
UCST behavior of polyampholytes based on stoichiometric RAFT copolymerization of cationic and anionic monomers†
Qilu
Zhang
and
Richard
Hoogenboom
*
Supramolecular Chemistry Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: richard.hoogenboom@ugent.be
First published on 28th October 2014
Abstract
Polyampholytes with controlled equimolar ratio of charges were synthesized by reversible addition–fragmentation chain transfer (RAFT) copolymerization of cationic and anionic monomers. The resulting charge-neutral polyampholytes exhibit upper critical solution temperature (UCST) thermoresponsive behavior in ethanol–water and methanol–water solvent mixtures based on electrostatic attraction. Finally, the temperature induced self-assembly of a polyampholyte with oligo(ethylene glycol) side chains into defined nanoparticles below the UCST transition is demonstrated.
Thermoresponsive polymers represent polymers that undergo a reversible phase transition at the lower critical solution temperature (LCST) or the upper critical solution temperature (UCST). The most commonly studied and firstly reported thermoresponsive polymer in aqueous solution is poly(N-isopropylacrylamide) (PNIPAM) with an LCST of ca. 32 °C,1,2 which is close to human body temperature. More recently, poly(oligoethylene glycol (meth)acrylate)s,3–7 poly(2-oxazoline)s,8–13 poly(vinyl ether)s14 and polypeptides15,16 have also been widely studied and have found various applications as smart materials. However, the reversed behavior, that is polymers that become soluble upon increasing temperature, would be more desirable for certain biological applications, such as auto-regulated drug release upon increasing body-temperature. Compared with polymers that undergo an LCST transition, such polymers exhibiting UCST behavior in aqueous solution have been much less documented as it is much more challenging to achieve this behavior in aqueous solutions requiring strong interpolymer attraction in combination with high hydrophilicity.17 Alcohol–water solvent mixtures have also been considered as promising solvents to obtain UCST behavior for various polymers arising from the presence of complex hydration shells around the ethanol molecules and a decrease in solvent polarity upon heating,18–20 including poly(methyl methacrylate)s (PMMA),21–28 poly(2-alkyl-2-oxazoline)s,24,29,30 PNIPAM31–33 and poly[N-(4-vinylbenzyl)-N,N-dialkylamine]s.33,34
Polyampholytes are polymers bearing both cationic and anionic repeat units.35–37 Amongst the different types of polyampholytes, polyzwitterions with cationic and anionic groups bound to the same monomer unit are mostly studied, also referred to as polybetaines, mostly driven by the interest in their UCST behavior in water resulting from strong interpolymer attraction.36 In contrast, only few reports describe polyampholytes prepared by controlled copolymerization of cationic and anionic monomers, although this type of polymers can show special properties by tuning of the ratio and/or distance of the two charges.38 The development of polyampholytes based on cationic and anionic monomers with UCST behavior is extremely challenging as both monomers need to be incorporated in stoichiometric amounts. An imbalance in the ratio of the monomers will lead to excess charges on the polymer leading to strong repulsion of the polymer chains obstructing their UCST behavior.
The development of controlled radical polymerization techniques (CRP), such as nitroxide-mediated polymerization (NMP),39 atom transfer radical polymerization (ATRP),40–42 and reversible addition–fragmentation chain transfer (RAFT) polymerization,43–45 has provided new tools for the synthesis of well-defined (co)polymers. RAFT polymerization has been reported to provide good control over the direct radical polymerization of monomers with tertiary amine or carboxylic acid groups.45 Nonetheless, the controlled stoichiometric copolymerization of a tertiary amine functionalized monomer and carboxylic acid functionalized monomer has rarely been reported.46 Georgiou et al.47 have reported the synthesis of 2-(dimethylamino)ethyl methacrylate (DMAEMA)–methacrylic acid (MAA) star copolymers via group transfer polymerization (GTP), although the acidic monomer had to be protected to make it compatible with GTP.
