
Sustained delivery of bioactive neurotrophin-3 to the injured spinal cord†
Irja
Elliott Donaghue
ab,
Charles H.
Tator
cde and
Molly S.
Shoichet
*ab
aDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, 200 College Street, Toronto, ON, Canada M5S 3E5. E-mail: molly.shoichet@utoronto.ca
bInstitute of Biomaterials and Biomedical Engineering, 164 College Street, Toronto, ON, Canada M5S 3G9
cDivision of Genetics and Development, Toronto Western Research Institute, University of Toronto, 399 Bathurst Street, Toronto, ON, Canada M5 T 2S8
dKrembil Neuroscience Centre, Toronto Western Hospital, University Health Network, ON, Canada
eDepartment of Surgery, Division of Neurosurgery, University of Toronto, ON, Canada
First published on 12th September 2014
Abstract
Spinal cord injury is a debilitating condition that currently lacks effective clinical treatment. Neurotrophin-3 (NT-3) has been demonstrated in experimental animal models to induce axonal regeneration and functional improvements, yet its local delivery remains challenging. For ultimate clinical translation, a drug delivery system is required for localized, sustained, and minimally invasive release. Here, an injectable composite drug delivery system (DDS) composed of biodegradable polymeric nanoparticles dispersed in a hyaluronan/methyl cellulose hydrogel was injected into the intrathecal space to achieve acute local delivery to the spinal cord after a thoracic clip compression injury. NT-3 was encapsulated in the DDS and released in vitro for up to 50 d. With a single injection of the DDS into the intrathecal space of the injured spinal cord, NT-3 diffused ventrally through the cord and was detectable in the spinal cord for at least 28 d therein. Delivery of NT-3 resulted in significant axon growth with no effect on the astroglial response to injury in comparison with vehicle and injury controls. NT-3 treatment promoted functional improvements at 21 d according to the Basso Beattie Bresnahan locomotor scale in comparison with the DDS alone. The sustained delivery of bioactive NT-3 to the injured spinal cord achieved in this study demonstrates the promise of this DDS for central nervous system repair.
Introduction
Spinal cord injury is a permanently disabling condition that substantially reduces a patient's life expectancy and quality of life.1 The majority of spinal cord injuries are impact/compressive injuries,2 and after the initial mechanical tissue damage a secondary injury cascade follows which results in neuronal and glial apoptosis, inflammation, axon degeneration, and the formation of a fluid-filled cystic cavity surrounded by a glial scar.1,3 Since endogenous repair mechanisms in the central nervous system (CNS) are insufficient to promote recovery,3 many treatment strategies currently under investigation focus on neuroprotection or tissue repair with the goal of promoting neurological function. These strategies include: endogenous stem cell stimulation, stem cell transplantation, and the delivery of therapeutics.4 Currently, only methyl prednisolone (an anti-inflammatory corticosteroid) is administered clinically; no approved clinical therapies for SCI exist that can induce tissue regeneration and repair.A promising area of research is the delivery of neurotrophic factors.5 Neurotrophic factors, such as brain-derived neurotrophic factor, glial-derived neurotrophic factor, and neurotrophin-3 (NT-3), which are essential for CNS development, have also been shown to induce neuronal growth and tissue repair following injury.6–8 In particular, NT-3 has been demonstrated to induce axon sprouting and functional recovery.8–13 However, the challenge for clinical translation of NT-3 and other therapeutic proteins is their sustained and localized delivery to the injured spinal cord.14,15 Previous methods of delivering NT-3 include genetically modified transplanted fibroblasts,12 viral transfection,16 intrathecal infusion via a catheter/minipump,10 and direct injection into the spinal cord.11 Genetically modified cells and viral transfection result in variable dosing, and these methods are currently not approved for clinical use. Osmotic minipump delivery provides sustained and controlled delivery of NT-3, yet is highly invasive and can result in infections and increased scarring around the injury site, hampering recovery.17,18 Direct injection carries the risk of causing further tissue injury. A minimally invasive, sustained delivery system is therefore required to effectively deliver NT-3 to the injured spinal cord.
Previously, a minimally invasive drug delivery system (DDS) capable of providing tunable delivery of proteins to the injured CNS was developed.19–21 The DDS of PLGA nanoparticles alone (without encapsulated therapeutic) dispersed in hyaluronan/methyl cellulose (HAMC) hydrogel is well-tolerated after intrathecal injection, having a minimal inflammatory response.18In vitro release of encapsulated NT-3 in the PLGA/HAMC DDS was demonstrated over a 7–28 day period.22 Here we study, for the first time, the in vivo sustained release of bioactive NT-3 from this composite DDS and the tissue and functional benefits of its delivery in a clinically relevant model of compressive SCI.
Materials and methods
NT-3 encapsulation and characterization
All chemicals were purchased from Sigma Aldrich (Oakville, CA) unless otherwise stated. NT-3 was encapsulated in PLGA nanoparticles by double emulsion-solvent evaporation, as previously described for other proteins.19 Briefly, PLGA (50/50, 7–17 kDa, acid-terminated, Sigma Aldrich) was dissolved in dichloromethane (DCM, Caledon Labs, Georgetown, CA) at 13 w/v% with 0.05 w/v% Pluronic NF-127 (BASF, Mississauga, CA) and 0.4 w/v% magnesium carbonate. NT-3 was dissolved in artificial cerebrospinal fluid (aCSF) with 10 w/v% bovine serum albumin (BSA). The aCSF solution was added to the DCM solution and the mixture vortexed for 10 s and sonicated on ice for 10 min at 20% amplitude (Vibra-Cell, Sonics, Newtown USA) to create the primary emulsion. A solution of 2.5 w/v% of poly(vinyl alcohol) (PVA) in double-distilled water was added, and the mixture vortexed for 10 s and sonicated on ice for a further 10 min at 30% amplitude to create the secondary emulsion. The solution was stirred at room temperature for 16 h to allow the dichloromethane to evaporate, then centrifuged and washed with double-distilled water four times, followed by lyophilization for 3 d. The resulting nanoparticles were stored at −20 °C.The diameter of the NT-3 encapsulated PLGA nanoparticles was determined by dynamic light scattering (Malvern Zetasizer, etc.) at a concentration of 1.5 mg mL−1 in double-distilled water after 5 min sonication. NT-3 loading and encapsulation efficiency was determined by first dissolving PLGA nanoparticles in 0.05 M sodium hydroxide for 1 h. Samples were then diluted in aCSF and the concentration of NT-3 was determined by enzyme-linked immunosorbent assay (ELISA, R&D Systems, Minneapolis, USA).
