
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic SNAr of unactivated aryl chlorides†
James W.
Walton
*ab and
Jonathan M. J.
Williams
a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: james.walton@durham.ac.uk
First published on 6th October 2014
Abstract
We present nucleophilic aromatic substitution of unsubstituted aryl chlorides via a mechanism that is catalytic in [CpRu(p-cymene)]PF6 and involves a Ru(η6-arylchloride) intermediate. From the spectroscopic evidence we infer that arene exchange is the rate limiting step in this process and develop several new Ru(II) complexes that lower the activation barrier to arene exchange.
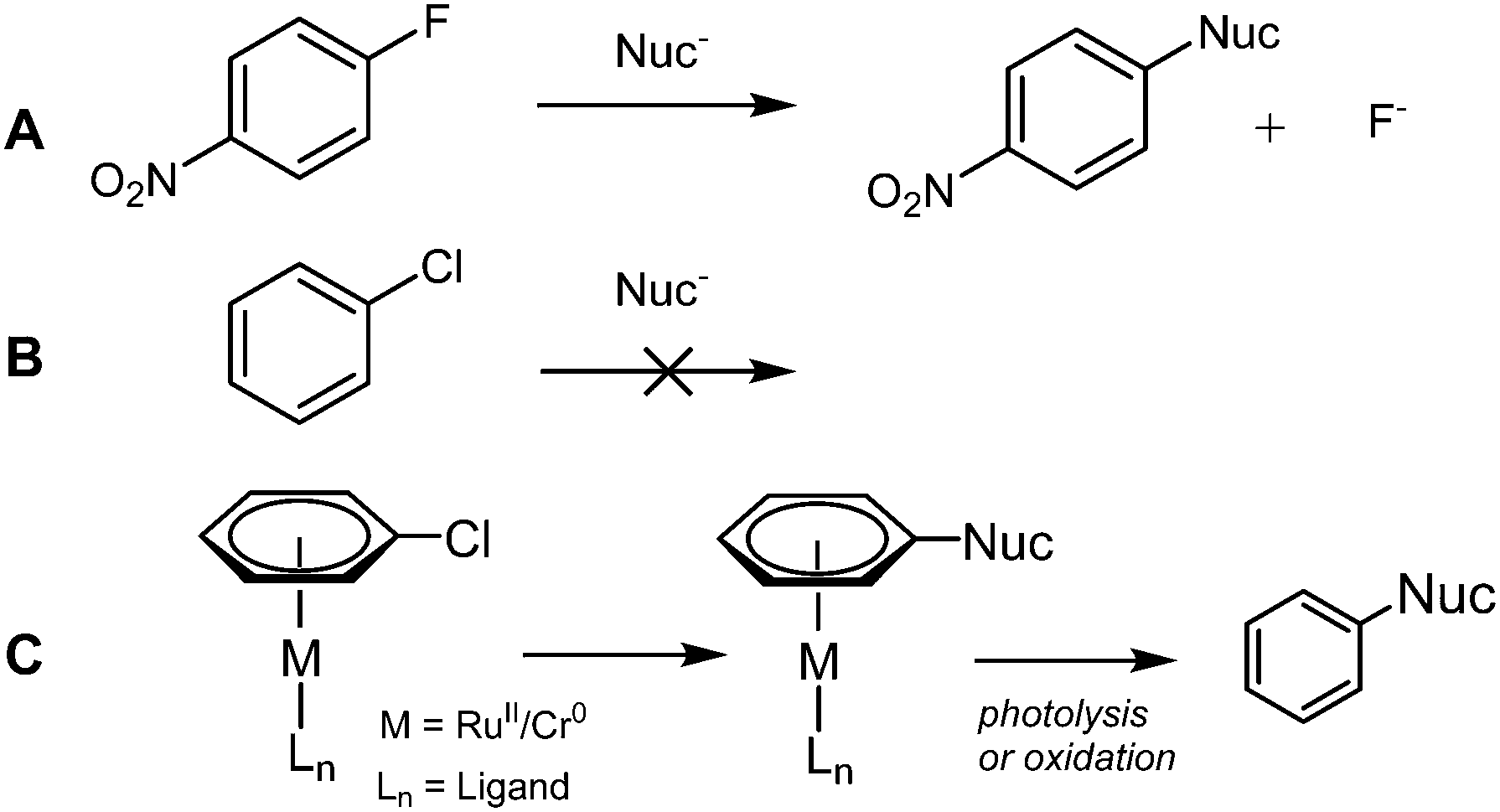
Nucleophilic aromatic substitution (SNAr) is one of the building blocks of synthetic chemistry, and is used far and wide in industry and academia. However, limitations restrict the use of SNAr to certain substrates. Activated arenes are required (incorporating an electron withdrawing group to stabilise the negatively charged intermediate, Scheme 1A) and the C–X bond must be polarised; the rate of reaction follows the order X = F > Cl ≫ Br. Alternative arylation methods include metal1a,b and non-metal1c catalysed coupling reactions of Ar–X and Ar–H.
