
Diarylethene based fluorescent switchable probes for the detection of amyloid-β pathology in Alzheimer's disease†
Guanglei
Lv
a,
Baiping
Cui
b,
Haichuang
Lan
a,
Ying
Wen
a,
Anyang
Sun
*b and
Tao
Yi
*a
aDepartment of Chemistry & Innovation Center of Chemistry for Energy Materials, Fudan University, 220 Handan Road, Shanghai 200433, P. R. China. E-mail: yitao@fudan.edu.cn; Fax: +86-21-55664621
bLaboratory of Neurodegenerative Diseases and Repair, Yancheng Institute of Health Sciences, 263 South Jiefang Road, Yancheng 224005, Jiangsu, P. R. China. E-mail: anyangsun@hotmail.com
First published on 4th November 2014
Abstract
Two fluorescent switchable diarylethene derivatives which exhibit high affinity for amyloid-β aggregates with the increase of fluorescence intensity were reported. Moreover, the probes show excellent photochromic and anti-photobleaching properties both in vitro and in vivo.
Reactive fluorescence labeling technology has been becoming increasingly popular for the monitoring of internal cellular processes due to several advantages including low cost, no exposure to radioactivity, availability and the ease of operation.1–4 Fluorescent probes that can detect simple cations and anions have been widely developed.5–7 However, rational design of a probe with high specificity towards macro-biomolecules, such as proteins and DNA, remains a major challenge due to the complexity and the lack of specific active sites of these macro-biomolecules. Amyloid-β (Aβ) is the major component of senile plaques, stemming from the consecutive cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase.8,9 It is well established that Aβ plays an important role in the pathophysiology and progression of Alzheimer's Disease (AD), so it is of immediate and practical importance to design imaging probes to detect Aβ. To date, the most widely applied Aβ imaging probes utilize positron emission tomography (PET) and single photon emission computed tomography (SPECT) using Congo Red (CR) and Thioflavin T (ThT),10–13 respectively. However these imaging modalities are hindered by their intrinsic high cost and time-consuming nature, and also result in patient exposure to radiation.
Fluorescence-based imaging of Aβ has emerged as a potential alternative to other techniques.14–17 Some fluorescent probes showing specific binding to Aβ have been reported. Theodorakis et al. developed a new family of fluorescent probes containing an aminonaphthalenyl-2-cyano-acrylate (ANCA) motif, which exhibited great specificity and affinity for Aβ (Kd = 1.4 ± 0.2 μM) deposits in tissue.18 Ran et al. synthesized curcumin-based near-infrared (NIR) fluorescent probes for detecting Aβ species, which showed significant fluorescence changes upon mixing with Aβ species in vitro and in vivo.19 However, clinical application of these fluorescent probes for Aβ detection is hampered by poor anti-photobleaching.
Conventional probes for fluorescence imaging are easily photobleached and therefore can only respond irreversibly to one event. In contrast, fluorophores that can respond reversibly constitute a simple and powerful tool for regional optical marking and tracing dynamic process of a bio-event and are thus potentially more valuable for use as fluorescent labels.20 Diarylethene (DAE) derivatives, which can reversibly photocyclise upon irradiation with UV and visible light, are the most promising candidate fluorescent switch systems because of their fast response, excellent thermal stability and outstanding fatigue resistance.21–23 These outstanding characteristics of DAE enable long time and real time operation in bioimaging. Compared with always-on probes, switchable DAE readily avoids background interference. More importantly, fluorescent switchable diarylethene has potential applications in super resolution imaging.24 However, far fewer reports on the biological applications of DAEs have been published.23 Irie reported a fluorescent photochromic DAE for labeling biomolecules.25 Branda first demonstrated that light-induced reactions of a photoresponsive DAE could be reversibly triggered in a living organism.26 Very recently, we have reported a thiazole orange linked DAE for detection of DNA and the intracellular nucleus.27 To the best of our knowledge, there is no report of photochromic compounds for fluorescence imaging of Aβ. Herein, by connecting the targeting unit (ANCA) of an Aβ deposit to a photochromic DAE unit on one and two sides (T1 and T2, respectively, in Scheme 1), we have developed a fluorescent switch for detecting Aβ deposits for the first time. The probes showed high affinity for Aβ aggregates in both solution and brain section with both a remarkable increase in the fluorescence intensity and a slight blue shift. The probes exhibited excellent photochromic properties with the fluorescence intensity undergoing reversible changes upon alternating irradiation of UV and visible light both in solution and in brain tissue, further highlighting the remarkable anti-photobleaching capability of the probes in tissue.
