
RApid Parallel Protein EvaluatoR (RAPPER), from gene to enzyme function in one day†
L. T.
Quertinmont
,
R.
Orru
and
S.
Lutz
*
Department of Chemistry, Emory University, 1515 Dickey Drive, Atlanta, GA 30322, USA. E-mail: sal2@emory.edu
First published on 4th November 2014
Abstract
Cell-free transcription–translation systems offer an effective and versatile platform to explore the impact of genetic variations on protein function. We have developed a protocol for preparing linear, mutagenic DNA templates for direct use in the PURE system, enabling the fast and semi-quantitative evaluation of amino acid variations on catalytic activity and stereo-selectivity in native and engineered variants of Old Yellow Enzyme.
Tailoring biocatalysts by directed evolution or protein design is traditionally performed through manipulation of the corresponding nucleic acid sequence. The mutated gene is then transformed into a host organism for heterologous expression, followed by purification and functional evaluation via enzyme assay. Overall, the established process is robust but laborious and time-consuming. Cell-free transcription–translation systems offer an attractive alternative, not only eliminating the need for DNA transformation, culture growth and induction but also offering easy control over reaction conditions and the ability to supplement the reaction mixture.1,2 The use of PURE (Protein synthesis Using Recombinant Elements), a chemically defined transcription–translation system first reported by Ueda and coworkers,3,4 is particularly attractive as it minimizes side-reactions and reduces background activity. Previously, we have successfully employed PURE for the evaluation of circular permutation (CP) libraries of the NADPH-dependent flavoprotein Old Yellow Enzyme from Saccharomyces pastoranius (OYE1).5 These studies relied on synthetic gene libraries, cloned in a circular DNA vector (pET-14b), as templates for protein synthesis in PURE. To further accelerate the discovery process for novel biocatalysts, we adapted our experimental protocol to directly use linear DNA products from polymerase chain reactions as templates for protein synthesis, hence allowing for assembly of (mutant) genes and their subsequent biocatalytic assessment in one day. We herein report on the validation of our new protocol named RAPPER (RApid Parallel Protein EvaluatoR) with established OYEs, as well as an initial exploration of OYE variants for improved catalytic activity and altered stereo-selectivity.