 | ||
| Scheme 1 | ||
Fluorobenzene can undergo SNAr with strong alkoxide nucleophiles2 but chlorobenzene does not undergo SNAr (Scheme 1B). One method by which unactivated aryl halides (i.e. those without an electron withdrawing group) can undergo SNAr is via η6-coordination to a transition metal (e.g. Ru(II),3 Cr(0),4 Fe(II),5 Mn(I),6Scheme 1C). It is well established that π-complexation of an arene to a transition metal increases its reactivity towards nucleophilic attack7 and deprotonation.8 More recently, activation of a Cr(0) η6-aryl C–H bond towards Pd insertion and subsequent arylation has been achieved.9 The drawback to these reactions is that the η6-arene–metal bond is strong and stoichiometric metal is required; liberation of the aryl product is carried out by photolysis10 or oxidation.11
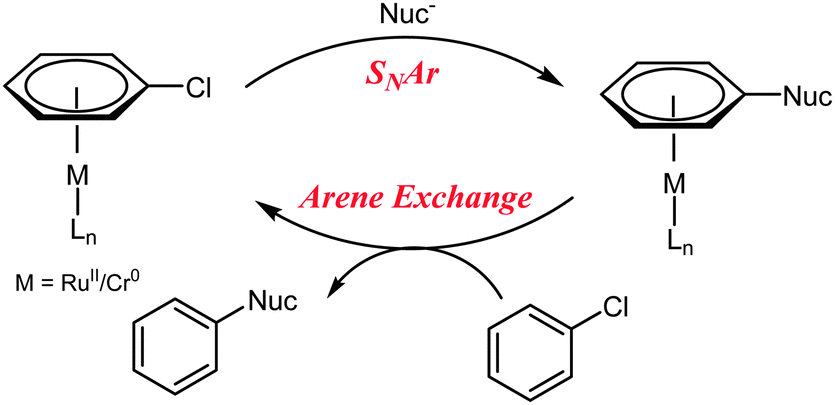
Semmelhack et al. propose a catalytic cycle in which an unactivated aryl chloride binds η6 to Cr(0), facilitating SNAr, before exchange between the bound product and free aryl chloride takes place, completing the catalytic cycle (Scheme 2).12 Despite demonstrating an increase in the rate limiting step (arene exchange), no catalytic SNAr was reported.
 | ||
| Scheme 2 | ||
Rh(III) catalysed intramolecular SNAr based on this catalytic cycle has been reported but is limited to aryl fluorides.13 Intermolecular SNAr of aryl fluorides (Ru(II) catalysed) has also appeared14 but requires a large excess of arene and is unsuccessful with aryl chlorides. Anti-Markovnikov hydroamination of styrene via catalytic π-complexation to Ru(II) has also been reported.15
In this report we present the first example of catalytic SNAr of unactivated aryl chlorides. Experimental evidence suggests that our method proceeds via a η6-coordination mechanism. In an effort to reduce the high reaction temperature and long reaction time, several new catalysts were synthesised with a view to lowering the activation barrier of the rate limiting step.
