
Dextransucrase-catalyzed elongation of polysaccharide brushes with immobilized mono-/di-saccharides as acceptors†
Yan
Fang
a,
Jian
Wu
b and
Zhi-Kang
Xu
*a
aMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: xuzk@zju.edu.cn
bDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China
First published on 3rd November 2014
Abstract
A quartz crystal microbalance (QCM) was used to monitor dextransucrase (DSase)-catalyzed polysaccharide elongation on the glucose-/maltose-ended self-assembly monolayer (SAM) surfaces. Kinetic parameters of the enzymatic elongation indicate that maltose is a promising substrate acceptor for DSase.
Polysaccharides are the core components of glycocalyx on the cell membrane surface, which are important in protein trafficking,1 viral and bacterial infections,2,3 and cell migration.4 Polysaccharide brushes are therefore more favourable as biomimetic surfaces in biomedical science and technology.5–7 However, it is extremely difficult to construct well-defined polysaccharide brushes because there are a series of challenges to perfectly control the regio- and stereo-chemistry of the saccharide chain via the ‘‘traditional’’ chemo-glycosylation reactions.8–10 Alternatively, polysaccharide brushes were recently constructed by enzyme-catalyzed elongation in vitro. This is a biomimetic process and is becoming increasingly promising due to its unique advantages such as green characteristics and regio-/stereo-selectivity. Enzymes from the glycosyltransferase family are the most used ones in this biomimetic process. They usually require activated substrates (nucleotides, saccharides and saccharide-1-phosphates) as saccharide donors and oligosaccharides (maltoheptaose/iso-maltoheptaose and oligo-dextran) as saccharide acceptors.11–16 However, the activated substrates are commonly unstable at ambient temperature and unimaginably expensive. The oligosaccharide acceptors are usually synthesized by a series of complicated reactions. Dextransucrase (DSase) is a kind of glycosyl-transferase that can catalyze the transfer of the D-glucose moiety from sucrose to a broad range of saccharide acceptors, including normal saccharides (mono-, di-, tri-, and oligo-saccharides) and unconventional ones (saccharide derivatives).17,18 It is clear that all these saccharides (as both donors and acceptors to this enzyme) are common and widely commercialized. Nevertheless, the DSase-catalyzed D-glucose moiety transfer and saccharide elongation are usually carried out in solution and the saccharide acceptors have a great effect on the enzymatic activity.17–20 Nihira and co-workers studied the DSase-catalyzed saccharide elongation on gold surfaces immobilized with dextran as an acceptor.11 They focused their attention mainly on monitoring the enzymatic process using a quartz crystal microbalance (QCM). Despite its acknowledged importance, the effect of saccharide acceptors has rarely been manipulated experimentally on the model or practical surfaces. Furthermore, the structures and properties of the resulted polysaccharide brushes are also not well understood. Therefore, we use the QCM to study the DSase-catalyzed saccharide elongation from the mono- and di-saccharide acceptors terminated self-assembly monolayer (SAM) on the gold surface. It is expected that the resulting polysaccharide brushes will have more defined structures than those obtained using dextran as an acceptor. At the same time, we aim to give detailed results to demonstrate that the enzymatic activity depends on the nature of the immobilized saccharide acceptors.21