The overall strategy for RAPPER is outlined in Fig. 1 and consists of three generic steps: mutagenesis, protein synthesis, and enzyme assay. For our specific study of OYE1 variants, the initial step consisted of a primer overlap extension PCR6 to introduce one or more mutations in the template DNA. Next, the modified DNA served as template for in vitro transcription–translation by PURE. Finally, the PURE reaction was mixed directly with a stock solution containing substrate, as well as necessary cofactors and buffer salts. Following a predetermined incubation time, the enzyme assay was quenched and product formation was detected by GC analysis.
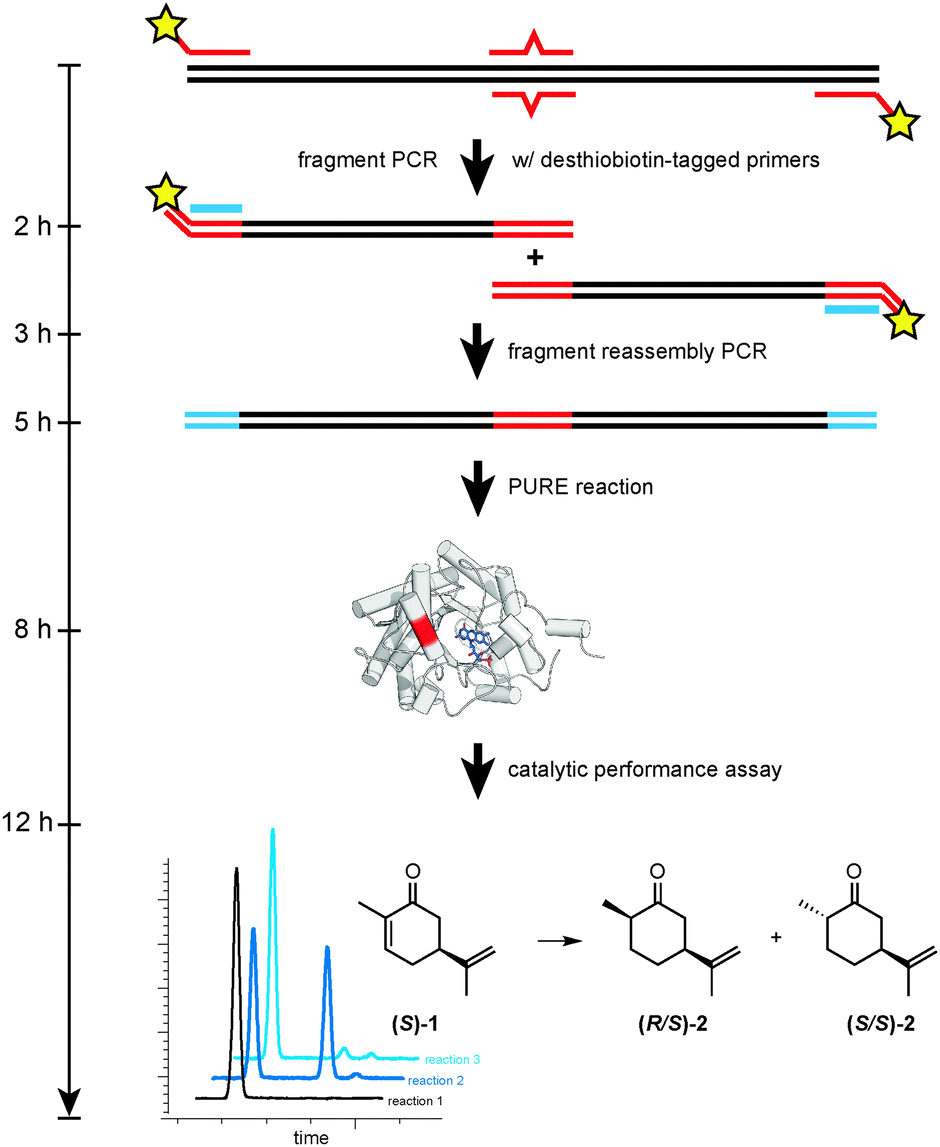 | ||
| Fig. 1 Overview of key steps (mutagenesis, protein synthesis, and enzyme assay) for the RAPPER protocol. | ||
To implement the three-step protocol of RAPPER, we first explored the effects of the regions flanking the gene of interest. Minimizing the size of up and downstream regions in the linear DNA template is desirable to simplify the PCR conditions, yet poses the risk of eliminating control elements for effective transcription and translation of the target protein. To probe for the shortest DNA template without diminishing effects in the subsequent enzyme activity assay, we prepared a series of five truncated DNA sequences based on the pET14b-OYE1 vector (Fig. 2). These linear DNA templates ranged in size from ∼5.9 kb (res. 2424–2424) to ∼1.5 kb (res. 401–678). The efficiency of protein synthesis with these linear DNA fragments by PURE was assessed via OYE reductase activity assay, measuring the percent conversion of (S)-carvone (1) to (R/S) and (S/S)-dihydrocarvone (2). Our results show that the enzyme activity for linear DNA templates are only slightly lower, yielding 80 to 90% conversion relative to circular DNA (+control). Furthermore, the data indicates no significant difference in the production of active enzyme as a function of DNA template size. These findings suggest no critical DNA sequence elements controlling protein synthesis beyond the roughly 150 base pairs flanking the gene of interest which encode for known elements including the RBS, as well as the T7 promoter and termination sites. In summary, linear DNA templates encoding minimal transcriptional and translational control elements express OYE at levels comparable to plasmid templates.