 | ||
| Scheme 1 The structures of T1 and T2 and the photochromism of T2. | ||
The binding properties of T1 and T2 towards Aβ were studied by fluorescence spectroscopy in buffer solution. T1 and T2 show very weak fluorescence in PBS buffer solution (pH = 7.3) at corresponding wavelengths of 586 and 592 nm with absolute fluorescence quantum yields (ΦF) of 1.1% and 0.4%, respectively, under excitation with 430 nm light. This fluorescence intensity was sharply increased by corresponding factors of 8.2 and 12.7 for T1 and T2 (Fig. 1a and b), while the ΦF values increased to 2.1% and 2.6%, respectively, in the presence of Aβ aggregates (Table 1). In addition, the fluorescence emission maxima of T1 and T2 blue shifted from 586 to 550 nm and 592 to 563 nm in the presence of Aβ aggregates, respectively. T2 exhibited a longer emission wavelength and a larger fluorescence shift before and after the addition of Aβ aggregates, showing superior probing properties towards Aβ aggregates than T1. The concentration-dependent fluorescence spectral changes of T1 and T2 showed that the intensity gradually increased along with increasing concentration of Aβ aggregates, accompanied by a slight blue-shift in λmax (Fig. 1c and Fig. S1, ESI†). A linear relationship between Aβ aggregate concentration and fluorescence intensity of the probes was observed in the concentration range of 1–13 μM (R2 = 0.98 for both T1 and T2, Fig. S2, ESI†), indicating that the concentration of Aβ aggregates may be quantitatively estimated by the probes. The dissociation constant (Kd) of T1 and T2 to Aβ aggregates was measured to be 1.3 μM and 1.1 μM, respectively,28 which was comparative to or smaller than that of ANCA.18 The better binding affinity of T2 for Aβ aggregates compared with that of T1 may be due to its two binding sites.
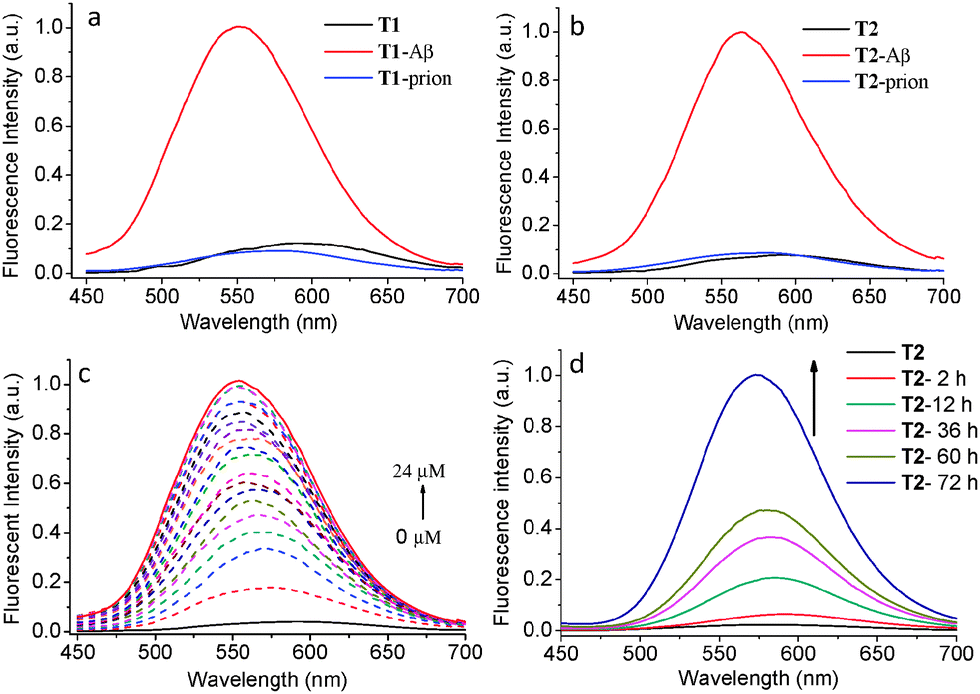 | ||
| Fig. 1 Normalized fluorescence spectra of (a) T1, T1 with Aβ aggregates (5.0 μM), T1 with prion aggregates (5.0 μM), and (b) T2, T2 with Aβ aggregates (5.0 μM), T2 with prion aggregates (5.0 μM); the fluorescence spectral change of T2 at (c) different concentrations of Aβ aggregates ranging from 0 to 24 μM and (d) different incubation time of the Aβ monomer over 72 h. The concentration of T1 and T2 is 2.0 μM (5% DMSO in PBS), λex = 430 nm. | ||
| λ em (nm) | E (%) | Φ o–c (%) | λ ab (nm) PSS | Φ F (%) | ΔF/FTb | |
|---|---|---|---|---|---|---|
| E = photocyclic transition efficiency; Φo–c = cyclization quantum yield; ΦF = absolute fluorescence quantum yields measured by using the calibrated integrating sphere.a Maximum emission wavelength with and without Aβ aggregates.b Fold change of the fluorescence intensity with Aβ aggregates, ΔF = FT+Aβ − FT. | ||||||
| T1 | 586 | 71 | 20.6 | 645 | 1.1 | |
| T1 + Aβ | 550 | 21.9 | 643 | 2.1 | 8.2 | |
| T2 | 592 | 63 | 14.2 | 687 | 0.4 | |
| T2 + Aβ | 563 | 15.8 | 679 | 2.6 | 12.7 | |