The overall strategy is schematically shown in Fig. 1. Glucose and maltose are chemically attached to the SAM fabricated on the gold surface by a typical procedure (ESI,† Scheme S1).21–23 The average density is 1.27 nm−2 and 1.30 nm−2 for the immobilized mono- and di-saccharide acceptors, respectively (ESI,† Fig. S1). And the water contact angles are 36° ± 2° and 35° ± 2° for these saccharide-immobilized SAM surfaces (ESI,† Fig. S2). The DSase-catalyzed saccharide chain elongation is then monitored in real time using the QCM with sucrose as the donor. DSase from Leuconostoc mesenteroides catalyzes the transfer of the glucose moiety from sucrose to a saccharide acceptor and then to resulting dextran with 95% of α, 1–6 bonds and only 5% of α, 1–3 bonds.24 Two steps in the enzymatic elongation process are monitored continuously from the time dependencies of QCM frequency changes, which include (1) recognition and then binding of DSase to the immobilized glucose/maltose acceptors and (2) enzymatic elongation of the complementary saccharide chains in the presence of sucrose (ESI,† Fig. S3 and S4).25,26Fig. 2 shows the QCM response curves for the recognition and then the binding of DSase to the glucose- and maltose-immobilized SAM surfaces, respectively. It can be seen that an immediate decrease appears in the QCM frequency when the enzyme was injected over the surfaces. This frequency decrease is due to the specific binding of DSase to the saccharide acceptors, and it depends both on the enzyme concentration and the acceptor structure. For the glucose-immobilized SAM surface, the bound DSase increases from 0.15 to 1.16 pmol cm−2 (Fig. 2(a)) with the enzyme concentration ranging from 20 nM to 100 nM. At the same time, it increases from 1.54 to 7.73 pmol cm−2 (Fig. 2(b)) on the maltose-immobilized SAM surface. We can calculate the rate constant of enzyme binding (kon) and dissociation (koff) from these curves. The dissociation constant (Kd) is then obtained from the ratio of koff to kon (ESI,† Fig. S5 and S6 and eqn (S1)–(S4)). It is known that a low dissociation constant indicates a high specificity between the acceptor and the enzyme.27,28Table 1 summarizes the results. Compared with the glucose-immobilized surface (63.78 nM), Kd is obviously lower for DSase on the maltose-immobilized surface (35.09 nM). The values indicate that the affinity between DSase and the maltose acceptor is greater than that between DSase and the glucose acceptor.
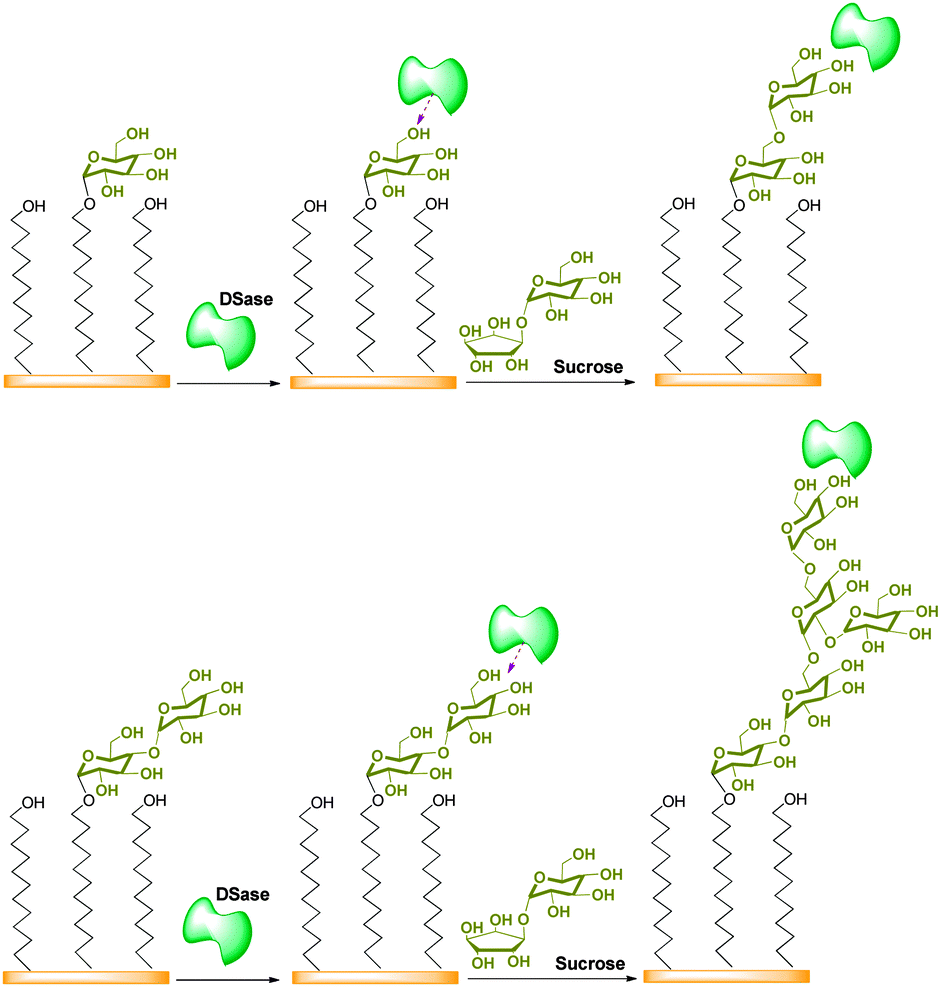 | ||
| Fig. 1 Overall strategy for the DSase-catalyzed elongation of polysaccharide chains to form brushes on the gold surface assembled with a monolayer and then immobilized with different saccharide acceptors (glucose/maltose). | ||
 | ||