In vitro characterization of the composite PLGA/HAMC DDS
HAMC was prepared by sequential dispersion of MC (6 w/v%, 300 kg mol−1, Shin-Etsu, Tokyo, Japan) and HA (2.8 w/v%, 4–1.8 × 106 g mol−1, NovaMatrix, Sandvika, Norway) in aCSF by a dual asymmetric centrifugal mixer. NT-3 nanoparticles were dispersed in aCSF by bath sonication, then mixed into HAMC for a final concentration of 15 w/v% nanoparticles, 1.4 w/v% HA and 3 w/v% MC with an additional 0.25 μg μL−1 of NT-3 mixed into the HAMC.NT-3 release from the PLGA/HAMC composite in vitro was analyzed by incubating 100 μL of composite with 900 μL of aCSF at 37 °C. At regular intervals, the aCSF was sampled with complete replacement. Samples were stored at −20 °C before analysis by ELISA. At the end of the release period, the composite was dissolved in 0.05 M NaOH and any remaining NT-3 was extracted into aCSF for analysis by ELISA.
Animal surgeries
All animal work was performed according to the guidelines established by the Canadian Council on Animal Care and all protocols were approved by the University Health Network's Animal Care Committee. Adult female Sprague-Dawley rats were anaesthetized with isofluorane and a laminectomy was performed at the T1–2 vertebral level. A moderate compressive injury was induced by a 26 g modified aneurysm clip for 1 min.23In vivo delivery of NT-3 DDS
For the delivery of NT-3 in the PLGA/HAMC DDS vs. the vehicle control of BSA encapsulated in PLGA nanoparticles in HAMC, a durotomy was performed with a 30 G angled beveled needle immediately caudal to the injury site and immediately after the injury was made. 10 μL of the nanoparticle/HAMC composite was injected into the intrathecal space with a 30 G angled, blunt-tipped needle over 30 s and the needle was held in place for an additional 1 min to allow gelation to occur. Animals in the injury-only control group did not receive a durotomy or injection. Following injury and injection, the overlying muscles and fascia were sutured closed and rats were placed under a heat lamp for recovery. Buprenorphine was administered post-surgery for 3 d to manage pain. Clavamox was administered orally for 5 d following surgery to prevent infection.In vivo quantification and tissue processing for ELISA
For the quantification of NT-3 delivery in vivo, animals were sacrificed at 1 d, 7 d, 14 d, or 28 d (n = 4 per time point). Animals receiving only SCI were used to control for endogenous NT-3, and were sacrificed at the same time points (n = 2 per time point). A 1.5 cm section of spinal cord at the T1–2 vertebral level was harvested and flash frozen in isopentane. The tissue was subsequently cut into 1 mm parasagittal slices using a tissue chopper (McIlwain, Redding, USA) and the medial two slices were sectioned dorsoventrally into 500 μm thick longitudinal sections. This allowed for the analysis of NT-3 concentration by ELISA at 500 μm intervals throughout the depth of the spinal cord (approximately 3 mm in diameter at the T1–2 vertebral level). Before ELISA analysis, tissue sections were suspended in a modified Tris-HCl buffer24 and homogenized with a bead-beater.Immunohistochemistry
In a separate study, animals receiving SCI alone (n = 7), SCI + vehicle DDS (n = 7), or SCI + NT-3 DDS (n = 13) were sacrificed 28 d after injury. Animals were transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS). A 1.5 cm section of spinal cord surrounding the lesion was removed and cryoprotected in 30% sucrose in PBS. Tissue was embedded in Cryomatrix (Thermoscientific, Nepean CA) and sectioned parasagittally into 20 μm slices.Tissue sections were stained with 200 kDa neurofilament (NF200, Sigma-Aldrich) for axons and glial fibrillary acidic protein (GFAP, Cell Signaling Technologies, Danvers, USA) for astrocytes. Tissue sections were imaged with an Olympus FV1000 confocal microscope. Image J was used for image analysis. For quantification of axon density, a 10 mm long area of tissue rostrocaudally centered around the lesion site was imaged, converted to a binary image, set to an automatic threshold, and the pixel count was measured. This count was divided by the total tissue area to yield % axon density. We also assessed axon density for uninjured animals that received a sham injection of aCSF (n = 3, 6 sections per animal). For axon counting, the number of NF200+ fibers across the mediolateral plane were counted at the centre of the excised cord (corresponding to the lesion epicentre in injured animals) and 4–5 mm rostral and caudal. An average of these 3 areas was used as the total axon count for the section. GFAP staining was used to quantify the lesion area. The GFAP-positive perimeter of the lesion was manually traced, and the lesion area measured. The density of GFAP staining was assessed in the same manner as the axon density. For each analysis, a minimum of 4 tissue sections per animal were used, distributed across the spinal cord. All tissues were analyzed blindly to the treatment.
In vivo behavioural analysis
Weekly behavioural testing was performed on all animals used in the 28 d survival study. The Basso–Beattie–Bresnahan rat score (BBB) was used to analyze locomotor function.25 All animals were scored blindly by two independent observers.Statistical analysis
All results are reported as mean ± standard deviation. Statistical testing was performed in GraphPad Prism 6. The differences in neurofilament density and lesion area between treatment groups were assessed by one-way analysis of variance (ANOVA) with Bonferroni's post-hoc at 95% confidence intervals. To analyze the BBB scoring, a repeated measures ANOVA was first performed to determine the effects of time and treatments.26 Differences between treatments at 21 d and 28 d were then separately assessed by one-way ANOVA with Bonferroni's post-hoc at 95% confidence intervals.Results
Sustained release of NT-3 in vitro and in vivo
NT-3 was encapsulated in PLGA nanoparticles by double emulsion-solvent evaporation. The nanoparticles had a volume-weighted average diameter of 220 nm. The encapsulation efficiency was determined to be 47 ± 2%, corresponding to a loading of 1.03 ± 0.04 μg mg−1 nanoparticle. An additional 0.025 μg μL−1 of soluble NT-3 was physically mixed into the nanoparticle/HAMC composite to increase the total delivered dose.NT-3 was released from the nanoparticle/HAMC DDS over 50 d in vitro (Fig. 1A). The release profile was triphasic, with a minimal burst and slow release for the first 10 d, followed by a period of faster release from 10–28 d and finally a slower release from 28–60 d. After 28 d, 83 ± 8% of the encapsulated NT-3 was released. A total of 1.72 ± 0.6 μg of NT-3 was released from 10 μL of composite DDS. Interestingly, the additional NT-3 added to the nanoparticle/HAMC DDS did not result in a burst release.