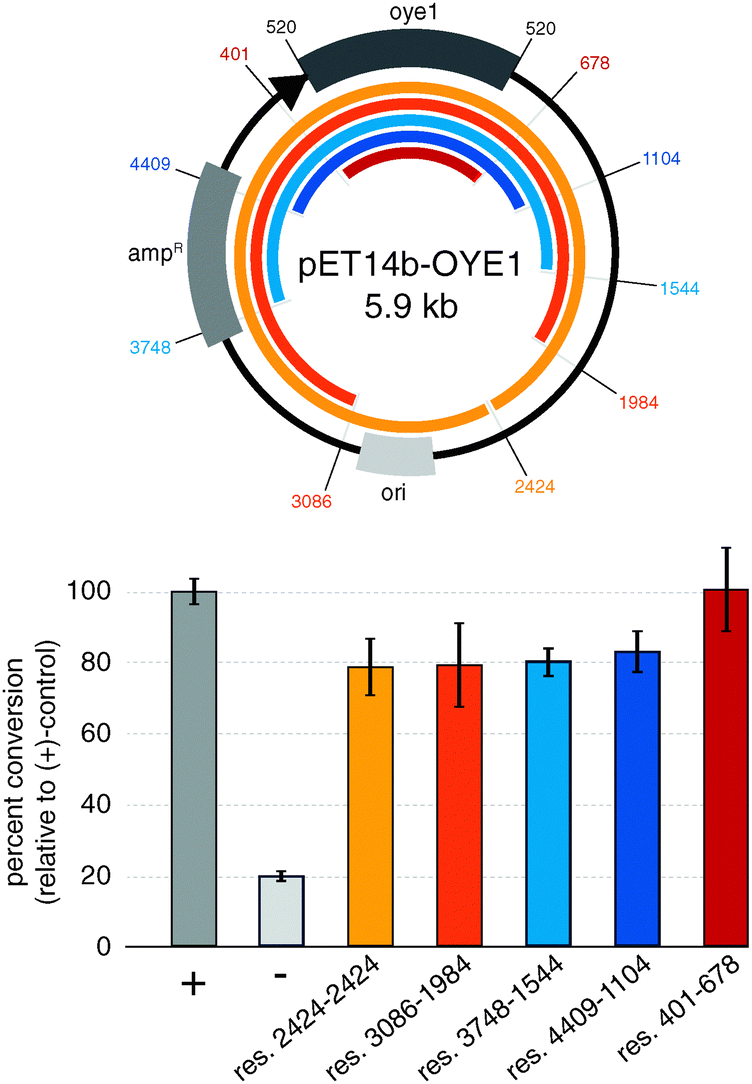 | ||
| Fig. 2 Impact of truncated 5′ and 3′-regions in linear DNA templates on transcription–translation efficiency. (+) control: circular pET14b-OYE1; (−) control: pET-DHFR. | ||
Next, we addressed compatibility issues between the mutagenesis step and PURE protocol. Dramatically fluctuating enzyme activity in replica experiments of individual OYE variants were noted in early experiments. Interestingly, the problem was not seen with native DNA templates or libraries generated by random mutagenesis (data not shown). The poor reproducibility was traced back to the agarose gel purification step in the primer overlap extension protocol. Gel purification of DNA fragments after the first PCR is necessary to remove the full-length DNA template, yet causes sample contamination with inhibitors of translation (NEB, personal communication). The use of desthiobiotin-tagged primers offered a simple alternative for effectively removing the DNA template by washing the PCR products over immobilized streptavidin (Fig. 1). Quantitative recovery of the desthiobiotin-tagged DNA fragments was possible by rinsing with elution buffer containing biotin.
Following the adjustments to the RAPPER protocol, we prepared a series of OYE variants to validate the method and to explore the functional impact of a few specific amino acid substitutions in native OYE1 and cpOYE303. cpOYE303 is a circular permutated OYE variant whose new protein sequence termini are located in the loop β6 region near the active site.5 Its enhanced ene-reductase activity is thought to derive from increased local backbone flexibility which improves active site accessibility and benefits rate-limiting conformational changes. Single and double mutations at amino acid positions 116, 250, 295, and 375 (all residue numbering based on OYE1) were introduced by primer overlap extension PCR. For validation purposes, aliquots of each PCR were directly processed by the PURE system, followed by detection of enzyme activity based on reduction of 1 (Fig. 3, “yellow” data). Separately, samples of the PCR products were also cloned into pET-14b and transformed into E. coli BL21(DE3) for heterologous expression. Following protein isolation, the purified enzymes were characterized by the same ene-reductase activity assay (Fig. 3, “blue” data). The catalytic conversion and diastereo-selectivity data for OYE variants based on RAPPER and purified enzymes are summarized in Fig. 3. Overall, the results from the two enzyme activity assays are consistent, supporting the notion that RAPPER is a reliable predictor of catalytic performance for individual variants. Similarly, our cell-free screening system accurately captures the diastereo-selectivity. Measuring reduction of 1 to either (R/S)-2 or (S/S)-2, the performance of OYE variants detected with RAPPER and the classical enzyme assay matches for all but one enzyme variant. We attribute the discrepancy in the data for cpOYE303(Y375F) to the enzyme's poor overall activity which results in low signal-to-noise and inaccurate peak integration.