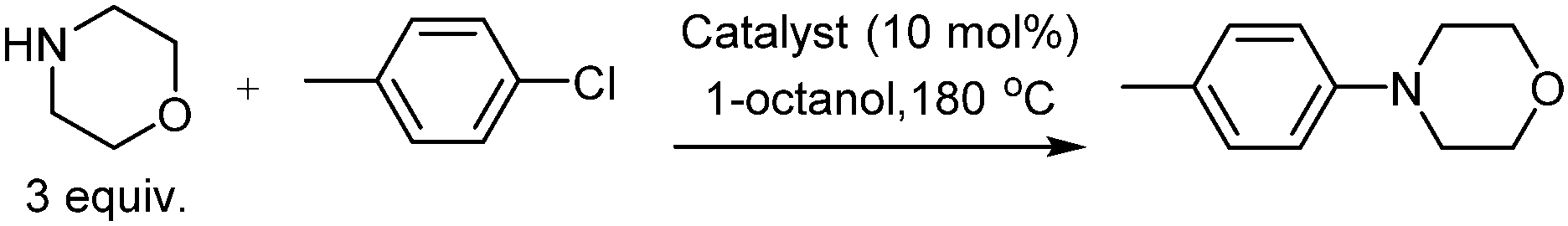
Our initial investigation focussed on the reaction between morpholine and chlorotoluene under a variety of reaction conditions (selected results are given in Table 1, see ESI† for a complete set of reaction conditions). In the absence of a catalyst no product formed, despite attempts over a range of temperatures and in several solvents (Table 1, entries 1–3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Time | Additive | Conversion (%) |
| 1 | — | MeCN | 80 | 18 h | — | 0 |
| 2 | — | Toluene | 110 | 18 h | — | 0 |
| 3 | — | Cyclohexanone | 150 | 18 h | — | 0 |
| 4 | [CpRu(MeCN)3]PF6 | Cyclohexanone | 150 | 18 h | — | 6 |
| 5 | [Cp*Ru(MeCN)3]PF6 | Cyclohexanone | 150 | 18 h | — | 5 |
| 6 | [CpRu(p-cymene)]PF6 | Cyclohexanone | 150 | 18 h | — | 10 |
| 7 | [CpRu(p-cymene)]PF6 | Cyclohexanone | 180 | 18 h | — | 18 |
| 8 | [CpRu(p-cymene)]PF6 | DMI | 180 | 18 h | — | 17 |
| 9 | [CpRu(p-cymene)]PF6 | Cyclohexanol | 180 | 18 h | — | 16 |
| 10 | [CpRu(p-cymene)]PF6 | p-Xylene | 180 | 18 h | — | 0 |
| 11 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 18 h | — | 25 |
| 12 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 18 h | Mol. sieves | 25 |
| 13 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 18 h | Et3N | 21 |
| 14 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 18 h | Na2CO3 | 11 |
| 15 | [Ru(p-cymene)Cl2]2 | 1-Octanol | 180 | 18 h | DPPPent | 0 |
| 16 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 4 d | — | 45 |
| 17 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 7 d | — | 75 |
| 18 | [CpRu(p-cymene)]PF6 | 1-Octanol | 180 | 14 d | — | 90 |

Based on the work of Woodgate et al.,3a we anticipated that catalysts incorporating a [CpRu]+ fragment (Cp = cyclopentadienyl) would expedite the SNAr reaction, via η6-coordination of chlorotoluene. A number of catalysts were investigated (Table 1, entries 4–6). Each catalyst led to conversion into the desired product, providing that high temperatures were employed, with [CpRu(p-cymene)]PF6 performing best. In a solvent screen at 180 °C, 1-octanol was found to give the highest conversion (Table 1, entries 7–11). If the reaction mixture was left for 14 days the product conversion reached 90% (Table 1, entries 16–18). The effect of additives on the rate of reaction was examined (Table 1, entries 12–15). No increase in product conversion was observed for a variety of additives, including base, molecular sieves and ligands. In summary, catalytic SNAr between morpholine and chlorotoluene can be achieved with 90% yield in 1-octanol at 180 °C in 14 days.