The dynamic aggregation process of the Aβ monomer was followed by changes in fluorescence intensity of T1 or T2 with different incubation times up to 72 h. There was no detectable fluorescence change of T1 and T2 in PBS solution after the addition of the Aβ monomer, however both T1 and T2 showed fluorescence enhancement upon prolonged incubation times (Fig. 1d and Fig. S3, ESI†). As Aβ aggregation increases with monomer incubation time, this fluorescence enhancement further confirms that T1 and T2 exhibit high affinity for Aβ aggregates. Therefore these probes are potentially capable of tracing the Aβ aggregation process. The specificity of T1 and T2 toward Aβ aggregates was studied by comparing with prion 106–126 peptides (an aggregation-prone peptide containing 21 amino acid residues from prion protein associated with prion disease).29 No significant increase in the fluorescence intensity of both probes was observed when the probes were incubated with prion, indicating the excellent selectivity of T1 and T2 toward Aβ aggregates (Fig. 1a and b).
To investigate the binding mechanism of T1 and T2 to Aβ aggregates (T2 taken here as an example), the fluorescence spectra of T2 with excitation at different wavelengths before and after the addition of Aβ aggregates were studied. When exciting T2 with a wavelength shorter than 350 nm light, T2 only shows weak fluorescence at about 440 nm belonging to DAE,23 whereas the fluorescence of ANCA was not observed (Fig. S4, ESI†). This indicates that the effective energy transfer between the DAE and ANCA groups has not happened. When red-shifting the excitation wavelength to 370 nm, the fluorescence of ANCA in T2 at about 560 nm was clearly observed. Both the fluorescence intensities at 440 nm and 560 nm was sharply increased upon addition of Aβ aggregates, indicating that both DAE and ANCA may contribute to the binding between T2 and Aβ aggregates. This was further proved by the study of the binding properties between Aβ aggregates and reference compounds 5 (ANCA with short polyether chain) and 7 (DAE with a short polyether chain) (Scheme S1, ESI†). Upon addition of Aβ aggregates to 7 or 5, both the fluorescence intensities were drastically increased (Fig. S5 and S6, ESI†), which further confirmed the synergistic binding effect of DAE and ANCA to the hydrophobic pockets of Aβ aggregates and restricted the mobility of T2, leading to a fluorescence increase.
The photochromic behaviour of T1 and T2 was investigated before and after the addition of Aβ aggregates. The maximum absorption of T1 in its open-ring form centered at 328 nm (ε = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L mol−1 cm−1) is ascribed to a π–π* transition. Upon irradiation with 365 nm UV light for 3 min, a new absorption band appeared at 645 nm, accompanied by a change in the colour from colourless to green in the photo-stationary state (PSS) caused by an increase in π-electron delocalization (Fig. 2a and Fig. S7, ESI†). The photochromic behaviour of T2 was similar to that of T1 with a decrease in absorption at 380 nm and a concomitant increase in absorption at 687 nm upon irradiation with UV light, slightly red-shifted compared with that of T1 (Fig. 2b and Fig. S8, ESI†). The reverse process was performed by irradiating the PSS of T1 or T2 with visible light (λ > 550 nm). The photocyclization efficiency of T1 was 71% in the PSS as measured by 1H NMR (Fig. S9, ESI†) and the photocyclization quantum yield (Φo–c)30 was determined to be 20.6% upon 365 nm light irradiation. The Φo–c and transformation efficiency of T2 were 14.2% and 63% (Fig. S10, ESI†), respectively, comparatively lower than that of T1.
