
Polymeric 3D nano-architectures for transport and delivery of therapeutically relevant biomacromolecules
G.
Gunkel-Grabole
,
S.
Sigg
,
M.
Lomora
,
S.
Lörcher
,
C. G.
Palivan
* and
W. P.
Meier
*
Department of Chemistry, University of Basel, Klingelbergstrasse 80, CH-4056 Basel, Switzerland. E-mail: cornelia.palivan@unibas.ch; wolfgang.meier@unibas.ch
First published on 14th August 2014
Abstract
A promising approach for addressing a range of diseases lies in the delivery of functional biomacromolecules such as nucleic acids or proteins to cells. Polymers, peptides and the different shapes accessible through self-assembly of polymeric and peptidic amphiphiles have been widely explored as carriers and as containers for reactions on the nanoscale. These building blocks are particularly interesting, because several essential parameters such as physical characteristics, conditions for degradation or biocompatibility can be tuned to suit specific requirements. In this review, different three-dimensional architectures ranging from dendrimers and hyperbranched molecules to micelles, vesicles and nanoparticles assembled from synthetic polymers and peptides are discussed. It is focused on their function as a carrier for biologically active macromolecules, highlighting seminal examples from the current literature and pointing out the remaining and upcoming challenges in this important area of research.
 G. Gunkel-Grabole | Gesine Gunkel-Grabole obtained her BSc and MSc degrees from Freie Universität Berlin, Germany. She subsequently pursued her PhD in the Melville Laboratory for Polymer Synthesis at the University of Cambridge, UK. After graduation in 2013 she joined the University of Basel as a postdoctoral researcher. Her main research interests include polymers at interfaces and the interactions of polymers with biological systems. |
 S. Sigg | Severin Sigg obtained his MSc degree from the University of Basel in 2011. Currently, he is a PhD candidate in the group of Prof. W. Meier at the University of Basel. For his doctoral studies he is working on the development of functional nano-carrier systems for gene- and drug delivery, self-assembled from amphiphilic peptides. |
 M. Lomora | Mihai Lomora obtained his MSc in Environmental Sciences from Transilvania University of Brasov, Romania in 2011. Afterwards he worked in the pharmaceutical industry. Since 2013, Mihai Lomora started to pursue his doctoral research in protein-polymer supramolecular assemblies for the design of antioxidant nanoreactors and processors, in Physical Chemistry, University of Basel, Switzerland. |
 S. Lörcher | Samuel Lörcher obtained his MSc in nano science from the University of Basel, Switzerland in 2011. Currently he is a PhD candidate in the polymer chemistry group of Prof. W. Meier and Prof. C. Palivan. His main interest is the development of novel tools to create building blocks which enable bio-inspired and exact nano-engineering. |
 C. G. Palivan | Cornelia G. Palivan studied Physics at the University of Bucharest and received, after a two years research stage at the University of Geneva, her PhD degree in Atomic and Molecular Physics in 1995. Appointed as a Lecturer at the University of Bucharest in 1997, she joined the Chemistry Department of the University of Basel in 1999, where she is currently a Professor in Physical Chemistry. She received several awards for her research. Her domain of interest is based on development and characterization of hybrid materials combining biomolecules with amphiphilic copolymers for medical, environmental, technological, and food science oriented applications. |
 W. P. Meier | Wolfgang Meier studied Chemistry at the University of Freiburg and received his PhD degree in Macromolecular Chemistry in 1992. In 1996 he was appointed as a lecturer in Physical Chemistry at the University of Basel where he received his ‘Habilitation’ in 1998. In 2001 he was appointed as a professor at the International University of Bremen and since 2003 he is a Professor of Chemistry at the University of Basel. He received several awards (Ruzicka-Price, 2001; Hermann-Staudinger-Price, 2006) for his research. His main research interests are in the field of hierarchical self-assembly of functional polymers, and polymer–protein hybrid materials. |
1. Introduction
The application of biomacromolecules to supplement or replace low molecular weight synthetic drugs is a promising idea to tackle a plethora of different diseases. Naturally occurring macromolecules such as proteins or nucleic acids can be used to treat medical conditions, which has several inherent advantages over conventional drugs: they are biocompatible, biodegradable, have a very specific function and typically do not induce immune responses.1 These therapeutic biomolecules can fulfil different functions ranging from vaccination to regulatory activities or diagnostics,1,2 while the delivery of genes can be applied to treat hereditary diseases.3 However, direct administration of these functional biomacromolecules to the patient is not feasible as both nucleic acids and proteins/enzymes are prone to degradation and clearance upon exposure to bodily fluids. This can significantly