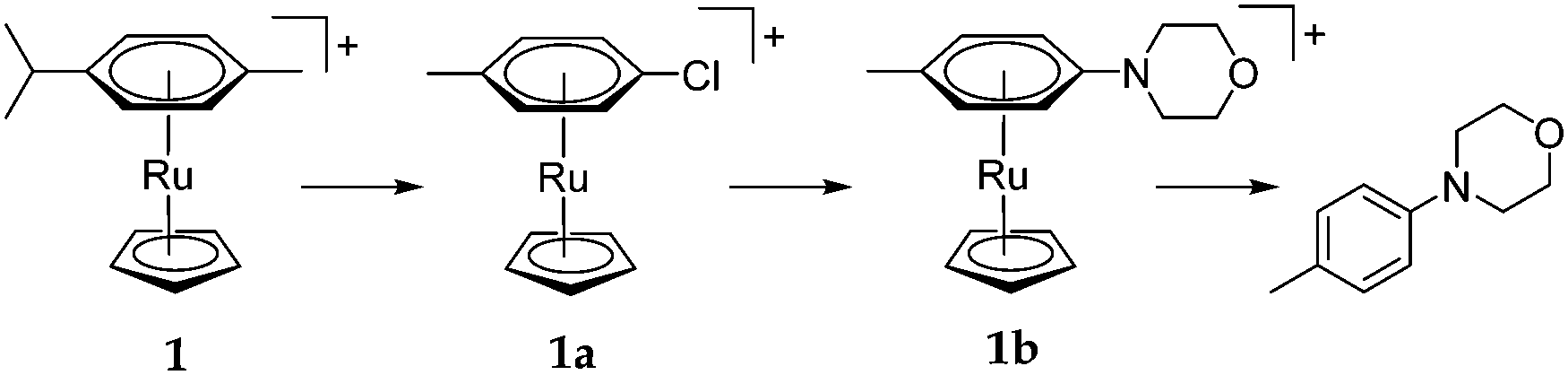
We sought to understand why such long reaction times and high reaction temperatures were required. Under the optimised reaction conditions, positive mode electrospray mass spectrometry (ESI-MS+) showed a peak at m/z = 344 with the characteristic Ru isotope pattern, corresponding to the η6-bound product (complex 1b in Scheme 3). 1H-NMR showed a pair of doublets in the range 5.5–5.7 ppm, also corresponding to 1b; no η6-bound chlorotoluene (1a) was observed by ES-MS+ or 1H-NMR. We postulate, therefore, that the reaction occurs via the mechanism shown in Scheme 2 and that arene exchange is the rate limiting step; SNAr is fast so that no bound chlorotoluene is observed but arene exchange is slow so that bound product is observed.
 | ||
| Scheme 3 | ||
The mechanism by which arene exchange takes place in [Cr(CO)3(η6-arene)] complexes has been well studied.16 It was determined that the rate-determining step is an initial change in arene bonding from η6 → η4 (Scheme 4). While similar analysis has not been carried out for RuII(η6-arene) complexes, we assume the same mechanism for arene exchange and propose that the success found for our catalytic system with 1-octanol as a solvent is due to coordination of the hydroxyl group during the slow η6 → η4 step. This maintains an 18 e− complex and lowers the activation energy. This theory is supported by the reasonable product conversion in solvents such as cyclohexanol and DMI (Table 1, entries 8 and 9) and the lack of conversion in the non-coordinating solvent p-xylene (Table 1, entry 10). It has previously been noted that addition of ketones, such as cyclohexanone, lowers the activation barrier to arene exchange in Cr(0) systems via coordination of the carbonyl lone pair during the η6 → η4 process.17
 | ||
| Scheme 4 | ||
To test this theory, several new catalysts were synthesised (2–6), each incorporating a tether, covalently bound to the Cp ring, capable of coordinating to Ru during the η6 → η4 step (Scheme 4).18 Our expectation was that intramolecular coordination of the tether to the Ru centre would lower the activation barrier to arene exchange and, subsequently, increase the rate of the SNAr reaction.