| Fig. 2 Effect of enzyme concentration on DSase recognition/binding onto the saccharide-immobilized surfaces. The curves were recorded by a QCM for the surfaces with different saccharide acceptors: (a) glucose; (b) maltose. | ||
| Acceptor | k on (10−3 M−1 S−1) | k off (10−4 S−1) | K d (nM) | K m (mM) | k cat (S−1) |
|---|---|---|---|---|---|
| Glucose | 2.54 | 1.62 | 63.78 | 18.30 | 0.0036 |
| Maltose | 3.99 | 1.40 | 35.09 | 8.09 | 0.021 |
| Dextran11 | 89.00 | 16.00 | 17.98 | 3.40 | 3.50 |
The acceptor-bound DSase is expected to transfer the glucose donor from sucrose and form dextran brushes on the surfaces. Fig. 3(a) and (b) show the effect of sucrose concentration on the enzymatic elongation of the immobilized acceptors. The frequency decrease reflects a mass increase because sucrose accesses the active sites of the bound enzymes and results in the enzymatic attachment of the glucose moiety at the nonreducing end of the immobilized acceptors.21 The amount of attached glucose increases upon increasing the sucrose concentration from 1 to 15 mM. It is from 12.56 to 62.78 ng cm−2 in the case of glucose as the acceptor and from 368.81 to 627.75 ng cm−2 in the case of maltose as the acceptor. These results indicate that polysaccharide brushes are formed by the enzymatic elongation process. FT-IR/MR and XPS were used to analyze the chemical composition information of the polysaccharide brushes (ESI,† Fig. S7–S9). It is obvious that the XPS peak of saccharide acetal (O–C–O) enhances significantly due to the formation of polysaccharide brushes (ESI,† Fig. S8 and S9). ToF-SIMS spectra (ESI,† Fig. S10) also show a series of specific secondary fragment ions from the polysaccharide brushes. Besides, the surface morphology changes measured by AFM further confirm the enzymatic elongation of polysaccharides on the mono-/di-saccharide immobilized surfaces (ESI,† Fig. S11 and S12). Fig. 3(c) presents the plots for the initial elongation rate (ν0) against sucrose concentration. It can be seen that ν0 increases with the increase of sucrose concentration, which is reasonable since more saccharide donors can access the active sites of DSases.21 And ν0 is much higher for the maltose acceptor than that for the glucose one. Importantly, the DSase-catalyzed saccharide elongation can simply be described by the Michaelis–Menten equation (ESI,† eqn (S5)–(S7)). Fig. 3(d) shows the reciprocal plots of ν0 against sucrose concentration. We can calculate the Michaelis constant (Km) and the catalytic rate constant (kcat) from the slope and the intercept of the plots. As can be seen from Table 1, the Km and kcat values are 18.30 mM and 0.0036 S−1 for the immobilized glucose acceptor, respectively. By contrast, the Km and kcat values are 8.09 mM and 0.021 S−1 for the immobilized maltose acceptor, respectively. The Km value for maltose is higher than that obtained on the glucose immobilized surface and is almost consistent with the Km value obtained on the dextran acceptor immobilized surface (3.40 mM, ref. 11). It is worth noting that the Km value for the immobilized dextran acceptor is well consistent with that obtained in solution (Km = 3.00 mM).29 