In this paper, we report the synthesis of a new type of charge-neutral polyampholytes by direct stoichiometric RAFT copolymerization of anionic and cationic monomers. The UCST thermoresponsive behavior of the resulting copolymers is described in alcohol–water solvent mixtures. Finally, the incorporation of a solvophilic neutral comonomer is demonstrated to lead to defined self-assembled temperature responsive nanoparticles.
The synthesis of polyampholytes was performed by RAFT copolymerization due to its high tolerance to functionalities. The copolymerization was first performed with identical equivalents of the two charged comonomers, namely MAA and DMAEMA, see Scheme 1, considering the fact that the two monomers bear similar reactive vinyl groups. However, a preliminary kinetic study with equimolar amounts of the monomers revealed a ratio of the polymerization rate constants of DMAEMA and MAA as high as 2.5. Hence, the copolymerization was then performed with an excess of MAA to obtain charge neutral polyampholytes. Fig. 1 shows the kinetics plots of the copolymerizations with feed ratios of MAA/DMAEMA at 100/40 and 733/200, respectively, relative to the CTA aiming for different target molecular weights. The kinetics of the two copolymerizations revealed linear first order kinetic plots for both monomers indicating a constant free radical concentration indicative for the absence of significant termination reactions. A linear increase of molecular weight with conversion as well as the relatively narrow molar mass distributions (Fig. S1, ESI†) further demonstrate good control over the copolymerizations of the two monomers.
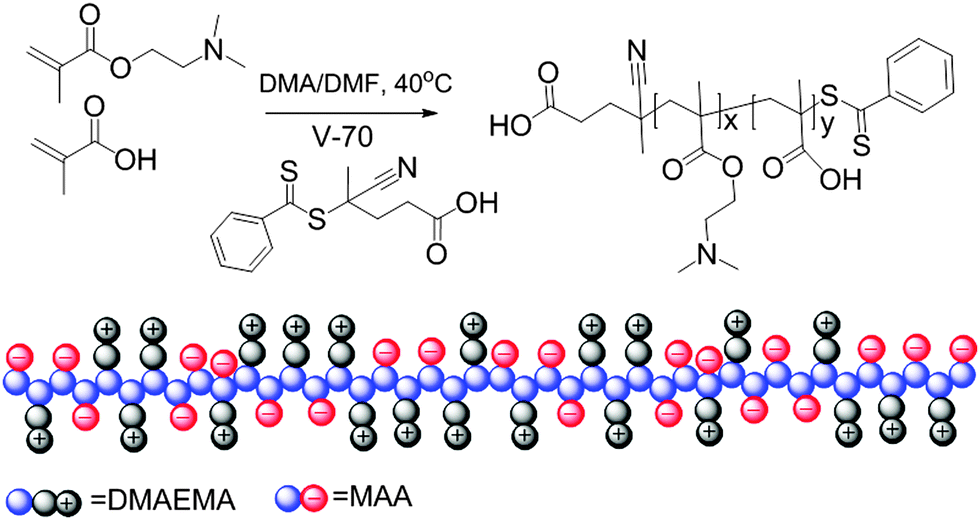 | ||
| Scheme 1 Schematic representation of the synthesis and structure of charge neutral polyampholytes prepared by stoichiometric RAFT copolymerization of anionic and cationic monomers. | ||
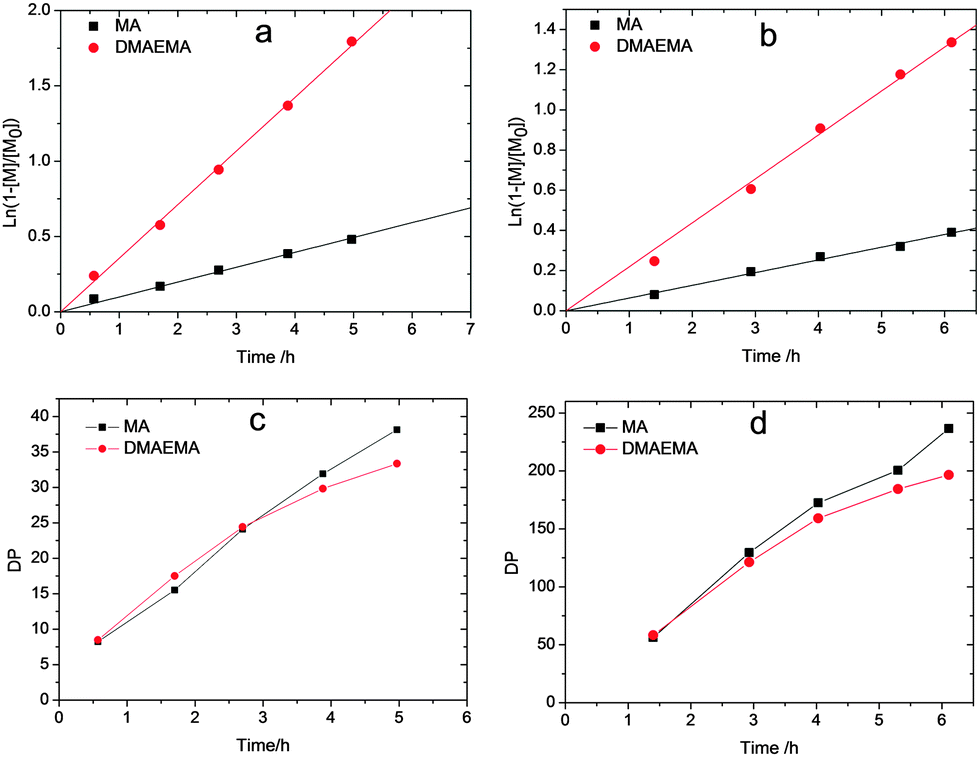 | ||
Fig. 1 (a, b) Kinetic plots and (c, d) DP versus reaction time plots for copolymerization of MAA and DMAEMA with feed ratio of MAA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMAEMA:CTA:V70 at (a, c) 100:40:1:0.1 and (b, d) 733:200:1:0.2, performed at 40 °C with V70 as initiator. :DMAEMA:CTA:V70 at (a, c) 100:40:1:0.1 and (b, d) 733:200:1:0.2, performed at 40 °C with V70 as initiator. | ||