reduce their efficiency, particularly if degradation occurs before reaching the intended target. In addition, biomacromolecules have a low capability to pass through biomembranes, which further decreases their delivery if penetration into the cell is required. Consequently, the functional molecules need to be protected from the surrounding environment until delivered to the target. Different architectures have been exploited for this purpose, ranging from natural, e.g. virus-based carriers to synthetic liposomes and peptide-based containers to those composed of entirely synthetic polymers.In recent years, research efforts towards superior carrier systems have increasingly focused on designing architectures composed of peptides and polymers as they have significant advantages over virus-based delivery vectors and liposomes. Most importantly, the risk of adverse side effects such as infections and immunogenicity can be drastically reduced.4 Moreover, the mechanic stability is increased and new paths for decoration with specific targeting moieties are opened up. Hence, in this review the recent advances and upcoming challenges for synthetic 3D architectures are discussed. The focus is laid on their application as delivery vehicles for biomacromolecules as well as nanoreactors and artificial organelles, while the delivery of small molecules and joint systems is reported elsewhere.5–8 The four predominant synthetic architectures, dendrimers and hyperbranched polymers, micelles, vesicles and nanoparticles (Scheme 1) are introduced and discussed in this review. The carriers differ in shape and size, dendrimers/hyperbranched polymers are the smallest with diameters in the range of 10–20 nm, while micelles, vesicles and nanoparticles are significantly larger with diameters ranging from under hundred nm to several hundred nm.9 In addition to the protection of encapsulated cargo, these carrier architectures fulfil other essential purposes as well. A range of different surface chemistries are available for decoration of the carrier surfaces in order to mediate the interaction with the surrounding environment, e.g. to avoid adsorption of blood proteins.10 The large pool of polymers and peptides available to generate carriers allows to address several important parameters and design the system to suit specific needs. The key prerequisites are biocompatibility, high encapsulation efficiency, “green” encapsulation (avoidance of organic solvents that may interfere with the cargo),11 conservation of activity, improved cellular uptake and sustained release. Past research has shown that it is challenging to combine all these properties in one carrier as they are influenced by different characteristics and some can even have conflicting outcomes. The introduction of positive charges for example can facilitate the complexation of negatively charged cargo such as nucleic acids or negatively charged proteins,12 and it can also aid the cellular uptake process through interactions with the negatively charged cell membrane.13 However, depending on ratio and amount, positive charges also have a detrimental effect as they are known to be highly cytotoxic.14 This example illustrates the trade-off that enhancing particular characteristics can have on other properties and underlines the importance and delicacy of designing carriers for functional biomacromolecules. The specifics of the cargo further determine the design of the carrier architecture. Individual loading strategies are required for DNA and siRNA/mRNA due to their difference in negative change density. Moreover, they have different intracellular destinations (nuclei and cytosol respectively), which requires distinct prerequisites to be considered during the design of a suitable nanocarrier. In this review, recent examples of successful carriers that combine some of the most important parameters are discussed.
 | ||
| Scheme 1 Illustration of the four major 3D synthetic architectures used as carriers and nanoreactors: dendrimers/hyperbranched polymers, spherical micelles, vesicles and nanoparticles (from left to right). Other shapes are also accessible and used, for simplicity only globular objects are drawn. | ||
The different carrier structures based on synthetic and natural polymers are introduced and their performance is evaluated. Moreover, the exciting application of these polymeric architectures as containers for enzymatic reactions on the nanoscale and very recently as artificial organelles is highlighted. In nanoreactor applications a different purpose is served as the contents should not be released. Instead, the polymeric assembly acts as a container for a functional molecule such as an enzyme, which is acting inside the three-dimensional assembly. The enzyme is retained while reactants travel in and out to enable reactions in the protected environment of a nano-architecture.15 In a functional artificial organelle, the nanoreactor remains active after uptake by cells and fulfils or supplements the tasks of naturally occurring organelles.16 The qualification of the different carrier types for these applications is described and explained on the basis of recently published examples.