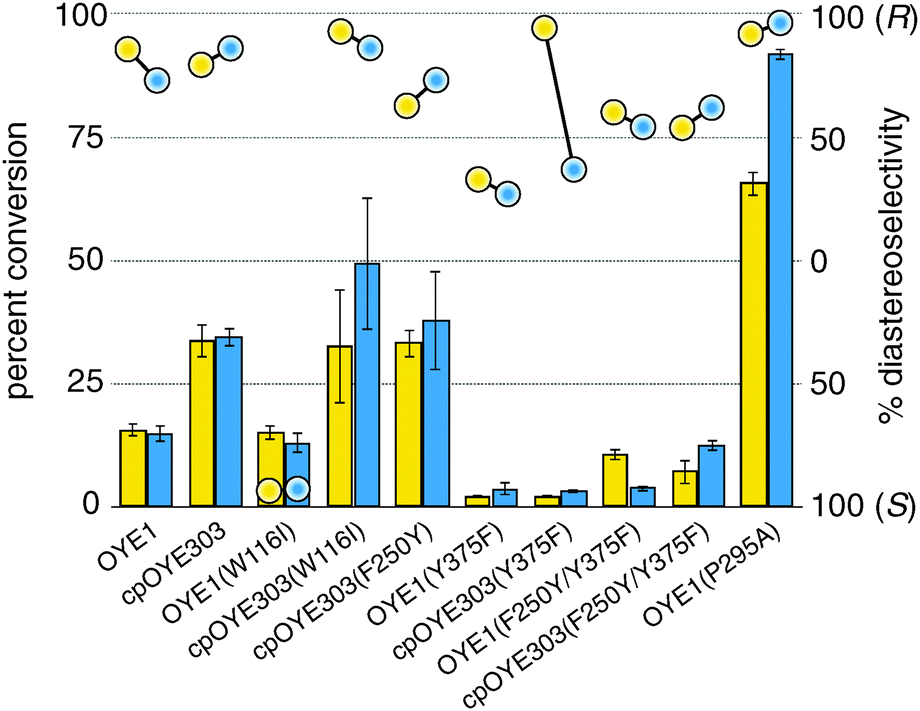 | ||
| Fig. 3 Comparison of activity (columns) and diastereo-selectivity (circles) data for reduction of 1 by selected OYE variants by RAPPER (yellow) and purified enzymes prepared by conventional methods using heterologous expression in E. coli host cells (blue). | ||
Beyond validation experiments, RAPPER also allowed for an initial evaluation of specific amino acid changes on the catalytic performance and diastereo-selectivity of OYE1 and cpOYE303. We first tested the effects of W116I on the biocatalytic performance of cpOYE303. In previous work, Stewart and coworkers showed a complete reversion of OYE1 diastereo-selectivity for the reduction of 1.7 While our assay accurately reproduced Stewart's results in OYE1, the same W116I change in cpOYE303 did not result in any detectable diastereo-selectivity change (Fig. 3). Crystallographic data for cpOYE303 suggest that slight conformational changes at Y375 might explain the selectivity shift.8 These findings also raised questions as to the possible role of neighboring residues in the active site and in the access tunnel, which can affect the pre-orientation of substrate. Hence we evaluated the effects of amino acid changes in positions F250 and Y375 (Fig. 4). Two single-site variants (F250Y and Y375F) and the double mutant (F250Y/Y375F) were prepared in OYE1 and cpOYE303, respectively. While the F250Y substitution showed little effect on activity and selectivity, Y375F proved highly detrimental to enzyme function. In contrast, combining the two amino acid changes resulted in recovery of activity at diminished diastereo-selectivity. Our initial screening data encourage the investigation of additional amino acid substitutions in these two positions to search for enzymes with altered selectivity without loss of catalytic activity.