Each complex was synthesised by reaction between [Ru(p-cymene)Cl2]2 and the corresponding Cp–tether adduct, in the presence of Na2CO3 and ethanol (Scheme 5).19 The reaction mixtures were treated with NH4PF6 to afford complexes 2–6. The formation of the Cp–tether adducts were specific to the various tethers. For complex 2, CpNa and BrCH2CO2Me were stirred in THF at 0 °C to give CpCH2CO2Me in quantitative yield20 before complexation with [Ru(p-cymene)Cl2]2.21 Similar protocols were used to synthesise the other Cp–tether compounds. In certain cases, it was necessary to synthesise the electrophilic component before reaction with CpNa. For example, in the synthesis of 5, 2-Py(CH2)2OH (2-Py = 2-pyridine) was converted into 2-Py(CH2)2OSO2Me, prior to reaction with CpNa. Each complex 2–6 was purified by column chromatography on silica and fully characterised. For full Experimental detail see ESI.†

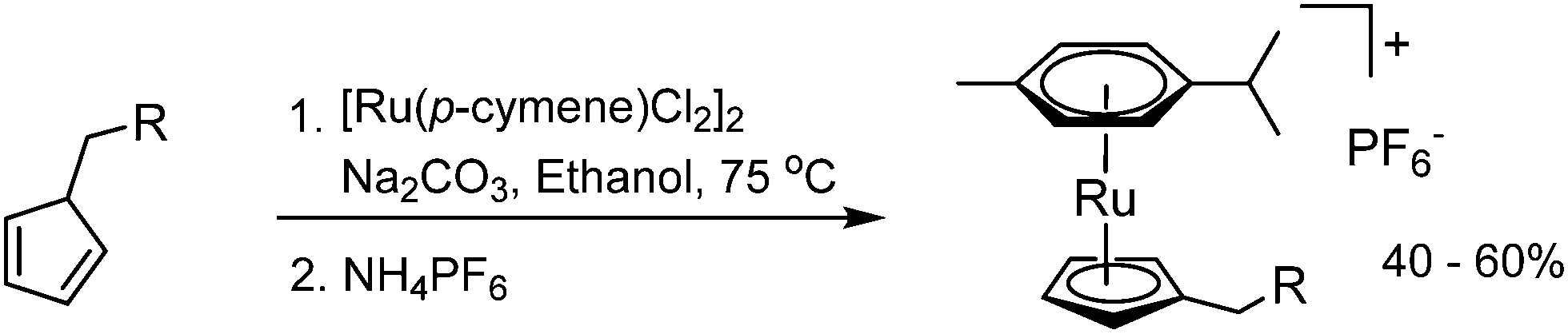 | ||
| Scheme 5 | ||
To investigate whether the presence of the various tethers would decrease the energy barrier for the η6 → η4 process, exchange experiments were carried out. Each complex was heated in either 1-octanol or cyclohexanone in the presence of 100 equivalents of hexamethylbenzene (C6Me6) for 16 h. The extent of exchange between p-cymene and C6Me6 was measured at 3 h and 16 h using ESI-MS+ (Table 2); calibration of the ESI-MS+ measurement was required to allow quantitative assessment (see ESI†). Relative to the parent complex, [CpRu(p-cymene)]PF6 (1), a moderate increase in the extent of arene exchange was observed for 3 and 6 in cyclohexanone after 16 h (51% and 50%, respectively versus 38% for 1). Most notable, however, was the complete arene exchange in cyclohexanone for complex 5, which incorporates a pyridyl tether. In the same solvent, 2 appeared to undergo ester hydrolysis and decarboxylation, giving [(CpMe)Ru(p-cymene)]+, indicated by a m/z peak at 315. This species underwent a similar amount of arene exchange to 1. Finally, exchange for 4 in cyclohexanone could not be determined due to a side reaction with the solvent. Similar exchange behaviour was found when 1-octanol was used as the solvent. Complex 5 again displayed the greatest amount of arene exchange. The tether components in 3 and 5 rapidly reacted with the solvent to form octyl esters, which exchanged at a similar rate to the parent complex (1). Once again, complex 2 decarboxylated whilst 6 showed a slight reduction in the rate of exchange compared to 1.