In addition, the monomer sequence was analyzed by plotting the degree of polymerization (DP, resulting from conversion determined by GC) versus time for both monomers. As shown in Fig. 1c and d, the DP of both monomers increased similarly during the copolymerization indicating efficient suppression of the gradient formation by controlling the feed ratio of the two monomers providing access to quasi-random charge neutral copolymers. On the basis of these copolymerization kinetics, a series of well-defined copolymers with different DP and equimolar MAA/DMAEMA ratios was prepared (Table 1).
Considering the strong ionic interaction of the two monomers as well as the reported examples of UCST behavior of polybetaines, the copolymers were expected to show an UCST phase transition in water. However, all the synthesized copolymers were found to be well soluble in water above 0 °C, even PA2 with the highest molecular weight, indicating insufficient supramolecular attraction for UCST behavior. Hence, less polar solvents, namely methanol and ethanol, were added to the aqueous solutions as co-nonsolvent leading to clouding of the solutions at room temperature.
Therefore, the UCST thermoresponsive behavior of the copolymers was investigated by turbidimetry in alcohol–water solvent mixtures at a concentration of 5 mg ml−1. For this purpose polymer solutions in alcohol–water solvent mixtures were heated and cooled between 2 and 80 °C (for ethanol–water solvent mixtures) or 60 °C (for methanol–water solvent mixtures) at a heating–cooling rate of 1 K min−1 while stirring. Cloud point temperatures (TCPs) were determined at 50% transmittance of light at wavelengths of 600 nm during cooling of the polymer solutions. During the cooling of the polymer solution, a sharp transition from high transmittance (about 100%) to low transmittance (about 20%) was detected indicating phase separation of the solution and the aggregation of the copolymer in the binary solvent (Fig. 2).