000 L mol−1 cm−1) is ascribed to a π–π* transition. Upon irradiation with 365 nm UV light for 3 min, a new absorption band appeared at 645 nm, accompanied by a change in the colour from colourless to green in the photo-stationary state (PSS) caused by an increase in π-electron delocalization (Fig. 2a and Fig. S7, ESI†). The photochromic behaviour of T2 was similar to that of T1 with a decrease in absorption at 380 nm and a concomitant increase in absorption at 687 nm upon irradiation with UV light, slightly red-shifted compared with that of T1 (Fig. 2b and Fig. S8, ESI†). The reverse process was performed by irradiating the PSS of T1 or T2 with visible light (λ > 550 nm). The photocyclization efficiency of T1 was 71% in the PSS as measured by 1H NMR (Fig. S9, ESI†) and the photocyclization quantum yield (Φo–c)30 was determined to be 20.6% upon 365 nm light irradiation. The Φo–c and transformation efficiency of T2 were 14.2% and 63% (Fig. S10, ESI†), respectively, comparatively lower than that of T1.
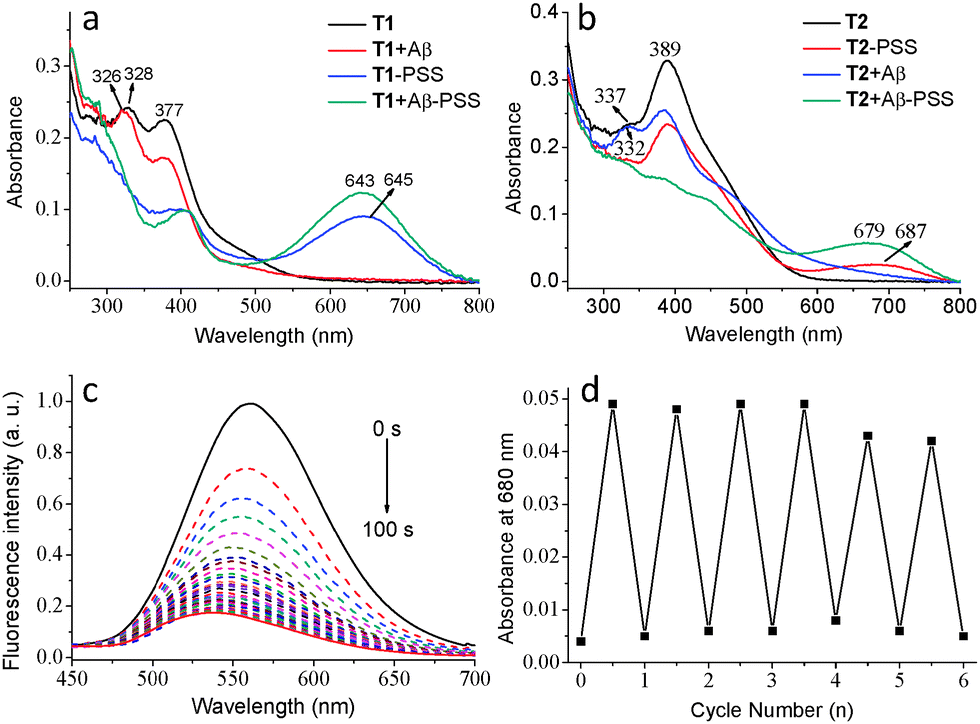 | ||
| Fig. 2 Absorption spectra of (a) T1 and (b) T2 in the open form and in the PSS with and without Aβ aggregates (CT = 1.0 × 10−5 M, 5% DMSO in PBS); (c) the fluorescence spectral change of T2 (2.0 × 10−6 M, 5% DMSO in PBS) in the presence of Aβ aggregates (5.0 × 10−6 M) upon irradiation with UV light; (d) absorbance changes at 680 nm of T2 + Aβ upon alternating irradiation of UV and visible light. | ||
The photochromic behaviours of T1 and T2 in the presence of Aβ aggregates were subsequently investigated (T2 taken here as an example). The wavelength of the absorption of the open form of T2 at 389 nm clearly decreased and the absorption band at 337 nm blue-shifted to 332 nm after the addition of Aβ aggregates (Fig. 2b). Upon irradiation of a solution containing T2 and Aβ aggregates with UV light, a new absorption band (687 nm) in the visible range emerged, accompanied by significant fluorescence quenching (>85%) in the PSS due to the energy transfer between ANCA and the closed form of DAE (Fig. 2c). The Φo–c of both T1 and T2 was increased after binding to Aβ aggregates. The reverse process was performed by irradiation of T2 + Aβ aggregates with visible light in its PSS (Fig. S11, ESI†), demonstrating typical photoswitching characteristics of DAE. This reversible photochromic cycle can be repeated many times with a low rate of fatigue (Fig. 2d). The solution containing T1 and Aβ aggregates showed similar reversible changes in absorption and fluorescence spectra to those of T2 under the same conditions (Fig. S13, ESI†).