|
|
||||
|---|---|---|---|---|
| Complex | Arene exchange (%) | |||
| Cyclohexanone | 1-Octanol | |||
| 3 h | 16 h | 3 h | 16 h | |
| a Values for decarboxylated 2 ([(MeCp)Ru(p-cymene)]+), which forms under exchange conditions. b Values for the octyl ester of 3, which forms in 1-octanol. c Complex 4 reacts with cyclohexanone, leading to invalid results. d Values for the octyl ester of 4, which forms under the exchange conditions. | ||||
| 1 | 6 | 38 | 17 | 92 |
| 2 | (6)a | (50)a | (15)a | (84)a |
| 3 | 9 | 51 | 13 (18)b | 90 (88)b |
| 4 | —c | —c | (17)d | (85)d |
| 5 | 44 | 100 | 36 | 100 |
| 6 | 12 | 50 | 12 | 65 |

Having established that arene exchange could be accelerated by the presence of a coordinating tether, we proceeded to calculate half lives (t1/2) for the initial p-cymene complexes of 1, 3, 5 and 6 under the exchange conditions described in Table 2, with cyclohexanone as the solvent. The results confirm that the pyridyl complex 5 exchanges an order of magnitude faster than the parent complex, 1 (t1/2 = 2.2 ± 0.1 h versus 34 ± 0.7 h, see ESI† for details). Complexes 3 and 6 each have shorter t1/2 than 1 (23 ± 0.3 h and 28 ± 0.4 h, respectively), confirming that the rate of arene exchange is accelerated by the coordinating tether.
Our hypothesis stated that faster arene exchange will lead to higher conversion in the SNAr reaction between morpholine and chlorotoluene. To test this, we carried out the SNAr reaction with three of the new potential catalysts, 1, 3 and 5 (Table 3). As a comparison, 1 with an equivalent of free pyridine was also tested.
|
|
|||
|---|---|---|---|
| Catalyst | Conversion (%) | ||
| 1 d | 4 d | 7 d | |
| 1 | 25 | 45 | 75 |
| 3 | 25 | 59 | 72 |
| 5 | 26 | 55 | 54 |
| 1 + Py | 24 | 59 | 65 |
After 24 h, each catalyst gave a similar conversion. After 4 d, the catalysts incorporating a tether showed a small improvement in conversion but after 7 d the parent compound returned the highest value, due, in the case of 5, to degradation of the catalytic species (indicated by loss of Ru species in the mass spectrum). Once again, mass spectrometric analysis after 24 h shows a m/z peak corresponding to the η6-bound product in each of the reactions. Ultimately, it appears that although the rate of arene exchange is accelerated in the p-cymene → C6Me6 experiment described above, this acceleration does not translate into a higher conversion in the SNAr reaction. Two explanations present themselves: (1) the η6-bound product is more kinetically inert than [CpRu(p-cymene)]+, so that arene exchange is slower, (2) the η6-bound product is more thermodynamically stable than [CpRu(η6-chlorotoluene)]+, leading to the build-up of the product bound complex and long reaction times. Of course, a combination of both factors may be present. [(η6-Dimethylaniline)Cr(CO)3] is known to be more thermodynamically stable than the chlorobenzene analogue16a,17 and a similar trend is likely in the case of [CpRu(η6-arene)]+ – the aryl ring in the product (e.g.1b in Scheme 3) is more electron rich than p-cymene. In an attempt to destabilise the η6-bound product, the nucleophile was changed from morpholine to 2,2,6,6-tetramethylmorpholine. Formation of product (either free or bound) was not observed, likely due to the steric influence of the nucleophile. Efforts are ongoing to understand and overcome the limiting factors of the reaction.
In conclusion, we have demonstrated for the first time catalytic SNAr of unactivated aryl chlorides, albeit at high temperatures and with long reaction times. In an attempt to move to milder reaction conditions, several new Ru complexes were synthesised, incorporating tethers capable of lowering the activation barrier to dissociation. Despite achieving an 18-fold increase in the rate of arene exchange, the rate of SNAr was not significantly increased in our test reactions. Once an efficient catalytic system based on η6-arene exchange is realised, we envisage many applications in efficient organic synthesis.
Notes and references
- (a) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) W. Lei, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef PubMed.
- (a) T. M. Koenig and D. Mitchell, Tetrahedron Lett., 1994, 35, 1339 CrossRef CAS; (b) J. R. Rodriguez, J. Agejas and A. B. Bueno, Tetrahedron Lett., 2006, 47, 5661 CrossRef CAS PubMed.
- (a) R. C. Cambie, G. R. Clark, S. L. Coombe, S. A. Coulson, P. S. Rutledge and P. D. Woodgate, J. Organomet. Chem., 1996, 507, 1 CrossRef CAS; (b) A. J. Pearson, J. G. Park, S. H. Yang and Y.-H. Chuang, J. Chem. Soc., Chem. Commun., 1989, 1363 RSC.