2. Dendrimers and hyperbranched polymers
Carriers based on dendrimers and hyperbranched polymers are composed of a single molecule, other than the self-assembled structures discussed in this review, which contain several macromolecules.Two different synthetic routes are available for the synthesis of monodisperse dendrimers, the divergent and the convergent method.17 In the former, the dendrimer is formed generation by generation starting from the multifunctional core, while the latter describes a method were the dendrons are first synthesised separately before attachment to a core in the final, dendrimer-forming reaction step. Hyperbranched polymers on the contrary are typically obtained through polymerisation of ABx-type monomers with dual functionality.18 The main advantage is their one-pot synthesis, and despite the polydispersity many characteristic properties are shared with their perfect counterparts. Owing to their highly branched structure, these molecules possess a globular shape. Typically, dendrimers and hyperbranched polymers reach up to 15 nm in diameter, which makes them well-suited candidates for delivery of biomedically relevant molecules.19 The predominantly used dendrimers are polyamidoamine (PAMAM), polypropylene imine (PPI) and polyethylene imine (PEI), polyglycerol (PG) and poly-L-lysine (PLLys), while PG and PEI/PPI are also applied as hyperbranched polymers.20 Commercially available PAMAM is the most used polymer and PPI received increased interest after significant improvements in its synthesis.21,22 These polymers have their cationic functional groups in common except for polyglycerol, which has a branched polyether structure without intrinsic cationic charges.23 Instead, it contains manifold hydroxyl groups on the surface that can be modified to cations used to complex the nucleic acid cargo.24 Moreover, the dendritic, globular structures contain inner cavities that can be explored for encapsulation of guest molecules; a concept that was introduced by Meijer an co-workers as a ‘dendritic box’.25,26 The cavities in dendrimers of higher generation are more closed due to the increased number of terminal groups, which form a dense surface layer. Consequently, a higher encapsulation efficiency as well as extended release times have been reported with small functional molecules as guests.27 In addition to the space inside dendrimers and hyperbranched polymers, their multivalent surface is also available for interactions with biomacromolecules (Scheme 2). Particularly large cargo such as nucleic acids is complexed through superficial functional groups. A multitude of functional groups that can be introduced at predefined regions, as for example realised in core-shell type dendrimers,28 allow to adjust the carrier specifically to the requirements of the cargo and thereby optimise the interaction.