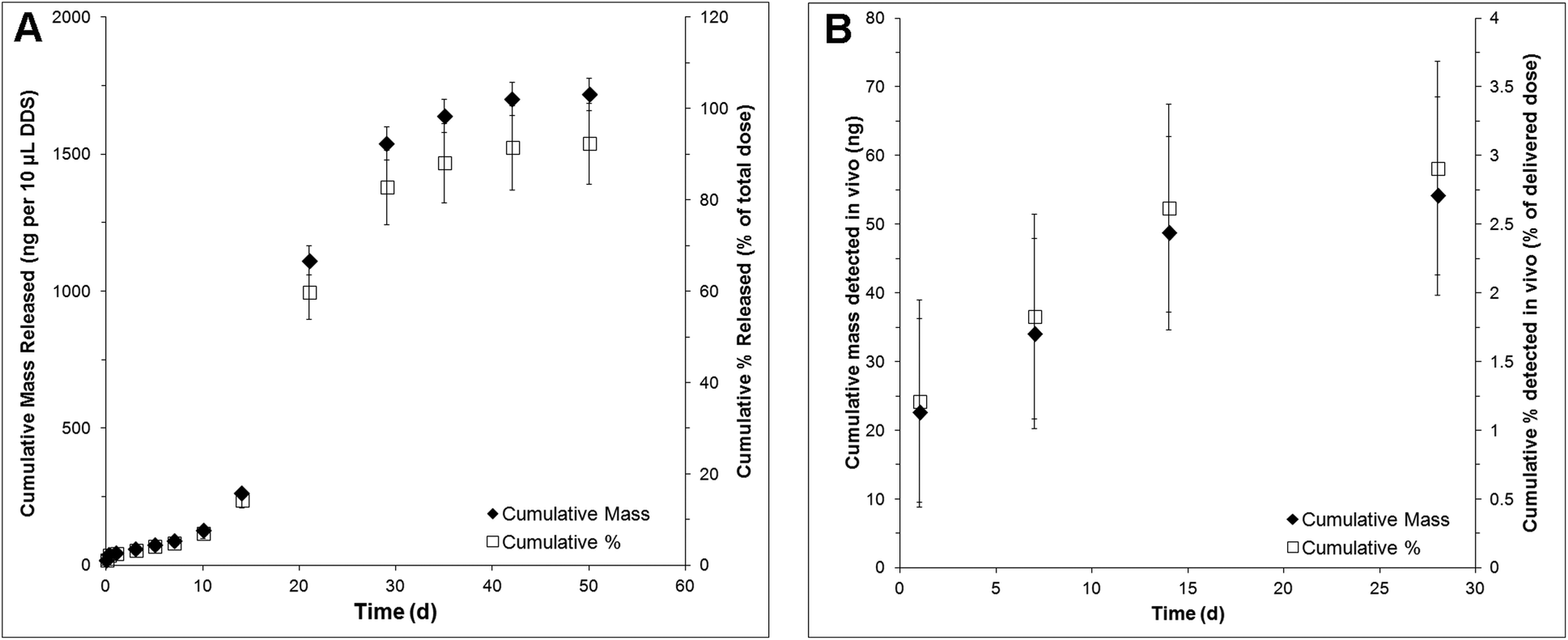 | ||
| Fig. 1 Release profile of NT-3 in terms of cumulative mass released and cumulative % released over 28 d as detected by NT-3 ELISA: (A) in vitro and (B) in vivo (n = 4, mean ± standard deviation). | ||
In vivo, NT-3 was detectable in the injured spinal cord for 28 d (Fig. 1B). The concentration of NT-3 in the spinal cord was measured at 1, 7, 14 and 28 d. The highest amount of NT-3 was detected at 1 d, with 22 ± 13 ng detected throughout the spinal cord, corresponding to 1.2 ± 0.7% of the total delivered NT-3. Cumulatively, 54 ± 14 ng of NT-3 (2.9 ± 0.8% of the delivered dose) was detected over all time points.
NT-3 was detected throughout the depth of the spinal cord, indicating that it was able to diffuse ventrally from the delivery site (Fig. 2). At 1 d, the profile is fairly linear, with the greatest concentration observed in the first 500 μm (Fig. 2A). At 7 d, the profile was flat, with a greater concentration observed at 1500 μm and below, indicating that an increased amount of NT-3 was able to reach the ventral half of the cord (Fig. 2B). Consistent with the in vitro release profile, a greater concentration of NT-3 was observed in the dorsal 500 μm of the spinal cord at 14 d (Fig. 2C), indicating that the release rate increased after day 7. At 28 d, NT-3 continued to be released into the spinal cord, with a similar concentration of NT-3 detected at all depths (Fig. 2D).
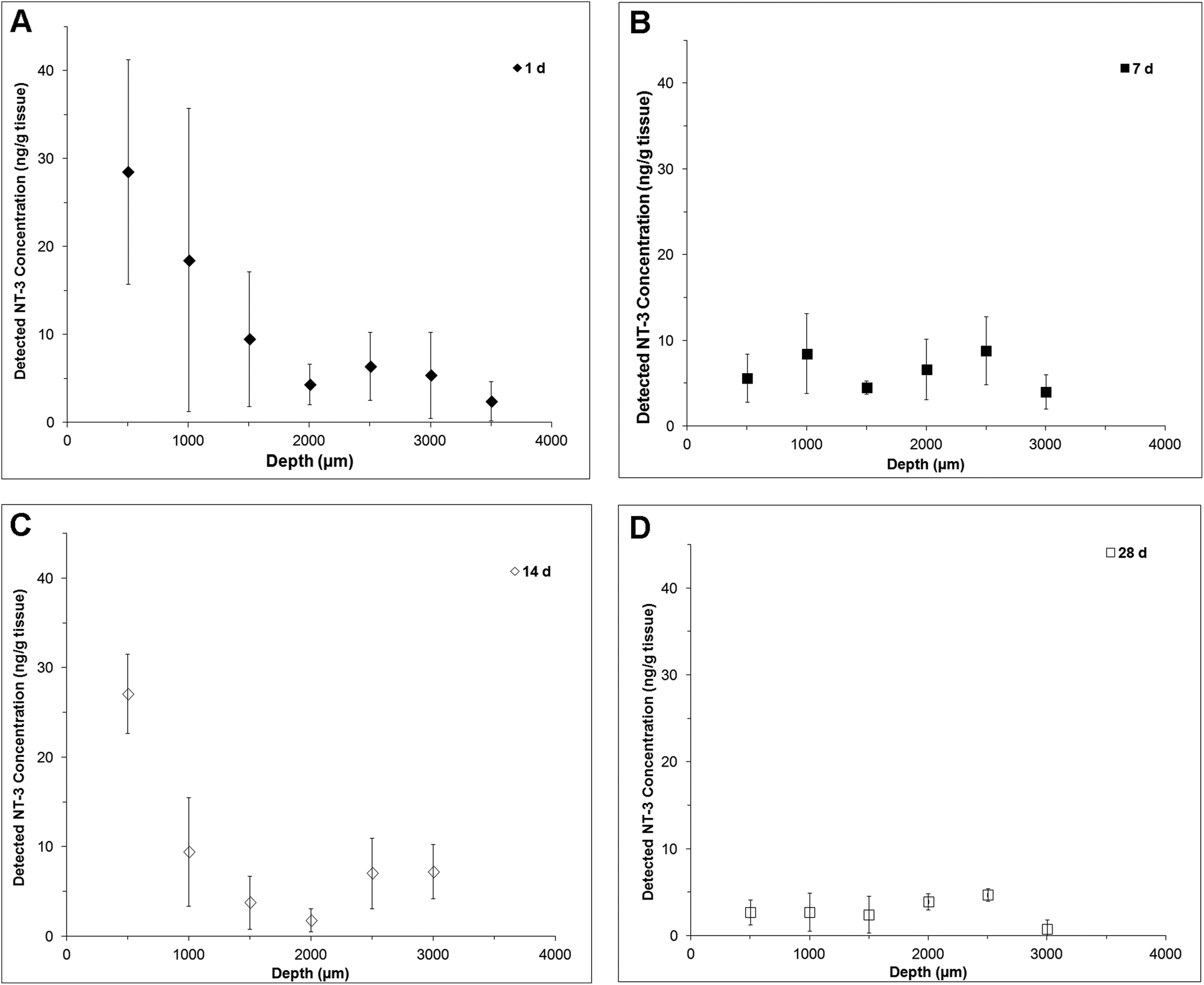 | ||
| Fig. 2 Depth profiles of released NT-3 detected in the injured spinal cord over 28 d by NT-3 ELISA (where injured tissue was spiked with known concentrations of NT-3 for the standard curve) at: (A) 1 d, (B) 7 d, (C) 14 d, and (D) 28 d (n = 4, mean ± standard deviation). | ||
Axon density and lesion size
The effects of NT-3 released from the PLGA/HAMC composite on spinal cord tissue were assessed 28 d after intrathecal injection and injury. The density of axons surrounding the lesion site was quantified with neurofilament staining (Fig. 3). Delivery of NT-3 resulted in significantly higher axon density surrounding the lesion site in comparison with the DDS control and SCI