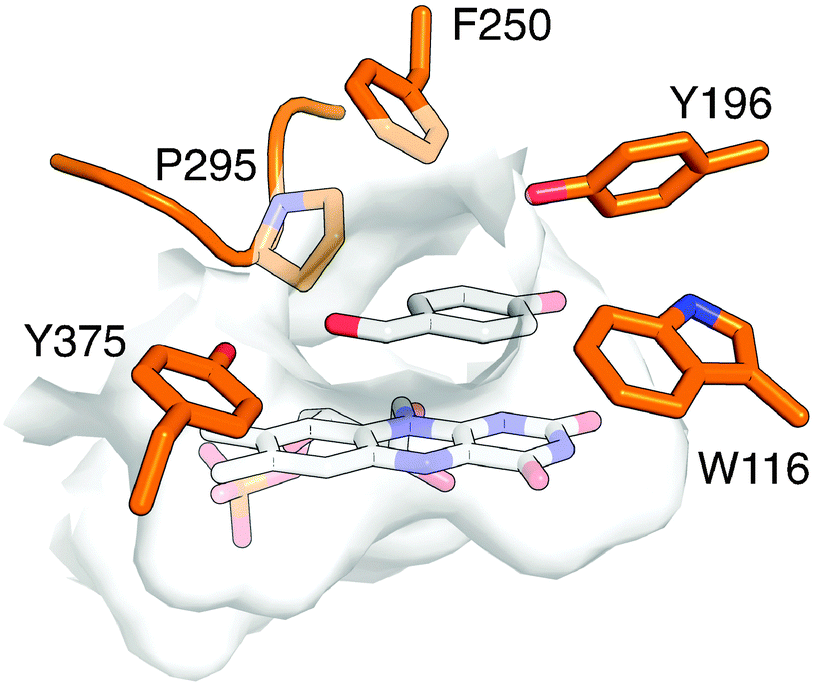 | ||
| Fig. 4 Active site of OYE1 (PDB: 1K03 (ref. 9)) with bound FMN and p-hydroxybenzaldehyde. Key active site residues are highlighted. | ||
In a final experiment, we used RAPPER to test an idea regarding the role of protein backbone flexibility in the loop β6 region of OYE1. Loop β6 forms a lid over the active site and is thought to be involved in the rate-limiting conformational change during biocatalysis. Consistent with this hypothesis, termini relocation into this region of the protein structure by circular permutation, which increases local conformational flexibility, resulted in over ten-fold improved catalytic activity for reduction of 1.5 An alternative strategy for probing the functional role of loop β6 is the substitution of alanine for proline 295, located at the tip of the loop. The amino acid change was predicted to reduce the region's conformational constrains, in turn translating into enhanced catalytic performance. Activity measurements in RAPPER and control experiments with the purified enzyme indicate significant functional gains that exceed even the increased activity observed for cpOYE303.
The modification of the primer overlap extension PCR protocol allows for direct use of linear DNA product as template in the PURE in vitro transcription–translation system. Such an adaptation allows for the rapid functional evaluation of one or more amino acid substitutions in target proteins. Our validation experiments with OYE variants demonstrate that the results from this approach are semi-quantitative in regard to catalytic activity and stereo-selectivity, hence presenting an effective method for functional prescreening of enzyme variants. RAPPER should be broadly applicable for proteins usually expressed in E. coli hosts. The method is also scalable for parallel screening of protein libraries in 96-well microtiter plate format. We consider RAPPER a useful tool to rapidly assess the function of enzyme variants without the need for lengthy procedures of cloning, transformation, culture growth and expression.
This work was supported in part by the US National Science Foundation (CBET-1159434). We thank the members of the Lutz lab for their critical and helpful comments and suggestions on the manuscript.
Notes and references
- Y. Shimizu, Y. Kuruma, B. W. Ying, S. Umekage and T. Ueda, Cell-free translation systems for protein engineering, FEBS J., 2006, 273, 4133–4140 CrossRef CAS PubMed.
- E. D. Carlson, R. Gan, C. E. Hodgman and M. C. Jewett, Cell-free protein synthesis: applications come of age, Biotechnol. Adv., 2012, 30, 1185–1194 CrossRef CAS PubMed.
- T. Ueda, Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa and K. Nishikawa, Cell-free translation reconstituted with purified components, Nat. Biotechnol., 2001, 19, 751–755 CrossRef PubMed.
- B. W. Ying and T. Ueda, Protein generation using a reconstituted system, The Protein Engineering Handbook, Wiley-VCH, Weinheim, 2009, vol. 2 Search PubMed.
- A. B. Daugherty, S. Govindarajan and S. Lutz, Improved biocatalysts from a synthetic circular permutation library of the flavin-dependent oxidoreductase old yellow enzyme, J. Am. Chem. Soc., 2013, 135, 14425–14432 CrossRef CAS PubMed.
- R. M. Horton, Z. L. Cai, S. N. Ho and L. R. Pease, Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction, Biotechniques, 1990, 8, 528–535 CAS.
- S. K. Padhi, D. J. Bougioukou and J. D. Stewart, Site-saturation mutagenesis of tryptophan 116 of Saccharomyces pastorianus old yellow enzyme uncovers stereocomplementary variants, J. Am. Chem. Soc., 2009, 131, 3271–3280 CrossRef CAS PubMed.
- A. B. Daugherty, J. R. Horton, X. Cheng and S. Lutz, 2014, manuscript submitted.
- B. J. Brown, J. Hyun, S. D. Duvvuri, P. A. Karplus and V. Massey, The role of glutamine 114 in old yellow enzyme, J. Biol. Chem., 2002, 277, 2138–2145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods. See DOI: 10.1039/c4cc08240k |
| This journal is © The Royal Society of Chemistry 2015 |