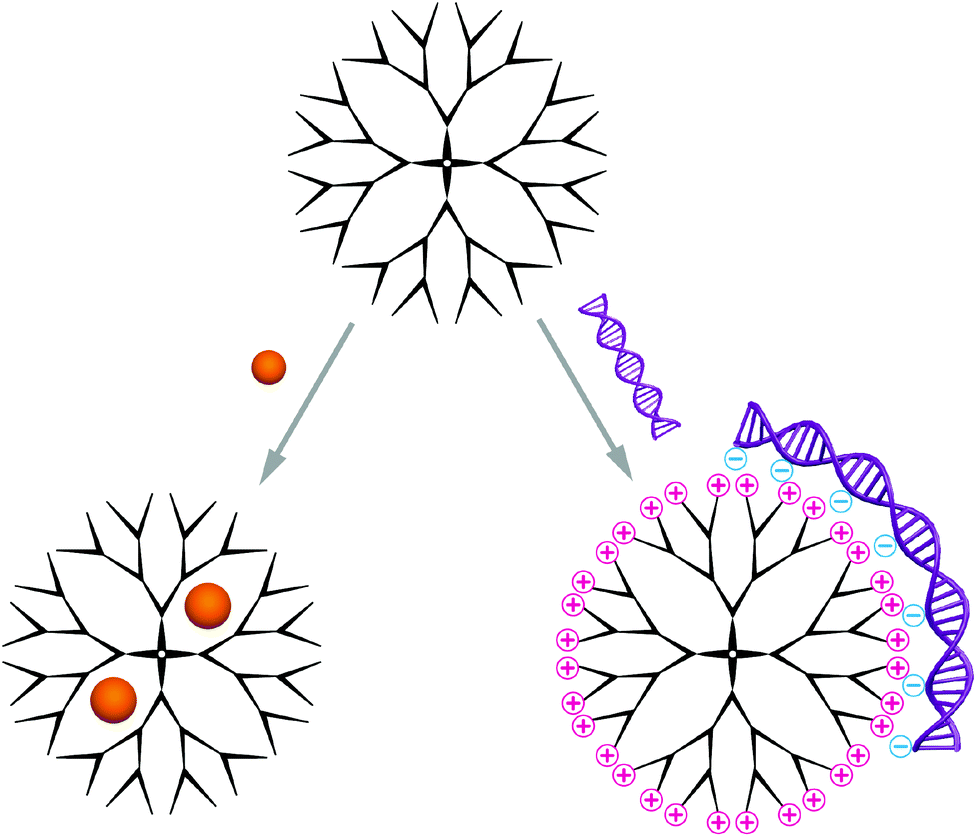 | ||
| Scheme 2 Schematic illustration of the encapsulation of compact guest molecules inside the branched polymers (left) and the complexation of large nuclear acid cargo via charged superficial functional groups (right). | ||
2.1 Delivery applications
2.2 Nanoreactors
Dendrimers are widely used as nanoreactors with entrapped inorganic catalysts, while their application as biological nanoreactors suffers from the same limitation encountered when entrapping enzymes for their delivery: the narrow dimensions of the inner cavity. Nonetheless, dendrimers have been successfully employed in intracellular nanoreactors containing small molecules as catalysts.58 In addition, an enzymatic nanoreactor was formed with laccase after envelopment with linear-dendritic copolymers to improve the stability and catalytic activity of this oxidative enzyme.593. Micelles
Micelles are nano-sized objects with a hydrophobic core and a hydrophilic corona, which can be formed by self-assembly of amphiphilic macromolecules in aqueous environment. Alternatively, reverse micelles are assembled in organic solvents yielding a hydrophilic core and a hydrophobic corona. As this review discusses the delivery and protection of cargo in aqueous environment, it is focused on micelles with hydrophilic exterior and the entrapment of biomacromolecules. Thus, formulations for cancer therapy are excluded and the reader is referred to a recently published review on polymeric micelle formulations in clinical trials.7Micelles are dynamic assemblies, because they are in thermodynamic equilibrium with solubilised macromolecule chains. The assemblies are stable above the critical micellar concentration (CMC), which strongly depends on the properties of the macromolecule and the chemical environment of the micelles. This dynamic nature of micelles calls for stabilising strategies to prevent chain abstraction or carrier dissolution upon dilution or exposure to physiological environments such as blood. Stabilisation can be achieved by addition of cross-linking molecules after the self-assembly process to covalently link building blocks in the corona or in the core. Various crosslinking methods can be applied, such as EDC/NHS coupling, azide alkyne click reactions, disulfide bond formation, thiolene or thiolyne click reactions, UV induced methacrylate crosslinking or maleimidethiol coupling. Ion complexation is a non-covalent alternative for micelle cross-linking.60 Aside from enhanced stability, cross-linking also