control (p ≤ 0.01). However, the axon density of uninjured tissue was significantly higher than that of all other groups (p ≤ 0.005). An increase in the number of axons in the area of the lesion was observed for NT-3-treated animals, but this method of analysis also introduced greater intra-group variability (p = 0.072, ESI Fig. 1†). The increase in axon density as a result of NT-3 delivery indicates that NT-3 bioactivity was maintained over 28 d in vivo and its delivery was efficacious. Moreover, the DDS alone did not affect axon density.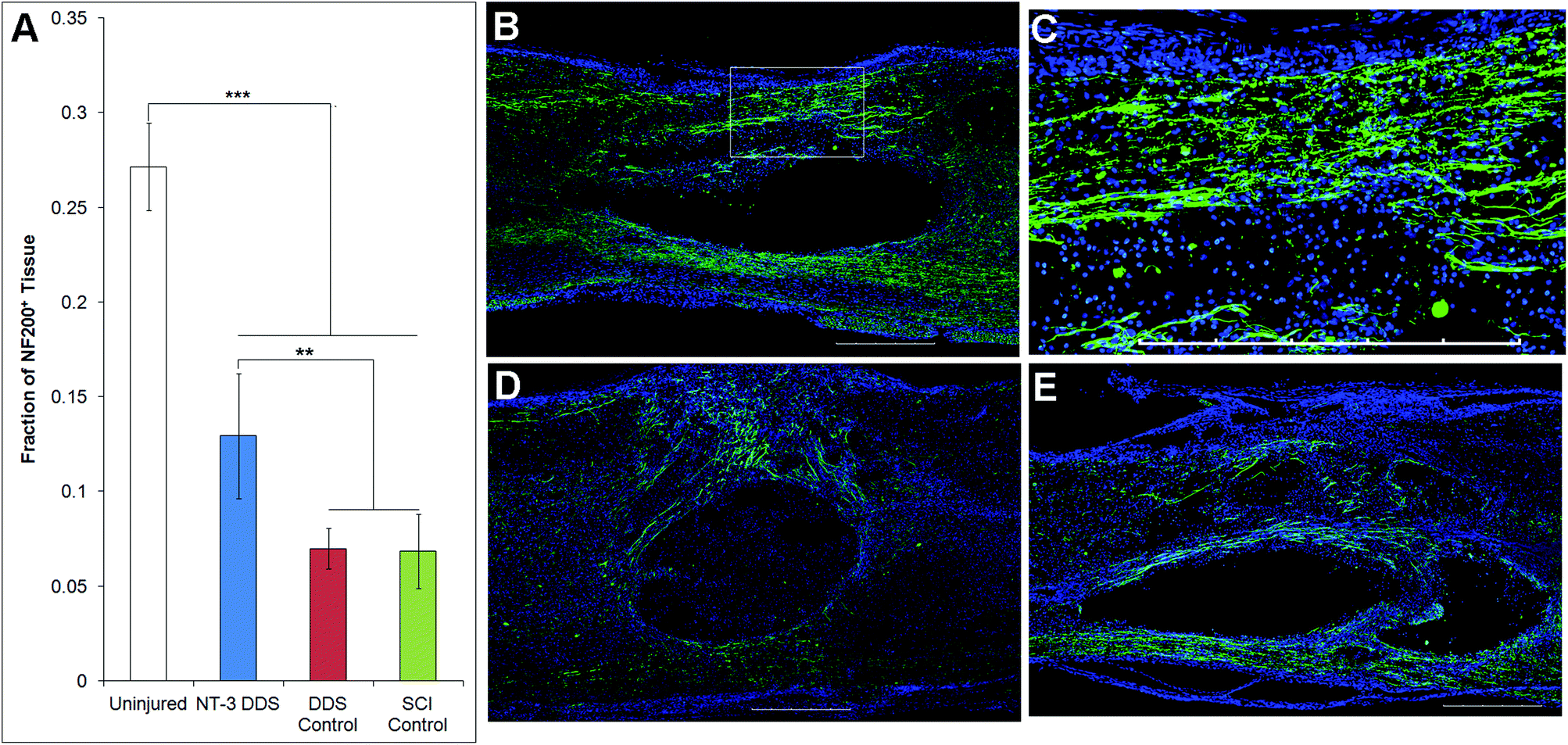 | ||
| Fig. 3 NF200 immunohistochemistry demonstrates that local sustained release of NT-3 promotes axonal growth. (A) A significantly greater fraction of tissue was stained with NF200 in animals that had local sustained release of NT-3 (n = 11) vs. vehicle alone (n = 4) or injury alone (n = 5) (p ≤ 0.01, mean ± standard deviation). Uninjured tissue had a significantly greater density of NF200 staining than all other groups (p ≤ 0.005, mean ± standard deviation). Representative images of immunohistochemical stained tissue for NF200 (green) and DAPI (blue) surrounding the injury site at 28 d for (B) NT-3 DDS and (C) enlarged view of lesion area outlined in (B); (D) DDS control and (E) SCI control. Scale bars = 500 μm. | ||
The lesion size at 28 d was analyzed to determine the impact of the DDS on tissue sparing and/or cavity formation (Fig. 4). The lesion area was quantified by tracing the GFAP+ border. As expected from previous characterization of similar systems,20,27 the DDS did not result in increased lesion size in comparison with the SCI control (Fig. 4A); however, there was also no decrease in lesion size with NT-3 delivery relative to vehicle and/or spinal cord injury alone controls, possibly due to the high degree of variability in the measured lesion area for the SCI control group. Representative images of immunohistochemically-stained tissues (Fig. 4B–E) reflect these quantitative data. Similarly, the density of GFAP staining in the area of the lesion for NT-3-treated animals or DDS-treated and untreated controls was not significantly different (ESI Fig. 2†).