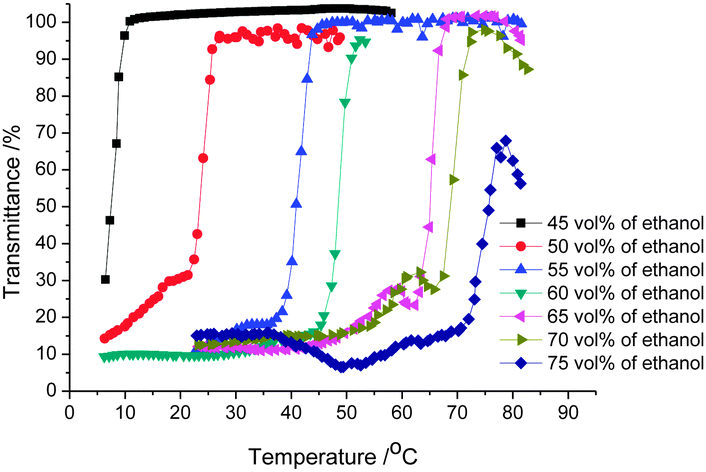 | ||
| Fig. 2 Transmittance versus temperature plots for 5 mg ml−1 PA1 solutions in different ethanol–water solvent mixtures. | ||
Fig. 3 displays the TCPs for PA1 and PA2 in alcohol–water solvent mixtures as a function of alcohol content and pH. Both of the copolymers show UCST thermoresponsive behavior in a wide range of ethanol content in the ethanol–water solvent mixtures. The TCPs for PA2 are higher than for PA1 in the same solvent mixtures at low content of ethanol indicating the expected molecular weight dependence of the UCST behavior, i.e. the higher the molar mass the stronger the interchain interaction the higher the TCP.17,33 With increasing ethanol content the TCPs increase due to the decrease in solvent polarity resulting in stronger electrostatic attraction. The slightly higher value of TCP obtained for PA1 than for PA2 at high ethanol content is within experimental error of the measurements since the copolymers show bad solubility in these solvent mixtures. The UCST behavior of PA1 was also investigated in methanol–water solvent mixtures, as shown in Fig. 3a. Higher alcohol content was needed for the polymer to show thermoresponsive behavior in methanol–water solvent mixture due to the higher polarity of methanol leading to better solvation of the polyampholyte. The influence of pH on the thermoresponsive behavior was also evaluated. As shown in Fig. 3b, a slight change of pH can greatly decrease the cloud point of PA2 in ethanol–water solvent mixtures since the solubility of PA is greatly increased with addition of either acid or base, which can increase the charge density of the copolymer.