Previous work pointed out that the enzymatic activity of DSase would not be affected by the dextran acceptor immobilized surface. Our values indicate that the enzymatic activity of DSase has been largely affected by the glucose acceptor immobilized surface, thus the enzymatic elongation rate is much lower than the reported values. This result may be due to the use of short and stiff undecyl spacers and the low affinity between the enzyme and the glucose acceptor.30,31 Meanwhile, it indicates that the enzymatic activity of DSase is not affected largely on the maltose acceptor immobilized surface. This is because the affinity between the enzyme and the maltose acceptor is high, and the decomposition rate of the enzyme–acceptor complex is very small (koff = 0.00014 S−1). DSase can quickly proceed to elongation (kcat = 0.021 S−1) after binding to maltose (consequent formation of the enzyme–acceptor complex). These features were also observed for the enzyme-catalyzed elongation of saccharides in solution.32,33
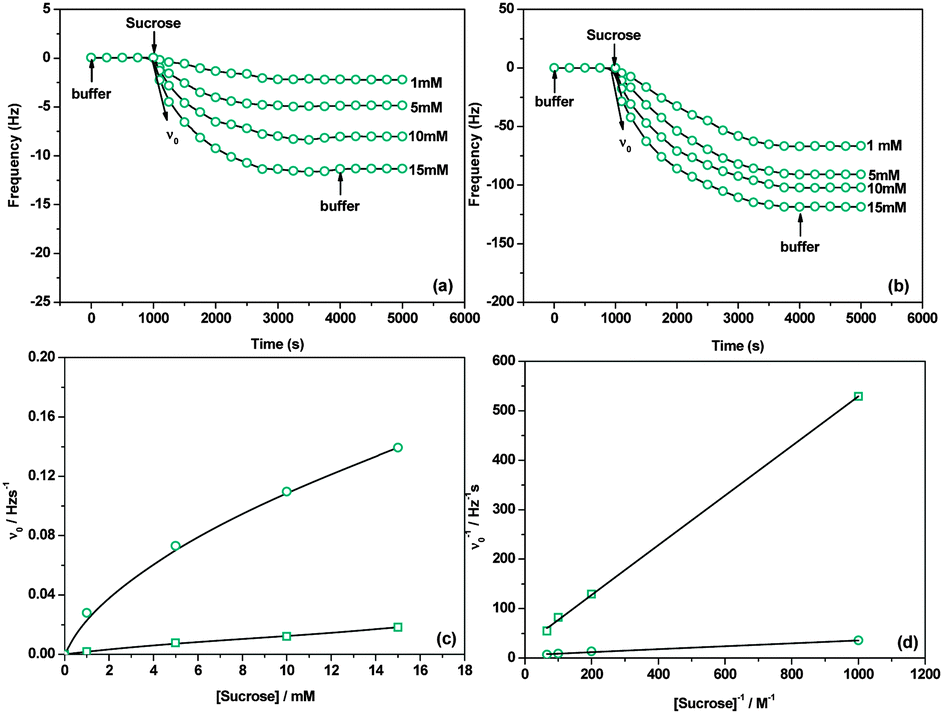 | ||
| Fig. 3 (a and b) QCM curves for the enzymatic elongation on the glucose/maltose-immobilized surfaces, (c) dependence of the initial elongation rate (ν0) on sucrose concentration, (d) reciprocal plots of ν0 against sucrose concentration ((□: glucose as the acceptor; ○: maltose as the acceptor). 0.78 pmol cm−2 of DSase was bound previously to the surface to induce the reaction). | ||
The synthesized polysaccharide (dextran) brushes are essentially linear polymers (α, 1–6 bonds) containing 5% of α, 1–3-linked branches with shorter side-chains.34,35 Thus, more nonreducing residues would be available for lectins to bind. Con A is a well-known mannosyl/glucosyl-specific lectin.36,37 An injection of Con A (20 μg mL−1) results in 162 Hz (7.97 pmol cm−2) and 165 Hz (8.12 pmol cm−2) binding response to the glucose and maltose acceptor immobilized surfaces, respectively. It increases to 200 Hz (9.80 pmol cm−2) and 360 Hz (17.72 pmol cm−2), 1.23-fold and 2.18-fold, for the surfaces after enzymatic elongation (Fig. 4). Obviously, more nonreducing glycosyl residues have been introduced onto the maltose acceptor immobilized surface. It further suggests that the maltose acceptor is more efficient than the glucose one in the DSase-catalyzed elongation of polysaccharide brushes. Besides, Fig. 4 also indicates that methyl α-D-mannopyranoside (MM) can be used to dissociate almost all lectins from the studied surfaces with an exception of the maltose acceptor immobilized surface after enzymatic elongation. Only about 40% Con A can be dissociated from this surface. It may be attributed to multivalent interaction between Con A and the polysaccharide brushes.38 These brushes show specific adsorption to Con A but high resistance to ricinus communis agglutinin 120 (RCA120) and bovine serum albumin (BSA) (ESI,† Fig. S13–S16). Fluorescence images of the surfaces also demonstrate the highly specific adsorption of Con A on the polysaccharide brushes (ESI,† Fig. S17 and S18). This indicates that our glycocalyx-like polysaccharide brushes are effective for detecting the saccharide–protein interaction.