To further evaluate whether these fluorescent probes could stain amyloid deposits in brain tissue, brain sections of AD mice (12 month-old APP/PS1 transgenic mice) were stained with T1 and T2. Both T1 and T2 can be practically applied for the detection of Aβ aggregates in brain tissues by employing confocal laser scanning microscopy (CLSM). As expected, the fluorescent probes could specifically highlight Aβ deposits in brain sections, whereas the brain sections without probes did not specifically highlight the aggregates (Fig. 3A and Fig. S14, ESI†). T2 exhibited a longer emission wavelength and higher local fluorescence intensity than T1, rendering T2 more suitable for in vivo detection of Aβ aggregates in brain tissue. To determine the location and specificity of the probes, the T2-loaded brain tissue (red channel, Fig. 3A) was further incubated with Aβ antibody (green channel, Fig. 3B) and tau protein antibody (green channel, Fig. S15, ESI†), respectively. The images of the brain section showed good colocalization of T2 and Aβ antibody (Fig. 3C), but different localization of T2 with tau protein, indicating that T2 specifically accumulated in Aβ deposits. These experiments clearly demonstrate that the probe can selectively mark the location of amyloid deposits in brain tissues.
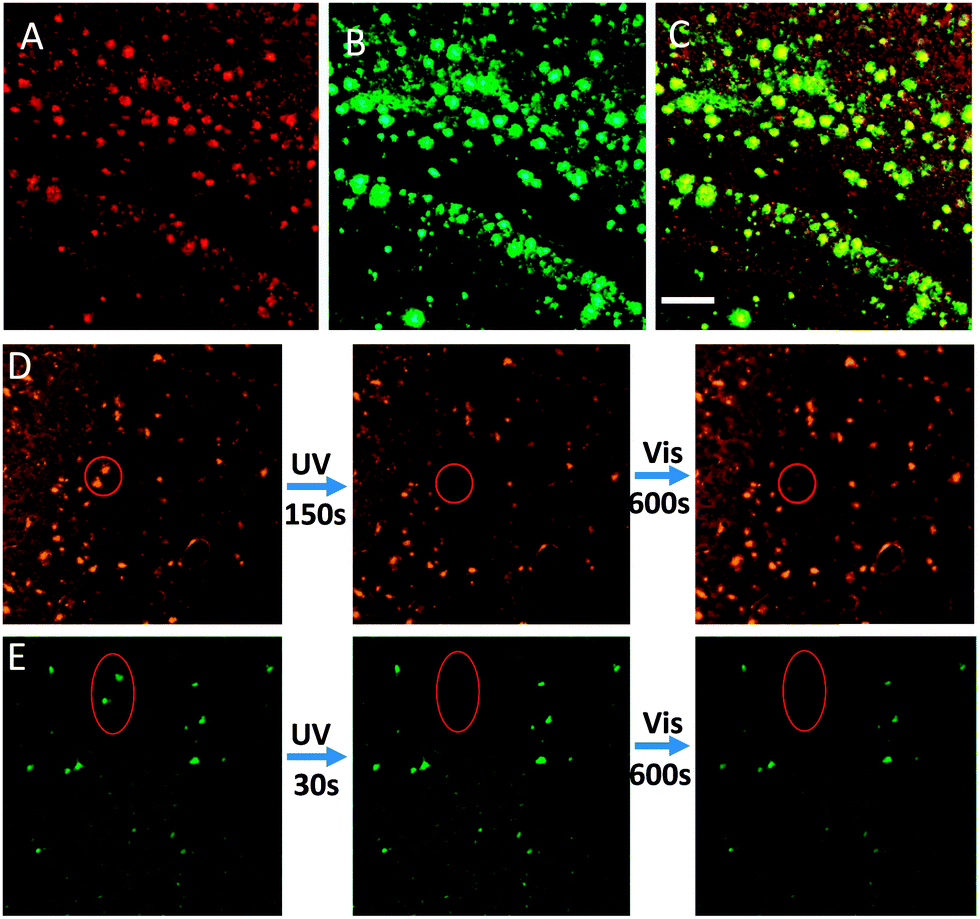 | ||
| Fig. 3 CLSM images of brain sections; (A) staining with 60 μM T2 (red channel: 575–625 nm); (B) staining with a β-amyloid antibody (green channel: 495–540 nm); (C) the merged image of A and B; scale bar: 20 μm. (D) Photochromic process of T2 (60 μM) in the brain section, left: original state; middle: irradiated by 405 nm light in the assigned circled region for 150 s; right: irradiated with 633 nm light over the entire region. (E) Contrast experiment stained with ThT (60 μM), left: original state; middle: irradiated by 405 nm light in the oval region for 30 s; right: irradiated with 633 nm light over the entire region. | ||
Importantly, the fluorescence of T2 in brain sections could also be tuned by varying the irradiation wavelength. Micro-fluorescence switching in brain sections was achieved by alternating between UV and visible light illumination. The selected T2-stained Aβ deposits (the circular region in Fig. 3D) were irradiated with 405 nm light for 150 s, and the fluorescence