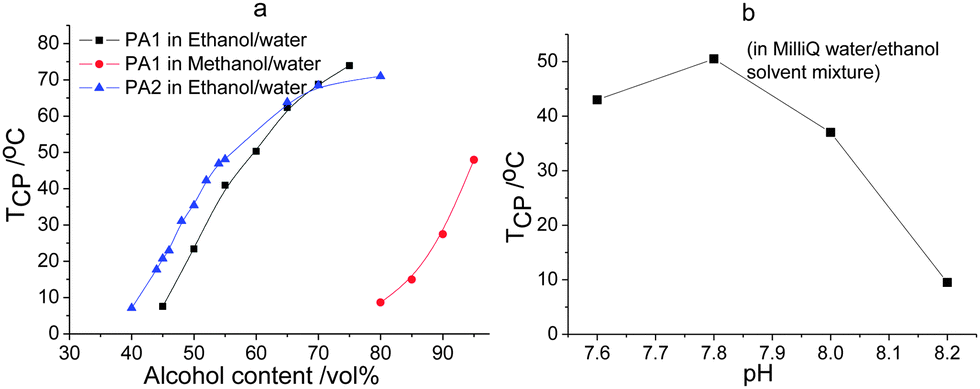 | ||
| Fig. 3 (a) UCST phase transition temperatures versus alcohol content for PA1 and PA2 in alcohol–water solvent mixtures (5 mg ml−1); and (b) UCST phase transition temperatures for PA2 versus pH in ethanol–water solvent mixtures with 60 vol% ethanol (4 mg ml−1) (lines added to guide the eye). | ||
This tunable thermoresponsive behavior of the charge-neutral polyampholytes resulting from strong interchain electrostatic attraction provides a platform for the preparation of more complex thermoresponsive copolymers. To explore this possibility, a brush copolymer polyampholyte was synthesized by copolymerization of the charged comonomers and a neutral oligo(ethylene glycol) methyl ether methacrylate (OEGMA, Mn = 480 Da) comonomer, as shown in Scheme 2. The statistical copolymer structure was employed in this report because of its much more facile synthesis compared to block copolymers, while self-assembly into well-defined structures is still possible as has been reported recently.48,49 The RAFT copolymerization was performed with a ratio of MAA:DMAEMA:OEGMA:CTA:V70 of 90:50:25:1:0.1. The use of this MAA to DMAEMA ratio of 1.8 led to a copolymer with identical DPs of MAA and DMAEMA, namely MAA38DMAEMA38OEGMAx. Unexpectedly, a previous trial with MAA to DMAEMA feed ratio of 2.5 did not lead to a charge balanced copolymer indicating a significant influence of the third comonomer on the copolymerization kinetics.
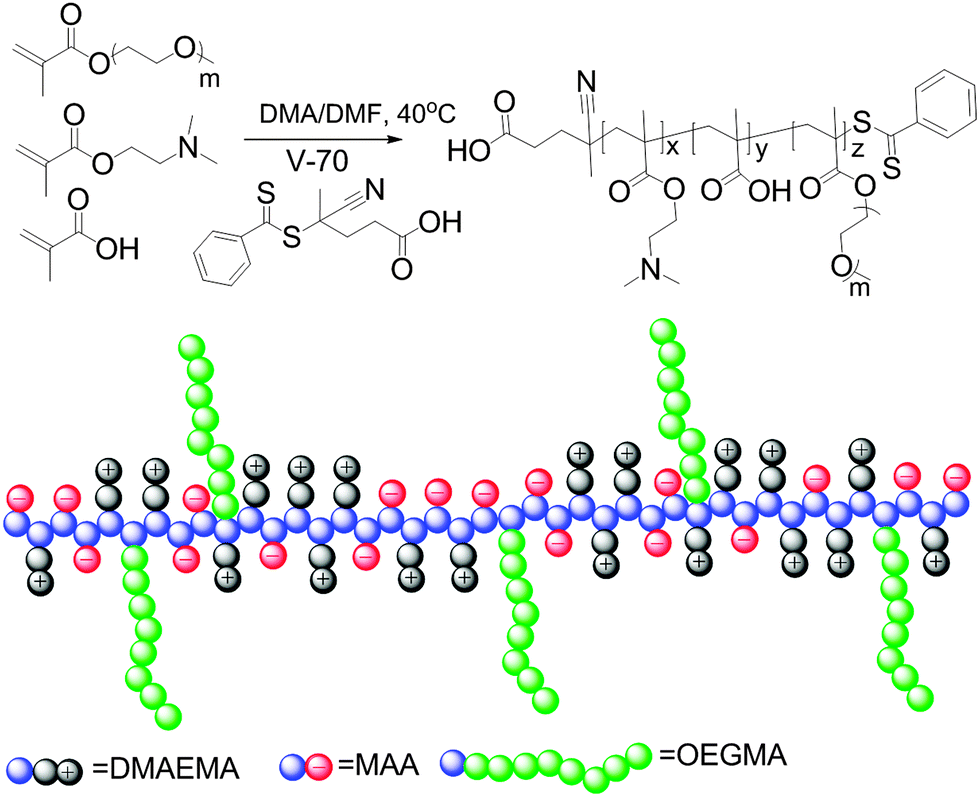 | ||
| Scheme 2 Schematic representation of the synthesis and structure of a polyampholyte with solvophilic side chains prepared by RAFT copolymerization of DMAEMA, MAA and mOEGMA. | ||