 | ||
| Fig. 4 QCM curves for the specific adsorption and the affinity elution of Con A on the fabricated surfaces. (□: the glucose acceptor immobilized surface; ○: the glucose acceptor immobilized surface after enzymatic elongation; △: the maltose acceptor immobilized surface; ◇: the maltose acceptor immobilized surface after enzymatic elongation.) (Con A: 20 μg mL−1, MM: 100 mM). | ||
In conclusion, we have verified that DSase-catalyzed polysaccharide elongation can be directly carried out on the mono-/disaccharide acceptors (glucose and maltose) immobilized SAM surfaces. The saccharide acceptors have a great effect on the enzymatic activity of DSase. Although the enzyme-catalyzed polysaccharide elongation is demonstrated on the model surface, it can be easily extended to other surfaces for practical applications. Polysaccharide brushes have great potential in biomedical technologies as biomaterials. Therefore, the described DSase-catalyzed polysaccharide elongation is also of significant promise in construction of glycocalyx-like biomimetic surfaces with specific saccharide–protein interaction.
Financial support from the National Natural Science Foundation of China (Grant No. 50933006) is acknowledged. The authors also thank discussion and suggestion from Dr Ling-Shu Wan in the early stage of this work.
Notes and references
- P. Sears and C. H. Wong, Cell. Mol. Life Sci., 1998, 54, 223 CrossRef CAS.
- B. B. Finlay and P. Cossart, Science, 1997, 276, 718 CrossRef CAS.
- G. M. Whitesides, Chem. Biol., 1996, 3, 97 CrossRef.
- S. C. Evans, A. Youakim and B. D. Shur, BioEssays, 1995, 17, 261 CrossRef CAS PubMed.
- W. T. E. Bosker, K. Patzsch, M. A. C. Stuart and W. Norde, Soft Matter, 2007, 3, 754 RSC.
- K. J. Rosenberg, T. Goren, R. Crockett and N. D. Spencer, ACS Appl. Mater. Interfaces, 2011, 3, 3020 CAS.
- W. T. E. Bosker, M. A. C. Stuart and W. Norde, Langmuir, 2013, 29, 2667 CrossRef CAS PubMed.
- W. T. E. Bosker, K. Ágoston, M. A. C. Stuart, W. Norde, J. W. Timmermans and T. M. Slaghek, Macromolecules, 2003, 36, 1982 CrossRef CAS.
- E. Sisu, W. Bosker, W. Norde, T. Slaghek, J. Timmermans, J. Peter-Katalinić, M. C. A. Stuart and A. Zamfir, Rapid Commun. Mass Spectrom., 2006, 20, 209 CrossRef CAS PubMed.
- W. J. Ma, X. B. Yuan, C. S. Kan, T. Su, X. Y. Yuan, P. Y. Pu and J. Sheng, Carbohydr. Polym., 2008, 72, 75 CrossRef CAS PubMed.
- T. Nihira, T. Mori, M. Asakura and Y. Okahata, Langmuir, 2011, 27, 2107 CrossRef CAS PubMed.
- J. v. d. Vlist, I. Schönen and K. Loos, Biomacromolecules, 2011, 12, 3728 CrossRef PubMed.
- R. Šardzík, A. P. Green, N. Laurent, P. Both, C. Fontana, J. Voglmeir, M. J. Weissenborn, R. Haddoub, P. Grassi, S. M. Haslam, G. Widmalm and S. L. Flitsch, J. Am. Chem. Soc., 2012, 134, 4521 CrossRef PubMed.
- C. Clé, A. P. Gunning, K. Syson, L. Bowater, R. A. Field and S. Bornemann, J. Am. Chem. Soc., 2008, 130, 15234 CrossRef PubMed.