of the irradiated deposits was turned off, due to T2 in this area changing from an open state to a closed state. Interestingly, the fluorescence of the Aβ deposits could be recovered by 633 nm light irradiation from the CLSM lamp for 10 min (Fig. 3D). The fluorescence intensity of T2 showed reversible changes upon alternating irradiation of UV and visible light, validating the photochromic properties of T2 and indicating that the probes show remarkable anti-photobleaching in tissues. As evident in Fig. S16 (ESI†), the optical switching of fluorescence can be repeated many times with little “fatigue” effects or photobleaching that were thought to be fatal disadvantages of the general fluorophore. In contrast, ThT (a common standard stain for Aβ plaques) stained brain sections were also irradiated with UV and visible light. The fluorescence intensity of ThT was rapidly quenched by UV light and did not recover under visible light (Fig. 3E). In addition, both T1 and T2 show low cytotoxicity by means of an MTT assay. The cellular viabilities were estimated to be greater than 85% after 2 h in the presence of 1–100 μM T1 or T2 (Fig. S17, ESI†). Thus, we concluded that T2 is a novel and superior fluorescent dye for use as a marker of Aβ deposits.
In summary, we designed and synthesized diarylethene-based fluorescent probes capable of specifically detecting Aβ aggregates. The fluorescence intensity dramatically increased in the presence of Aβ aggregates, accompanied by a blue shift. The fluorescence can be readily tuned under alternating irradiation of UV and visible light, exhibiting excellent fatigue resistance and anti-photobleaching. These probes can specifically target Aβ deposits in brain sections. The synergistic binding effect of DAE and ANCA to the hydrophobic pockets of Aβ aggregates contributes to the binding efficiency of the probes. The switchable fluorescence image operated by light in brain tissues may provide a new window for in vivo high resolution imaging. Further studies of these probes are ongoing in our laboratory.
The authors are grateful for the financial support from 973 (2013CB733700), NNSFC (21125104, 51373039), PIRTU (IRT1117) and Shanghai Sci. Tech. Comm. (12XD1405900).
Notes and references
- V. Ntziachristos, C. Bremer and R. Weissleder, Eur. J. Radiol., 2003, 13, 195 Search PubMed
.