The temperature responsive UCST-based self-assembly behavior of the resulting copolymer was first evaluated in pure ethanol. A preliminary test of the solubility in ethanol revealed that PA3 is fully soluble in ethanol at room temperature, while PA1 and PA2 are not, expectedly indicating enhanced solubility resulting from incorporation of the OEGMA comonomer. Fig. 4 displays the size distribution, obtained by dynamic light scattering (DLS), of PA3 dissolved in ethanol at 1 mg ml−1 during cooling from 15 °C to 5 °C. The copolymer was completely soluble in ethanol at temperatures higher than 10 °C as indicated by the small size of 6 nm corresponding to unimers. By cooling down to 5 °C, the particle size dramatically increased to about 45 nm, together with a relatively low polydispersity index (PDI of 0.28), indicating the formation of defined nanostructures. The UCST-like temperature induced self-assembly of PA3 can be ascribed to collapse of the copolymer due to strong electrostatic interactions of the positive and negative charges, while the solvophilic ethylene glycol side chains serve as solvophilic corona that stabilizes the nano-structures and prevent further agglomeration.
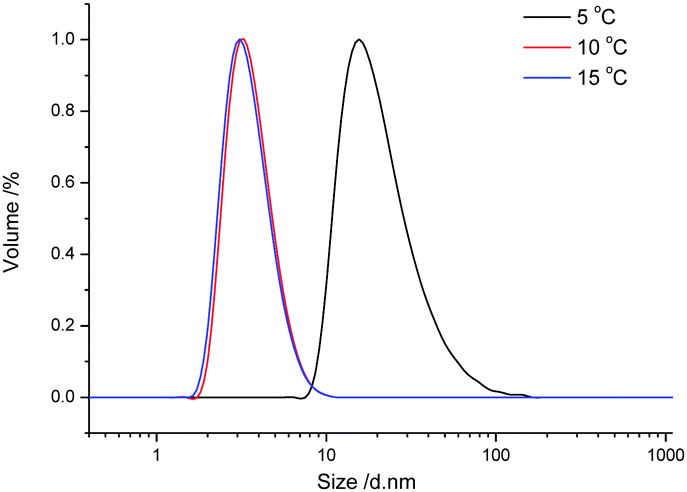 | ||
| Fig. 4 Size distribution at various temperatures of PA3 in ethanol (1 g L−1) determined by dynamic light scattering. | ||
The self-assembly behavior of copolymer PA3 was also investigated in isopropanol at 1 mg ml−1 upon cooling from 60 to 20 °C, as shown in Fig. 5. The aggregation of the copolymer in isopropanol happened at 50 °C, higher than in ethanol due to the lower polarity of the solvent. The particle size increased to about 35 nm when cooling down to 50 °C, together with the low PDI (0.17) indicating the formation of defined nanostructures. Interestingly, the size of the nanostructures gradually increased upon further cooling of the solution, which is most likely due to the formation of larger agglomerates due to further electrostatic assembly indicating that in isopropanol the steric stabilization by the OEG chains is insufficient to suppress the stronger electrostatic attraction. Noteworthy is that defined nanostructures are obtained at 45 °C and lower temperatures with PDIs below 0.10.
