-mediated homocoupling reaction to give vicinal diamine 2.
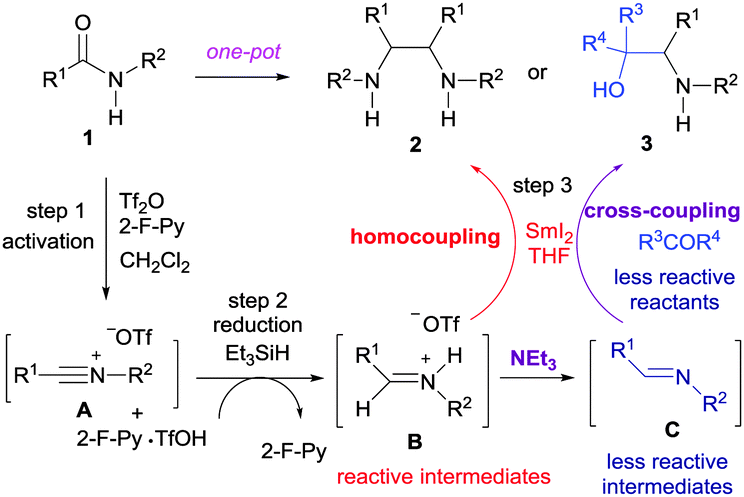 | ||
| Scheme 1 A plausible reaction mechanism for the coupling reactions. | ||
The predominance of 2 in the product profile of the reaction of 1 with cyclopentenone can also be attributed to the predisposition of the highly reactive intermediate B to undergo the reductive homocoupling reaction. This undesired homocoupling reaction is suppressed by triethylamine to convert B to its less reactive neutral form imine C. Having comparable reactivity, the SmI2-mediated cross-coupling reaction between imine C and a ketone is favoured to give vic-amino alcohol 3.
In summary, we have demonstrated for the first time that secondary vicinal diamines and vicinal amino alcohols can be synthesized efficiently from secondary amides through reductive coupling reactions. The method relied on the generation of α-amino carbon radicals from secondary amides through amide activation, controlled reduction, and SmI2-mediated single-electron transfer. The homocoupling of the α-amino radical and cross coupling with ketones afforded a variety of vicinal diamines and vicinal amino alcohols, respectively. The more challenging cross-coupling reaction required a careful control of the reactivity of the imine intermediate. Studies that employ other radical acceptors for the α-amino radicals to generate functionalized amines are currently underway and will be reported in due course.
The authors are grateful for financial support from the NSF of China (21332007 and 21472153) and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education, China.
Notes and references
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714 RSC.
- Secondary amides: (a) L. Huang, Q. Wang, W. Wu and H. Jiang, J. Org. Chem., 2014, 79, 7734 CrossRef CAS PubMed; (b) N. Armanino, M. Lafrance and E. M. Carreira, Org. Lett., 2014, 16, 572 CrossRef CAS PubMed; (c) L. Song, K. Liu and C. Li, Org. Lett., 2011, 13, 3434 CrossRef CAS PubMed ; tertiary amides: ; (d) B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718 CrossRef CAS PubMed; (e) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462 CrossRef CAS PubMed; (f) B. Peng, D. Geerdink and N. Maulide, J. Am. Chem. Soc., 2013, 135, 14968 CrossRef CAS PubMed; (g) V. Valerio, D. Petkova, C. Madelaine and N. Maulide, Chem. – Eur. J., 2013, 19, 2606 CrossRef CAS PubMed.
- (a) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477 CrossRef CAS PubMed; (b) L.-S. Zhang, K. Chen, G. Chen, B.-J. Li, S. Luo, Q.-Y. Guo, J.-B. Wei and Z.-J. Shi, Org. Lett., 2013, 15, 10 CrossRef CAS PubMed; (c) Q. Chen, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 428 CrossRef CAS PubMed; (d) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS PubMed.
- N. Z. Burn, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef PubMed.
- (a) V. Chaplinski and A. de Meijere, Angew. Chem., Int. Ed. Engl., 1996, 35, 413 CrossRef CAS; (b) V. Chaplinski, H. Winsel, M. Kordes and A. de Meijere, Synlett, 1997, 111 CrossRef CAS PubMed; (c) J. Lee and J. K. Cha, J. Org. Chem., 1997, 62, 1584 CrossRef CAS; (d) N. Ouhamou and Y. Six, Org. Biomol. Chem., 2003, 1, 3007 RSC.
- For reviews, see: (a) O. G. Kulinkovich and A. de Meijere, Chem. Rev., 2000, 100, 2789 CrossRef CAS PubMed; (b) V. Pace and W. Holzer, Aust. J. Chem., 2013, 66, 507 CAS; (c) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147 RSC.
- (a) G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, J. Org. Chem., 2006, 71, 704 CrossRef PubMed; (b) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592 CrossRef CAS PubMed; (c) H.-B. Zhou, G.-S. Liu and Z.-J. Yao, J. Org. Chem., 2007, 72, 6270 CrossRef CAS PubMed; (d) S.-L. Cui, J. Wang and Y.-G. Wang, J. Am. Chem. Soc., 2008, 130, 13526 CrossRef CAS PubMed.