- V. Ntziachristos, Annu. Rev. Biomed. Eng., 2006, 8, 1 CrossRef CAS PubMed
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626 CrossRef CAS PubMed
- K. Licha and C. Olbrich, Adv. Drug Delivery Rev., 2005, 57, 1087 CrossRef CAS PubMed
- M. Royzen, A. Durandin, V. G. Young, N. E. Geacintov and J. W. Canary, J. Am. Chem. Soc., 2006, 128, 3854 CrossRef CAS PubMed
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536 CrossRef CAS PubMed
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and L. Mosca, J. Am. Chem. Soc., 2013, 135, 6345 CrossRef CAS PubMed
- D. J. Selkoe, Ann. N. Y. Acad. Sci., 2000, 924, 17 CrossRef CAS PubMed
- W. Annaert and B. De Strooper, Annu. Rev. Cell Dev. Biol., 2002, 18, 25 CrossRef CAS PubMed
- L. Cai, R. B. Innis and V. W. Pike, Curr. Med. Chem., 2007, 14, 19 CrossRef CAS
- H. Quigley, S. J. Colloby and J. T. O'Brien, Int. J. Geriatr. Psychiatry, 2011, 26, 991 CrossRef PubMed
- C. C. Rowe and V. L. Villemagne, J. Nucl. Med., 2011, 52, 1733 CAS
- C. A. Mathis, N. S. Mason, B. J. Lopresti and W. E. Klunk, Semin. Nucl. Med., 2012, 42, 423 CrossRef PubMed
- Y. O. Lee, J. W. Shin, C. Yi, Y. H. Lee, N. W. Sohn, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 5741 RSC
- K. Liu, T. L. Guo, J. Chojnacki, H.-G. Lee, X. Wang, S. L. Siedlak, W. Rao, X. Zhu and S. Zhang, ACS Chem. Neurosci., 2012, 3, 141 CrossRef CAS PubMed
- A. Aaslund, C. J. Sigurdson, T. Klingstedt, S. Grathwohl, T. Bolmont, D. L. Dickstein, E. Glimsdal, S. Prokop, M. Lindgren, P. Konradsson, D. M. Holtzman, P. R. Hof, F. L. Heppner, S. Gandy, M. Jucker, A. Aguzzi, P. Hammarstroem and K. P. R. Nilsson, ACS Chem. Biol., 2009, 4, 673 CrossRef CAS PubMed
- E. E. Nesterov, J. Skoch, B. T. Hyman, W. E. Klunk, B. J. Bacskai and T. M. Swager, Angew. Chem., Int. Ed., 2005, 44, 5452 CrossRef CAS PubMed
- K. Cao, M. Farahi, M. Dakanali, W. M. Chang, C. J. Sigurdson, E. A. Theodorakis and J. Yang, J. Am. Chem. Soc., 2012, 134, 17338 CrossRef CAS PubMed
- X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397 CrossRef CAS PubMed
- L. Zhu, W. Wu, M.-Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524 CrossRef CAS PubMed
- M. Irie, Chem. Rev., 2000, 100, 1683 CrossRef CAS PubMed
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603 CrossRef CAS PubMed
- Y. Zou, T. Yi, S. Xiao, F. Li, C. Li, X. Gao, J. Wu, M. Yu and C. Huang, J. Am. Chem. Soc., 2008, 130, 15750 CrossRef CAS PubMed
- M. Bates, B. Huang, G. T. Dempsey and X. Zhuang, Science, 2007, 317, 1749 CrossRef CAS PubMed
- N. Soh, K. Yoshida, H. Nakajima, K. Nakano, T. Imato, T. Fukaminato and M. Irie, Chem. Commun., 2007, 5206 RSC
- U. Al-Atar, R. Fernandes, B. Johnsen, D. Baillie and N. R. Branda, J. Am. Chem. Soc., 2009, 131, 15966 CrossRef CAS PubMed
- K. Liu, Y. Wen, T. Shi, Y. Li, F. Li, Y. L. Zhao, C. Huang and T. Yi, Chem. Commun., 2014, 50, 9141 RSC
- J. Sutharsan, M. Dakanali, C. C. Capule, M. A. Haidekker, J. Yang and E. A. Theodorakis, ChemMedChem, 2010, 5, 56 CrossRef CAS PubMed
- B. Kurganov, M. Doh and N. Arispe, Peptides, 2004, 25, 217 CrossRef CAS PubMed
- M. Irie, T. Lifka, K. Uchida, S. Kobatake and Y. Shindo, Chem. Commun., 1999, 747 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details and additional spectra. See DOI: 10.1039/c4cc07656g |
| This journal is © The Royal Society of Chemistry 2015 |