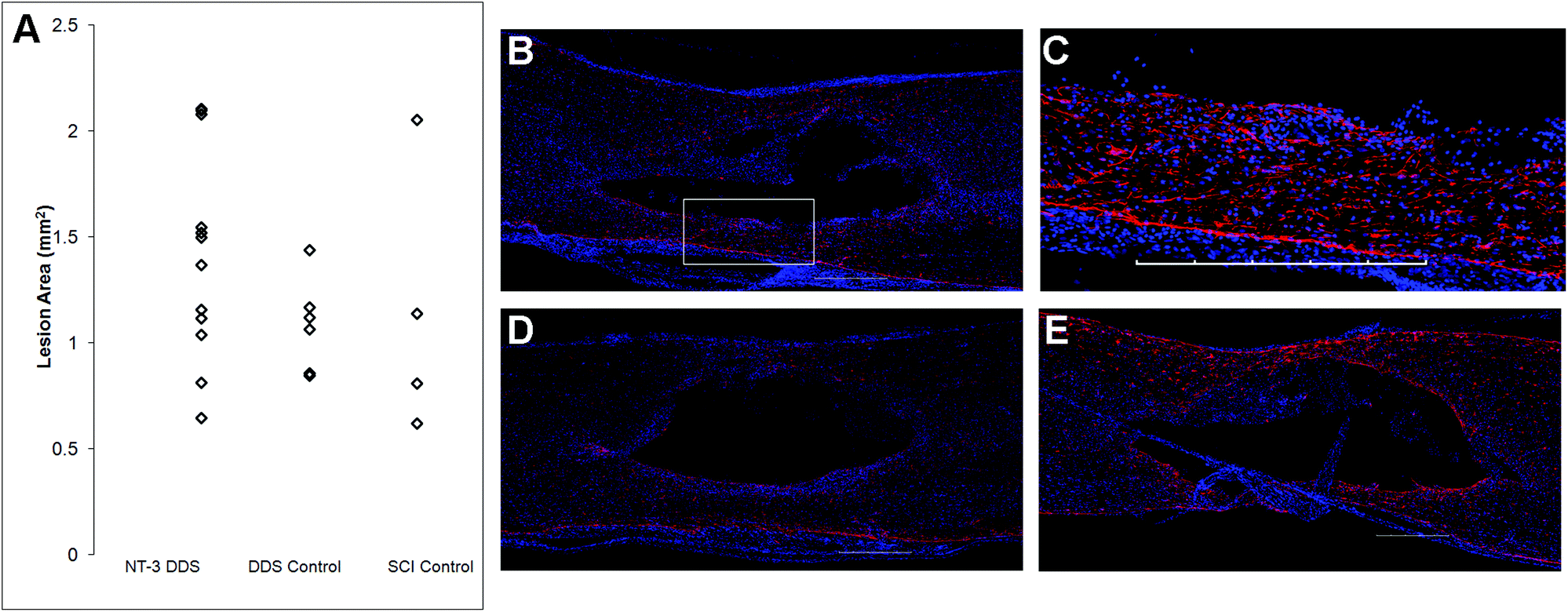 | ||
| Fig. 4 GFAP immunohistochemistry shows no significant difference in astroglial response among the three groups, demonstrating that neither local delivery of NT-3 nor vehicle alone affects the lesion area vs. spinal cord injury alone. (A) The lesion area in the injured spinal cord is not significantly different between animals that had NT-3 delivered in the DDS (n = 11) vs. DDS alone (n = 4) vs. injury alone (n = 5) (mean ± standard deviation). Representative images of immunohistochemical stained tissue for GFAP (red) and DAPI (blue) surrounding the injury site at 28 d for (B) NT-3 DDS and (C) enlarged view of lesion area outlined in (B); (D) DDS control; and (E) SCI control. Scale bars = 500 μm. | ||
Behavioural recovery
The locomotor function following SCI was assessed using the BBB score. In all animals, the BBB score improved over the 28 d study period, consistent with moderate compressive injury (Fig. 5A). At 21 d, NT-3-treated animals had a significantly higher BBB score in comparison with the DDS alone (p ≤ 0.05). Due to higher intra-treatment variability, this effect was not significant at 28 d. Interestingly, a greater percentage of NT-3 animals were able to achieve a BBB score of 13 or higher, indicative of frequent to consistent forelimb–hindlimb coordination, in comparison with both DDS vehicle controls and SCI controls (Fig. 5B). Thus, sustained NT-3 delivery was able to elicit modest effects on locomotor function.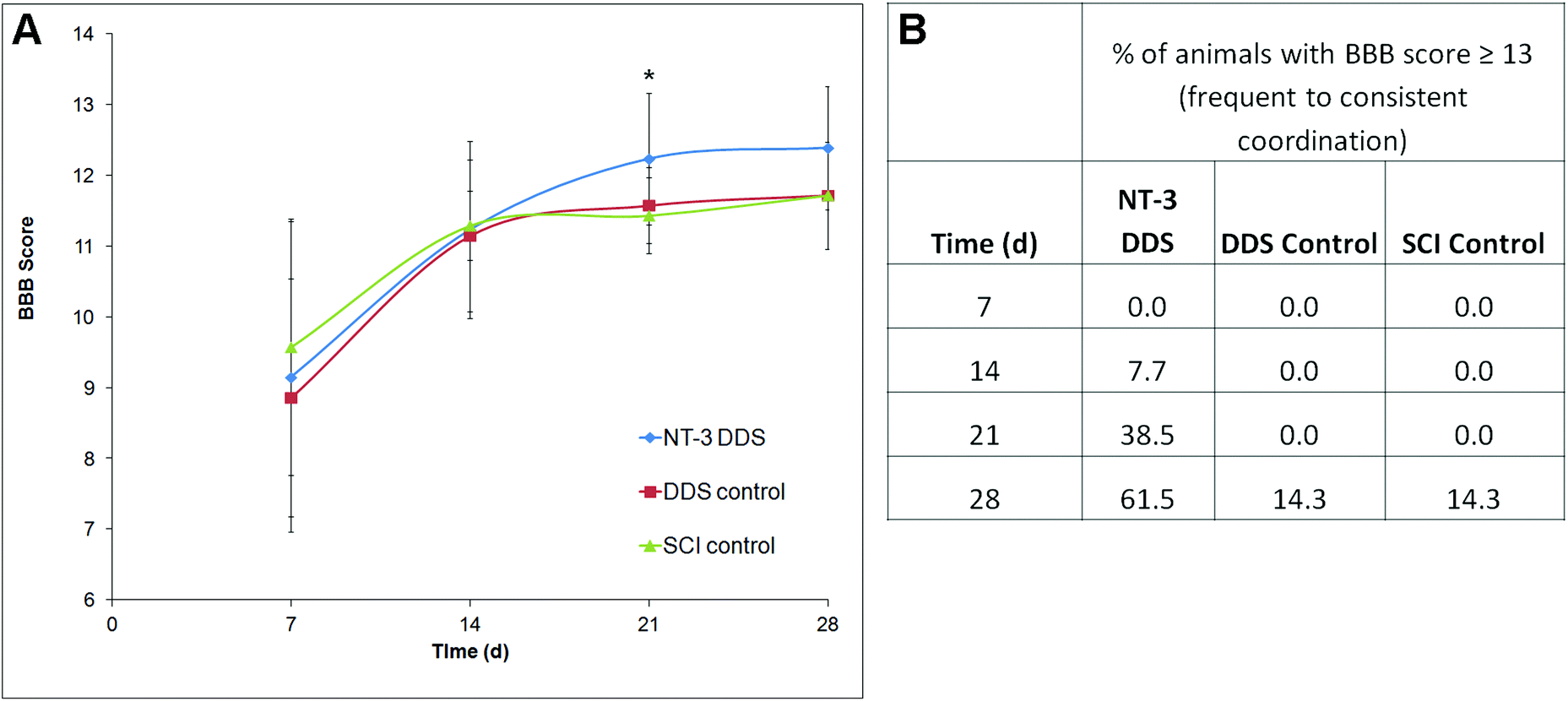 | ||
| Fig. 5 The locomotor function of rats was followed by the Basso, Beattie Bresnahan scoring method weekly for 28 days. (A) At 21 d, NT-3-treated animals had a significantly greater BBB score in comparison with DDS control (p ≤ 0.05, n = 13 for NT-3-treated, n = 7 for DDS control, SCI control). (B) At 14, 21, and 28 d, a greater percentage of NT-3-treated animals exhibited frequent to consistent coordination, as reflected by the percentage of animals that had BBB scores greater than 13. | ||