- (a) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037 CrossRef CAS PubMed; (b) Y. Oda, T. Sato and N. Chida, Org. Lett., 2012, 14, 950 CrossRef CAS PubMed; (c) S.-Y. Huang, Z. Chang, S.-C. Tuo, L.-H. Gao, A.-E Wang and P.-Q. Huang, Chem. Commun., 2013, 49, 7088 RSC; (d) P.-Q. Huang, W. Ou, K.-J. Xiao and A.-E Wang, Chem. Commun., 2014, 50, 8761 RSC.
- (a) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228 CrossRef CAS PubMed; (b) K.-J. Xiao, A.-E Wang, Y.-H. Huang and P.-Q. Huang, Asian J. Org. Chem., 2012, 1, 130 CrossRef CAS; (c) K.-J. Xiao, Y.-H. Huang and P.-Q. Huang, Acta Chim. Sin., 2012, 70, 1917 CrossRef CAS; (d) K.-J. Xiao, A.-E Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314 CrossRef CAS PubMed.
- (a) A. Ogawa, N. Takami, M. Sekiguchi, I. Ryu, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1992, 114, 8729 CrossRef CAS; (b) A. Ogawa, N. Takami, T. Nanke, S. Ohya and T. Hirao, Tetrahedron, 1997, 53, 12895 CrossRef CAS; (c) X.-L. Xu and Y.-M. Zhang, Tetrahedron, 2002, 58, 503 CrossRef CAS.
- (a) S. Kashimura, M. Ishifune, Y. Murai, H. Murase, M. Shimomura and T. Shono, Tetrahedron Lett., 1998, 39, 6199 CrossRef CAS; (b) C. E. McDonald, A. M. Galka, A. I. Green, J. M. Keane, J. E. Kowalchick, C. M. Micklitsch and D. D. Wisnoski, Tetrahedron Lett., 2001, 42, 163 CrossRef CAS; (c) K. Selvakumar and J. F. Harrod, Angew. Chem., Int. Ed., 2001, 40, 2129 CrossRef CAS; (d) K. Rangareddy, K. Selvakumar and J. F. Harrod, J. Org. Chem., 2004, 69, 6843 CrossRef CAS PubMed; (e) M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2014, 136, 2268 CrossRef CAS PubMed (chemoselective reduction of primary, secondary, and tertiary amides to alcohols).
- (a) S. R. S. S. Kotti, C. Timmons and G. Li, Chem. Biol. Drug Des., 2006, 67, 101 CrossRef CAS PubMed; (b) Y. L. Bennani and S. Hanessian, Chem. Rev., 1997, 97, 3161 CrossRef CAS PubMed.
- D. Lucet, T. L. Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580 CrossRef CAS.
- (a) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS; (b) G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831 CrossRef CAS PubMed; (c) O. N. Burchak and S. Py, Tetrahedron, 2009, 65, 7333 CrossRef CAS PubMed.
- For a recent review on the chemistry of Tf2O, see: I. L. Baraznenok, V. G. Nenajdenko and E. S. Balenkova, Tetrahedron, 2000, 56, 3077 CrossRef CAS ; For recent progress, see: ref. 2d–g, 6b, 7–9, 16, 17, and references cited therein.
- J. W. Medley and M. Movassaghi, J. Org. Chem., 2009, 74, 1341 CrossRef CAS PubMed.
- G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817 CrossRef CAS PubMed.
- (a) D. N. Kursanov, Z. N. Parnes and N. M. Loim, Synthesis, 1974, 633 CrossRef CAS; (b) P.-Q. Huang, Synlett, 2006, 1133 CrossRef CAS PubMed.
- For SmI2-mediated coupling reactions, see: (a) S. F. Martin, C.-P. Yang, W. L. Laswell and H. Rüeger, Tetrahedron Lett., 1988, 29, 6685 CrossRef CAS; (b) E. J. Enholm, D. C. Forbes and D. P. Holub, Synth. Commun., 1990, 20, 981 CrossRef CAS; (c) T. Imamoto and S. Nishimura, Chem. Lett., 1990, 1141 CrossRef CAS; (d) F. Machrouhi and J.-L. Namy, Tetrahedron Lett., 1999, 40, 1315 CrossRef CAS; (e) M. Kim, B. W. Knettle, A. Dahlén, G. Hilmersson and R. A. II Flowers, Tetrahedron, 2003, 59, 10397 CrossRef CAS PubMed; (f) Y.-W. Zhong, Y.-Z. Dong, K. Fang, K. Izumi, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956 CrossRef CAS PubMed; (g) R.-H. Liu, K. Fang, B. Wang, M.-H. Xu and G.-Q. Lin, J. Org. Chem., 2008, 73, 3307 CrossRef CAS PubMed.
- During our investigations, we found that the reductive homocoupling reactions also proceeded smoothly in the absence of NiI2 (see ESI‡), although the addition of a catalytic amount of this additive led to better yields and more consistent results. For a comparison of more results of the reactions conducted in the presence or in the absence of NiI2, see Table S1 in the ESI‡.
Footnotes |
| † Dedicated to Professor Henri-Philippe Husson on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1019290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08330j |
| This journal is © The Royal Society of Chemistry 2015 |