assures circulation of intact carriers in the organism, however, it also impedes efficient cargo release. This can be overcome by using stimuli responsive cross linkers or introduce responsive moieties in different parts of the micelle building blocks.61,62 Encapsulation of biomacromolecules in micelles can occur before or after the assembly process and is facilitated by hydrophobic, hydrophilic or electrostatic interactions as well as covalent linking of the cargo molecules to the carrier forming amphiphiles (Scheme 3).63
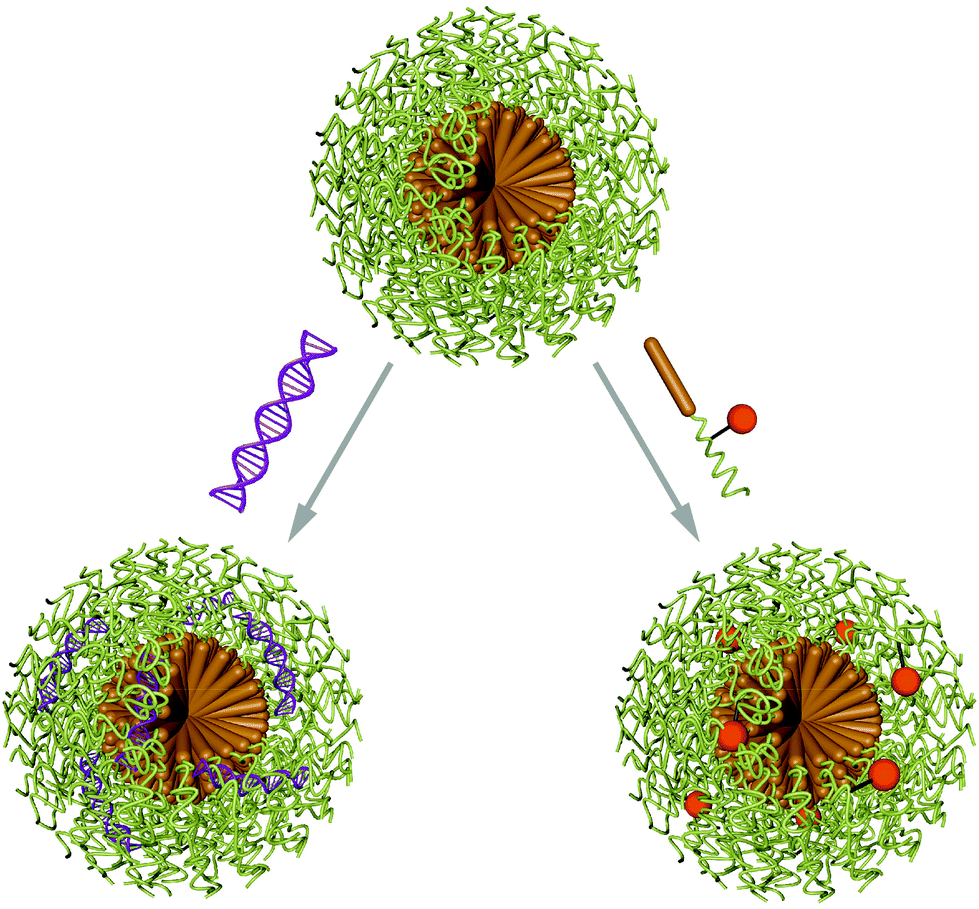 | ||
| Scheme 3 Illustration of a typical spherical micelle assembled from amphiphilic macromolecules (hydrophobic domain in brown, hydrophilic domain in green). Loading of cargo is achieved either pre- (orange sphere) or post-assembly (purple nucleic acid helix). | ||
The densely packed molecules and the lack of a cavity prevent the use of micelles as effective nanoreactors for biomacromolecules, while they have been successfully employed with small molecule catalysts.64–66 Furthermore, nearly all biomacromolecules are hydrophilic and consequently not suited for entrapment in the hydrophobic interior of a micelle.
3.1 Delivery applications
4. Vesicles
Polymer vesicles or polymersomes – as they were first named in 199979 – are micro- or nanosized, spherical and hollow 3D architectures. An aqueous solution is enclosed by a membrane composed of amphiphilic block copolymers (Scheme 4).80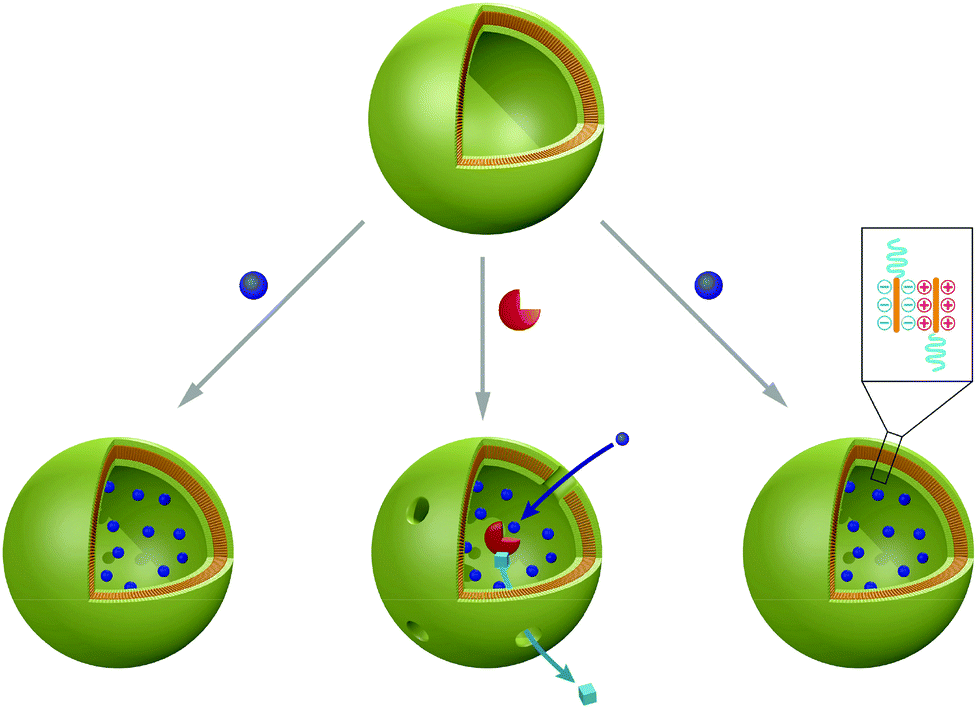 | ||
| Scheme 4 Schematic illustration of a synthetic vesicle (top) showing the possibility of entrapping biomacromolecules and its application as a nanocarrier (bottom, left and right) or as a nanoreactor (bottom, middle). These structures can be obtained by the self-assembly in aqueous solutions of amphiphilic block-copolymers, amphiphilic oligopeptides or oppositely charged macromolecules. | ||
These structures are similar to liposomes (lipid-based vesicles), but possess a membrane composed of higher molecular weight species, which improves the stability to external stress factors such as mechanical shear forces.81 Important parameters affecting the formation of polymer vesicles are the critical micellar concentration (CMC) and the volume ratio of the hydrophilic to hydrophobic block of the amphiphilic copolymer. The self-assembly process is strongly influenced by the equilibrium between the free energy contributions to the self-assembly and kinetic factors.82,83 Hence, introduction of additional blocks or variation of the block lengths induces morphological changes of the final vesicular structures (Fig. 1).84 Two general methods are known for polymersome preparation and they