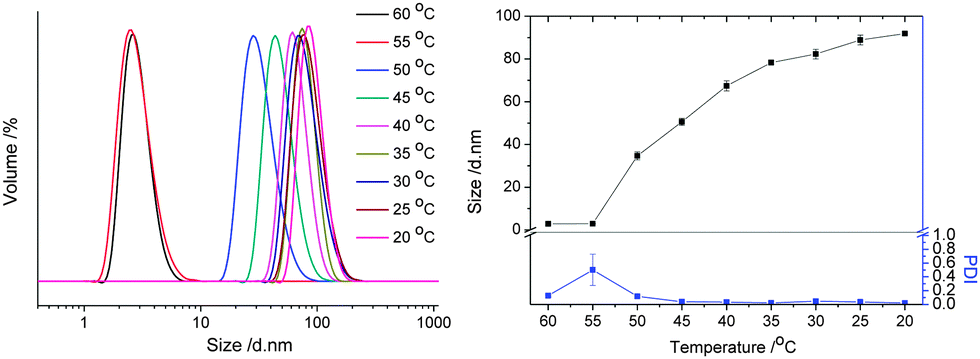 | ||
| Fig. 5 Left: size distribution at various temperatures, and right: Z-average size and PDI versus temperature of PA3 in isopropanol (1 g L−1) determined by dynamic light scattering (lines added to guide the eye). | ||
Direct RAFT radical copolymerization of cationic and anionic monomers revealed that the reactivity of the two monomers is severely influenced by their strong electrostatic interactions. However, fine-tuning of the feed ratio of the two monomers could suppress the gradient formation allowing the synthesis of quasi-random copolymers with equimolar amounts of the anionic and cationic charged monomers. The resulting charge neutral polyampholytes were found to show UCST behavior in various alcohol–water solvent mixtures. In addition, the UCST of the copolymers can be well tuned by varying the content of alcohol in the solvent mixtures. The temperature-dependent interchain electrostatic attraction of the copolymers can be used for UCST-based temperature controlled self-assembly. As a proof of concept, a polyampholyte with ethylene glycol side chains was synthesized that formed defined thermoresponsive nano-structures.
Q.Z. acknowledges Chinese Scholarship Council (2010629042) and Ghent University (BOF11/CHN/009) for the financial support; R.H. thanks FWO, Ghent University and the Interuniversity Attraction Pole Programme (P7/05) initiated by the Belgian Science Policy Office for financial support.
Notes and references
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074 CrossRef CAS PubMed.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459 CrossRef CAS.
- J.-F. Lutz, Adv. Mater., 2011, 23, 2237 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS PubMed.
- J.-F. Lutz and A. Hoth, Macromolecules, 2005, 39, 893 CrossRef.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978 CrossRef CAS PubMed.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686 CrossRef CAS PubMed.
- R. Obeid, E. Maltseva, A. F. Thuenemann, F. Tanaka and F. Winnik, Macromolecules, 2009, 42, 2204 CrossRef CAS.
- P.-F. Caponi, X.-P. Qiu, F. Vilela, F. M. Winnik and R. M. Ulijn, Polym. Chem., 2011, 2, 306 RSC.
- H. Schlaad, C. Diehl, A. Gress, M. Meyer, A. L. Demirel, Y. Nur and A. Bertin, Macromol. Rapid Commun., 2010, 31, 511 CrossRef CAS PubMed.
- R. Luxenhofer, Y. C. Han, A. Schulz, J. Tong, Z. J. He, A. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613 CrossRef CAS PubMed.
- S. Aoshima and S. Kanaoka, Adv. Polym. Sci., 2008, 169 CAS.
- A. Chilkoti, M. R. Dreher and D. E. Meyer, Adv. Drug Delivery Rev., 2002, 54, 1093 CrossRef CAS.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373 RSC.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898 CrossRef CAS PubMed.