Discussion
Controlled protein delivery to the injured CNS has been most commonly studied with hydrogel systems.28,29 However, release from hydrogels has been typically controlled by simple diffusion or non-covalent interactions, and is usually characterized by a high burst release.29 In this work, the PLGA/HAMC DDS was able to provide sustained release of NT-3 for up to 50 d in vitro and 28 d in vivo. The in vitro release profile showed slow release, with a minimal burst, in the first 10 d even with the addition of soluble NT-3. This suggests either hydrophobic or electrostatic interactions between the NT-3 and the PLGA nanoparticles.In vivo, NT-3 released from the DDS was able to diffuse ventrally through the spinal cord from the delivery site and was detectable for 28 d. After implantation of the DDS, NT-3 can diffuse into the spinal cord tissue or into the CSF. Once in the spinal cord tissue, NT-3 can be eliminated before reaching its target, the TrkC+ axons located primarily in the corticospinal and rubrospinal tracts30 (Fig. 6). Importantly, the depth profile, determined by ELISA, indicates that NT-3 released from this DDS can reach the entire thickness of the cord. However, the low concentration of detected NT-3 at each time point in comparison with the delivered dose (less than 5%) indicates that the NT-3 is rapidly eliminated from the spinal cord. Clearance mechanisms that are responsible for the elimination of NT-3 from the spinal cord tissue include intra- and extracellular degradation, internalization and transport of the NT-3-TrkC complex, and diffusion into the CSF and vasculature.31 While a mass balance analysis is desirable, it was not possible to remove the PLGA/HAMC DDS to determine the amount of remaining NT-3 after sacrifice due to limitations associated with tissue preparation. The data collected in vivo therefore represent only a snapshot of the total NT-3 that was released from the DDS at each time point.