- K. Kamikawa, S. Kinoshita, M. Furusyo, S. Takemoto, H. Matsuzaka and M. Uemura, J. Org. Chem., 2007, 72, 3394 CrossRef CAS PubMed.
- (a) R. C. Cambie, S. J. Janssen, P. S. Rutledge and P. D. Woodgate, J. Organomet. Chem., 1991, 420, 387 CrossRef CAS; (b) A. J. Pearson, J. G. Park and P. Y. Zhu, J. Org. Chem., 1992, 57, 3585 Search PubMed.
- A. J. Pearson and H. Shin, J. Org. Chem., 1994, 2314 CrossRef CAS.
- (a) E. L. Velarde, R. A. Stephen, R. N. Mansour, L. T. Hoang and D. J. Burkey, J. Am. Chem. Soc., 2003, 125, 1188 CrossRef CAS PubMed; (b) Y. K. Chung, H. S. Choi, N. G. Connelly and D. A. Sweigart, J. Am. Chem. Soc., 1982, 104, 4245 CrossRef CAS.
- (a) A. Schlüter, K. Bieber and W. S. Sheldrick, Inorg. Chim. Acta, 2002, 340, 35 CrossRef; (b) S. Chen, V. Carperos, B. Noll, R. J. Swope and M. R. DuBois, Organometallics, 1995, 14, 1221 CrossRef CAS.
- P. Ricci, K. Krämer, X. C. Cambeiro and I. Larrosa, J. Am. Chem. Soc., 2013, 135, 13258 CrossRef CAS PubMed.
- (a) J. W. Janetka and D. H. Rich, J. Am. Chem. Soc., 1997, 119, 6488 CrossRef CAS; (b) F. C. Pigge, J. J. Coniglio and N. P. Rath, J. Org. Chem., 2003, 69, 1161 CrossRef PubMed.
- (a) W. Braun, B. Calmuschi-Cula, U. Englert, K. Höfener, E. Alberico and A. Salzer, J. Organomet. Chem., 2008, 2065 CAS; (b) R. P. Houghton, M. Voyle and R. Price, J. Organomet. Chem., 1983, 259, 183 CrossRef CAS.
- M. F. Semmelhack, A. Chlenov and D. M. Ho, J. Am. Chem. Soc., 2005, 127, 7759 CrossRef CAS PubMed.
- R. P. Houghton and M. Voyle, J. Chem. Soc., Perkin Trans. 1, 1984, 925 RSC.
- (a) M. Otsuka, K. Endo and T. Shibata, Chem. Commun., 2010, 46, 336 RSC; (b) M. Otsuka, H. Yokoyama, K. Endo and T. Shibata, Synlett, 2010, 2601 CAS.
- (a) J. Takaya and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5756 CrossRef CAS PubMed; (b) M. Otsuka, H. Yokoyama, K. Endo and T. Shibata, Org. Biomol. Chem., 2012, 10, 3815 RSC.
- (a) T. G. Traylor, K. J. Stewart and M. J. Goldberg, J. Am. Chem. Soc., 1984, 106, 4445 CrossRef CAS; (b) T. G. Traylor, K. J. Stewart and M. J. Goldberg, Organometallics, 1986, 5, 2062 CrossRef CAS.
- C. A. L. Mahaffy and P. L. Pauson, J. Chem. Res., Synop., 1979, 126 CAS.
- A similar approach has been shown to increase arene exchange in Cr(η6-arene) complexes, see ref. 2b and (a) E. P. Kündig, M. Kondratenko and P. Romanens, Angew. Chem., Int. Ed., 1998, 37, 3146 CrossRef; (b) M. F. Semmelhack, W. Seufert and L. Keller, J. Organomet. Chem., 1982, 226, 183 CrossRef CAS.
- B. M. Trost and C. M. Older, Organometallics, 2002, 21, 2544 CrossRef CAS.
- P.-H. Yeh, Z. Pang and R. F. Johnson, J. Organomet. Chem., 1996, 509, 123 CrossRef CAS.
- Transesterification to form the ethyl ester took place under the reaction conditions.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc07116f |
| This journal is © The Royal Society of Chemistry 2015 |