- H. S. Frank and M. W. Evans, J. Chem. Phys., 1945, 13, 507 CrossRef CAS PubMed.
- F. Franks and D. J. G. Ives, Q. Rev., Chem. Soc., 1966, 20, 1 RSC.
- S. Noskov, G. Lamoureux and B. Roux, J. Phys. Chem. B, 2005, 109, 6705 CrossRef CAS PubMed.
- S. Piccarolo and G. Titomanlio, Makromol. Chem., Rapid Commun., 1982, 3, 383 CrossRef CAS.
- J. Cowie, I. McEwen and M. Garay, Polym. Commun., 1986, 27, 122 CAS.
- J. Cowie, M. Mohsin and I. McEwen, Polymer, 1987, 28, 1569 CrossRef CAS.
- R. Hoogenboom, S. Rogers, A. Can, C. R. Becer, C. Guerrero-Sanchez, D. Wouters, S. Hoeppener and U. S. Schubert, Chem. Commun., 2009, 5582 RSC.
- R. Hoogenboom, C. R. Becer, C. Guerrero-Sanchez, S. Hoeppener and U. S. Schubert, Aust. J. Chem., 2010, 63, 1173 CrossRef CAS.
- C. Pietsch, R. Hoogenboom and U. Schubert, Polym. Chem., 2010, 1, 1005 RSC.
- Q. Zhang, P. Schattling, P. Theato and R. Hoogenboom, Polym. Chem., 2012, 3, 1418 RSC.
- Q. Zhang, P. Schattling, P. Theato and R. Hoogenboom, Eur. Polym. J. DOI:10.1016/j.eurpolymj.2014.06.029.
- R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Soft Matter, 2008, 4, 103 RSC.
- H. M. L. Lambermont-Thijs, H. P. C. Van Kuringen, H. P. C. J. P. W. Van de Put, U. S. Schubert and R. Hoogenboom, Polymers, 2010, 2, 188 CrossRef CAS PubMed.
- R. O. R. Costa and R. F. S. Freitas, Polymer, 2002, 43, 5879 CrossRef CAS.
- M. J. A. Hore, B. Hammouda, Y. Li and H. Cheng, Macromolecules, 2013, 46, 7894 CrossRef CAS.
- L. Liu, T. Wang, C. Liu, K. Lin, G. Liu and G. Zhang, J. Phys. Chem. B, 2013, 117, 10936 CrossRef CAS PubMed.
- Y. Su, M. Dan, X. Xiao, X. Wang and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4399 CrossRef CAS.
- S. E. Kudaibergenov and A. Ciferri, Macromol. Rapid Commun., 2007, 28, 1969 CrossRef CAS.
- S. Kudaibergenov, W. Jaeger and A. Laschewsky, Adv. Polym. Sci., 2006, 157 CrossRef CAS.
- P. J. Roth, J. Y. Quek, Y. Zhu, B. M. Blunden and A. B. Lowe, Chem. Commun., 2014, 50, 9561 RSC.
- K. S. Pafiti, M. Elladiou and C. S. Patrickios, Macromolecules, 2014, 47, 1819 CrossRef CAS.
- E. Malmström and C. J. Hawker, Macromol. Chem. Phys., 1998, 199, 923 Search PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS.
- K. Matyjaszewski and N. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- R. Rajan, M. Jain and K. Matsumura, J. Biomater. Sci., Polym. Ed., 2013, 24, 1767 CrossRef CAS PubMed.
- T. K. Georgiou and C. S. Patrickios, Biomacromolecules, 2007, 9, 574 CrossRef PubMed.
- K. Sui, X. Zhao, Z. Wu, Y. Xia, H. Liang and Y. Li, Langmuir, 2011, 28, 153 CrossRef PubMed.
- D. Hu, Z. Cheng, J. Zhu and X. Zhu, Polymer, 2005, 46, 7563 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c4cc07930b |
| This journal is © The Royal Society of Chemistry 2015 |