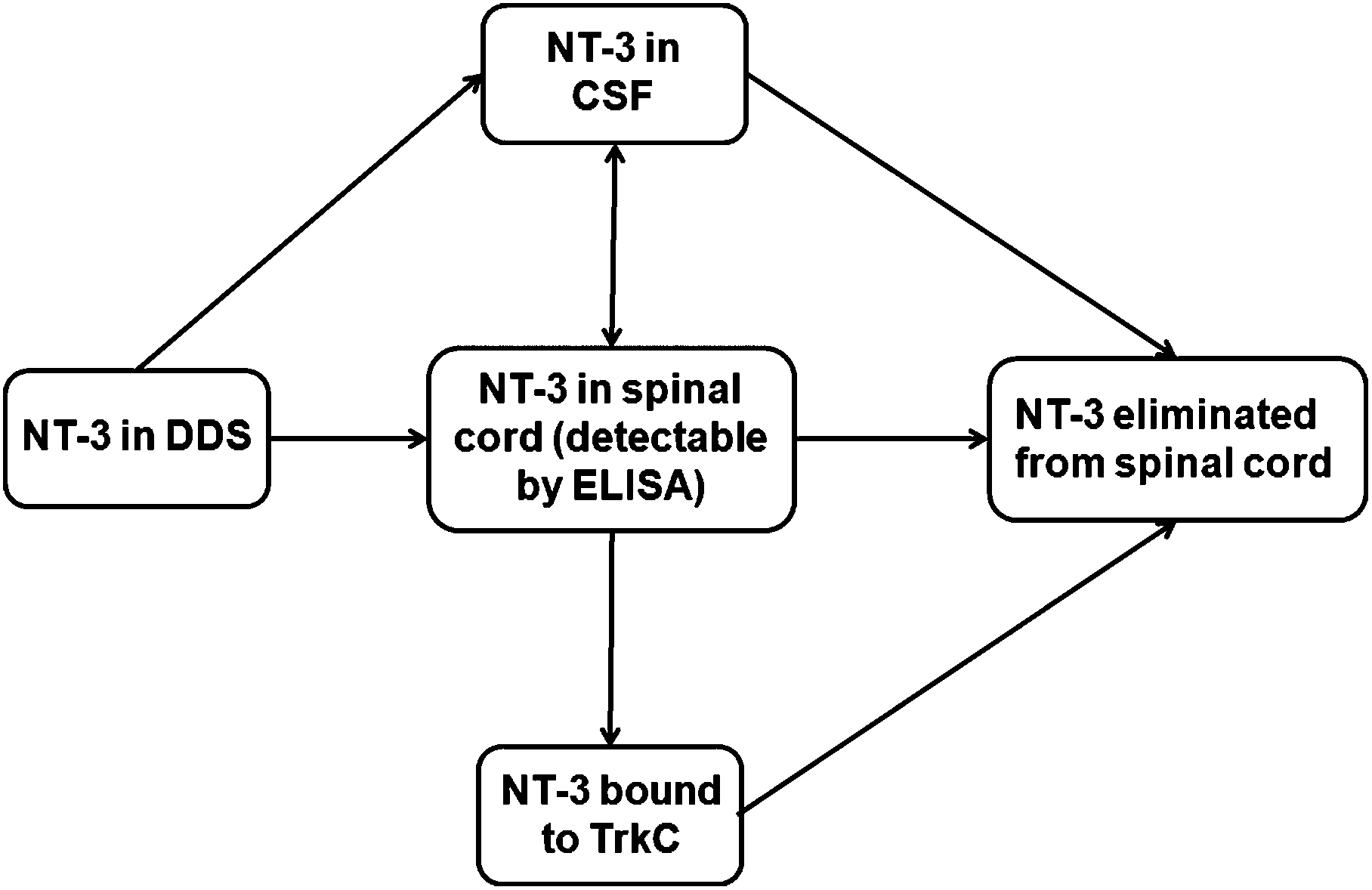 | ||
| Fig. 6 The pathways for NT-3 release, diffusion, and clearance from the spinal cord after intrathecal delivery in the DDS. NT-3 released from the DDS can diffuse into spinal cord tissue or into the CSF. NT-3 can also diffuse into and out of the spinal cord. In spinal cord tissue, NT-3 is detectable by ELISA until it binds to the receptor TrkC or p75NTR (not shown), or until it is eliminated from the cord by diffusion into vasculature, degradation, or cellular uptake. | ||
Few studies that examine NT-3 delivery to the injured spinal cord have quantified the delivered dose, or determined the in vivo release profile. Piantino et al. used a cross-linked poly(lactide)-block-poly(ethylene glycol)-block-poly(lactide) (PLA-b-PEG-b-PLA) hydrogel to deliver NT-3, and determined that in vivo concentrations of NT-3 peaked at 3 h after delivery, but were still elevated in comparison with controls at 14 d.13 A total of 1 μg of NT-3 was delivered, and the maximum detected concentration in the tissue was approximately 70 ng mL−1 g−1 tissue. The concentration of NT-3 detected was not normalized to the endogenous level of NT-3, which was determined to be approximately 10 ng mL−1 g−1 tissue. As a hemisection model was used by Piantino et al. for the PEG-PLA-PEG hydrogel, a greater surface area between the hydrogel and tissue may have resulted in faster diffusion. In addition, release from that hydrogel was diffusion-mediated, resulting in a high burst release.13 In contrast, our PLGA/HAMC DDS delivered NT-3 without a burst release, yet with a similar dose (1.72 ± 0.6 μg), but a maximum concentration of 28 ng g−1 tissue was detected. The lower detected dose in the current study may be due to these differences in injury models, drug delivery systems and detection methodology.
Previous studies of NT-3 delivery to the spinal cord have also used widely varying doses, from 4.2 ng![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 to 336 μg.10 Although the in vitro effective dose of NT-3 to induce axon growth has been determined to be 25 ng mL−1,33 the minimum required dose in vivo is not known. In the present study, a total of 1.72 μg of NT-3 was delivered, and was sufficient to induce axon growth after 28 d. While this dose elicited functional recovery that was significant at 21 d, it did not persist at 28 d. The increase in hindlimb–forelimb coordination observed for NT-3-treated animals in comparison with controls points to effects on fine motor function that could be further assessed in future studies with the ladder-walk behavioural task.34
32 to 336 μg.10 Although the in vitro effective dose of NT-3 to induce axon growth has been determined to be 25 ng mL−1,33 the minimum required dose in vivo is not known. In the present study, a total of 1.72 μg of NT-3 was delivered, and was sufficient to induce axon growth after 28 d. While this dose elicited functional recovery that was significant at 21 d, it did not persist at 28 d. The increase in hindlimb–forelimb coordination observed for NT-3-treated animals in comparison with controls points to effects on fine motor function that could be further assessed in future studies with the ladder-walk behavioural task.34
The effects of delivered NT-3 were assessed in this study in a compressive model of SCI whereas previous studies on NT-3 delivery have primarily investigated the effects of NT-3 on a hemisection SCI model, in which the corticospinal tract (CST) is completely severed. Axons observed in the injury site, therefore, have sprouted or regenerated from severed axons rostral or caudal to the injury site. In a compressive injury, some axons in or close to the injury site are spared, and thus quantification of axon regeneration due to NT-3 delivery is more difficult. Yet, for clinical translation, it is necessary to demonstrate efficacy in relevant animal models. The present study demonstrates that NT-3 delivered over 28 d from an intrathecal DDS can elicit significant axon growth in a clinically relevant model.
Our group has previously shown that the PLGA/HAMC DDS is well-tolerated by the injured spinal cord.20,27 The results of the present study are consistent with the previous findings where comparable lesion sizes amongst all groups, as determined by GFAP staining, underscore the minimal host response to this DDS. Importantly, we build on previous knowledge and show, for the first time, the controlled release of a growth factor to the injured spinal cord from this DDS, and the consequent tissue and functional benefit.