- T. Mori, M. Asakura and Y. Okahata, J. Am. Chem. Soc., 2011, 133, 5701 CrossRef CAS PubMed.
- K. Loos, V. v. Braunmühl and R. Stadler, Macromol. Rapid Commun., 1997, 18, 927 CrossRef CAS.
- A. M. Pereira, F. A. A. Costa, M. I. Rodrigues and F. Maugeri, Biotechnol. Lett., 1998, 20, 397 CrossRef CAS.
- K. Demuth, H. J. Jördening and K. Buchholz, Carbohydr. Res., 2002, 337, 1811 CrossRef CAS.
- C. Kubik, B. Sikora and S. Bielecki, Enzyme Microb. Technol., 2004, 34, 555 CrossRef CAS PubMed.
- G. Richard, S. Morel, R. M. Willemot, P. Monsan and M. R. Simeon, Carbohydr. Res., 2003, 338, 855 CrossRef CAS.
- Y. Fang, W. Xu, J. Wu and Z. K. Xu, Chem. Commun., 2012, 48, 11208 RSC.
- B. T. Houseman and M. Mrksich, Angew. Chem., Int. Ed., 1999, 38, 782 CrossRef CAS.
- F. J. Muñoz, J. Pérez, A. Rumbero, J. I. Santos, F. J. Cañada, S. André, H. J. Gabius, J. J. Barbero, J. V. Sinisterra and M. J. Hernáiz, Bioconjugate Chem., 2009, 20, 673 CrossRef PubMed.
- M. Naessens, A. Cerdobbel, W. Soetaert and E. J. Vandamme, J. Chem. Technol. Biotechnol., 2005, 80, 845 CrossRef CAS.
- H. Matsudo, K. Niikura and Y. Okahata, Chem. – Eur. J., 2001, 7, 3305 CrossRef.
- Y. Okahata, T. Mori, H. Furusawa and T. Nihira, Springer Ser. Chem. Sens. Biosens., 2007, 5, 341 CAS.
- A. Frankel, N. Yadav, J. Lee, T. L. Branscombe, S. Clarke and M. T. Bedford, J. Biol. Chem., 2002, 277, 3537 CrossRef CAS PubMed.
- Y. Fang, W. Xu, X. L. Meng, X. Y. Ye, J. Wu and Z. K. Xu, Langmuir, 2012, 28, 13318 CrossRef CAS PubMed.
- M. Kitaoka and J. F. Robyt, Carbohydr. Res., 1999, 320, 183 CrossRef CAS.
- N. Laurent, R. Haddoub and S. L. Flitsch, Trends Biotechnol., 2008, 26, 328 CrossRef CAS PubMed.
- M. Dols, M. R. Simeon, R. M. Willemot, M. Vignon and P. Monsan, Appl. Environ. Microbiol., 1998, 64, 1298 CAS.
- H. Nishino, A. Murakawa, T. Mori and Y. Okahata, J. Am. Chem. Soc., 2004, 126, 14752 CrossRef CAS PubMed.
- A. S. Ruiz, S. Serna, N. Ruiz, M. M. Lomas and N. C. Reichardt, Angew. Chem., 2011, 123, 1841 CrossRef.
- R. Irague, S. Massou, C. Moulis, O. Saurel, A. Milon, P. Monsan, M. R. Simeon, J. C. Portais and G. P. Véronesè, Anal. Chem., 2011, 83, 1202 CrossRef CAS PubMed.
- R. Irague, A. R. Sabaté, L. Tarquis, J. L. Doublier, C. Moulis, P. Monsan, M. R. Siméon, G. P. Véronesè and A. Buléon, Biomacromolecules, 2012, 13, 187 CrossRef CAS PubMed.
- O. Renaudet and P. Dumy, Org. Lett., 2003, 5, 243 CrossRef CAS PubMed.
- Z. Peia, H. Andersonb, T. Aastrup and O. Ramström, Biosens. Bioelectron., 2005, 21, 60 CrossRef PubMed.
- C. H. Liang, S. K. Wang, C. W. Lin, C. C. Wang, C. H. Wong and C. Y. Wu, Angew. Chem., Int. Ed., 2011, 50, 1608 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc06137c |
| This journal is © The Royal Society of Chemistry 2015 |