Conclusions
In this study, a minimally invasive DDS was investigated for the sustained delivery of NT-3 to the spinal cord after compression injury. NT-3 was released from the DDS over 28 d and induced the growth of axons surrounding the injury site. No adverse tissue effects were observed. Importantly, the delivered NT-3 induced statistically significant improvements in locomotor function. This study demonstrates that the PLGA/HAMC DDS is able to provide sustained, bioactive protein delivery in a clinically relevant model of SCI. This DDS is therefore promising for the application of protein therapeutics and combination therapies for CNS regeneration.Acknowledgements
We are grateful to Peter Poon for histological sectioning and Tobias Führmann for insightful comments on tissue analysis. We thank NSERC (PGSD to IED and Discovery Grant to MSS) and CIHR (Operating grant to MSS and CHT) for financial support. Congratulations to Michael Sefton, University Professor, on his 65thbirthday.References
- B. K. Kwon, L. H. Sekhon and M. G. Fehlings, Spine, 2010, 35, S263–S270 CrossRef PubMed.
- G. D. Carlson and C. Gorden, Spine J., 2002, 2, 116 CrossRef.
- F. J. Obermair, A. Schroter and M. Thallmair, Physiology, 2008, 23, 296–304 CrossRef PubMed.
- J. R. Wilson, N. Forgione and M. G. Fehlings, Can. Med. Assoc. J., 2013, 185, 485–492 CrossRef PubMed.
- N. K. Mohtaram, A. Montgomery and S. M. Willerth, Biomed. Mater., 2013, 8, 022001 CrossRef PubMed.
- L. B. Jakeman, P. Wei, Z. Guan and B. T. Stokes, Exp. Neurol., 1998, 154, 170–184 CrossRef CAS PubMed.
- A. M. Moore, M. D. Wood, K. Chenard, D. A. Hunter, S. E. Mackinnon, S. E. Sakiyama-Elbert and G. H. Borschel, J. Hand Surg., 2010, 35, 2008–2017 CrossRef PubMed.
- L. Schnell, R. Schneider, R. Kolbeck, Y.-A. Barde and M. E. Schwab, Nature, 1994, 367, 170 CrossRef CAS PubMed.
- R. Grill, K. Murai, A. Blesch, F. H. Gage and M. H. Tuszynski, J. Neurosci., 1997, 17, 5560–5572 CAS.
- E. J. Bradbury, S. Khemani, V. R. King, J. V. Priestley and S. B. McMahon, Eur. J. Neurosci., 1999, 11, 3873–3883 CrossRef CAS PubMed.
- J. Namiki, A. Kojima and C. H. Tator, J. Neurotrauma, 2000, 17, 1219–1230 CrossRef CAS.
- M. H. Tuszynski, R. Grill, L. L. Jones, A. Brant, A. Blesch, K. Low, S. Lacroix and P. Lu, Exp. Neurol., 2003, 181, 47–56 CrossRef CAS.
- J. Piantino, J. A. Burdick, D. Goldberg, R. Langer and L. I. Benowitz, Exp. Neurol., 2006, 201, 359–367 CrossRef CAS PubMed.
- D. Maysinger and A. Morinville, Trends Biotechnol., 1997, 15, 410–418 CrossRef CAS.
- W. Pardridge, NeuroRX, 2005, 2, 3–14 CrossRef PubMed.
- L. Taylor, L. Jones, M. H. Tuszynski and A. Blesch, J. Neurosci., 2006, 26, 9713–9721 CrossRef CAS PubMed.
- K. A. Follett, R. L. Boortz-Marx, J. M. Drake, S. DuPen, S. J. Schneider, M. S. Turner and R. J. Coffey, Anesthesiology, 2004, 100, 1582–1594 CrossRef PubMed.
- L. L. Jones and M. H. Tuszynski, Microsc. Res. Tech., 2001, 54, 317–324 CrossRef CAS PubMed.
- M. D. Baumann, C. E. Kang, J. C. Stanwick, Y. F. Wang, H. Kim, Y. Lapitsky and M. S. Shoichet, J. Controlled Release, 2009, 138, 205–213 CrossRef CAS PubMed.
- M. D. Baumann, C. E. Kang, C. H. Tator and M. S. Shoichet, Biomaterials, 2010, 31, 7631–7639 CrossRef CAS PubMed.
- Y. F. Wang, M. J. Cooke, N. Sachewsky, C. M. Morshead and M. S. Shoichet, J. Controlled Release, 2013, 172, 1–11 CrossRef CAS PubMed.
- J. C. Stanwick, M. D. Baumann and M. S. Shoichet, J. Controlled Release, 2012, 160, 666–675 CrossRef CAS PubMed.
- A. S. Rivlin and C. H. Tator, Surg. Neurol., 1978, 10, 39–43 Search PubMed.
- J. C. Petruska, B. Kitay, V. S. Boyce, B. K. Kaspar, D. D. Pearse, F. H. Gage and L. M. Mendell, Eur. J. Neurosci., 2010, 32, 997–1005 CrossRef PubMed.
- D. M. Basso, M. S. Beattie and J. C. Bresnahan, J. Neurotrauma, 1995, 12, 1–21 CrossRef CAS.
- S. W. Scheff, D. A. Saucier and M. E. Cain, J. Neurotrauma, 2002, 19, 1251–1260 CrossRef PubMed.
- D. Gupta, C. H. Tator and M. S. Shoichet, Biomaterials, 2006, 27, 2370–2379 CrossRef CAS PubMed.
- G. Perale, F. Rossi, E. Sundstrom, S. Bacchiega, M. Masi, G. Forloni and P. Veglianese, ACS Chem. Neurosci., 2011, 2, 336–345 CrossRef CAS PubMed.
- M. M. Pakulska, B. G. Ballios and M. S. Shoichet, Biomed. Mater., 2012, 7, 024101 CrossRef PubMed.
- I. Mocchetti and J. R. Wrathall, J. Neurotrauma, 1995, 12, 853–870 CrossRef CAS.
- R. G. Thorne and W. H. Frey, Clin. Pharmacokinet., 2001, 40, 907–946 CrossRef CAS PubMed.
- S. J. Taylor, E. S. Rosenzweig, J. W. McDonald and S. E. Sakiyama-Elbert, J. Controlled Release, 2006, 113, 226–235 CrossRef CAS PubMed.
- H. R. Widmer, D. R. Kaplan, S. J. Rabin, K. D. Beck, F. Hefti and B. Knusel, J. Neurochem., 1993, 60, 2111–2123 CrossRef CAS PubMed.
- G. A. Metz and I. Q. Whishaw, J. Neurosci. Methods, 2002, 115, 169–179 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00311j |
| This journal is © The Royal Society of Chemistry 2015 |