differ in their use or avoidance of organic solvents.85 The block copolymer can either be dissolved in an aqueous solution directly (direct dissolution) or via the hydration of a pre-casted copolymer film (film rehydration) using an aqueous solution under mechanical stirring. In the electroformation method, the application of an electrical field induces vesicle formation. Organic solvent based methods on the other hand involve the use of emulsion templates or dissolution of the polymers in a suitable organic solvent, followed by the injection of this solution into an aqueous phase (co-solvent or the phase inversion technique). It has to be taken into account, however, that residual organic solvent can impede the functionality of biomacromolecular cargo. Polymersomes are particularly attractive because they are able to entrap hydrophilic molecules in their inner cavity86 and at the same time they can insert hydrophobic compounds in their membrane.87 In addition, it is possible to generate hybrid polymer and lipid vesicles by alignment of amphiphilic compounds (for example lipids) at the hydrophobic–hydrophilic interface.88 The membrane of polymersomes is flexible and can undergo conformational changes in order to allow insertion of membrane proteins.89 Such membrane proteins serve as gates for transfer of products/substrate between the inner cavity and the surrounding environment of the vesicles. The release of encapsulated cargo can be achieved by degradation90 or by conformational changes91 when using stimuli-responsive block copolymers. Importantly, polymersomes designed to be used as nanoreactors or even artificial organelles need to be stable for long time periods upon internalisation in cells. To date, a maximum of 48 hours stability has been reported for artificial peroxisomes.92 Depending on the specific application as conventional drug delivery system, as nanoreactors or as artificial organelles, polymersomes have to fulfil different requirements regarding their stability, membrane permeability, biocompatibility and flexibility.
 | ||
| Fig. 1 Exemplary TEM images of 3D vesicular nanoarchitectures. The block copolymers used to assemble these structures are composed of glycerol monomethacrylate (G), 2-hydroxypropyl methacrylate (H), benzyl methacrylate (B) and ethylene glycol dimethacrylate (E). The top row of TEM micrographs represents an overview of the vesicle samples, while the images below provide details of single vesicles. On the left (group A) it is shown that the introduction of an additional B block and changing the polymer block length induces conformational changes in the structure. On the left (group B), similar behaviour is observed by replacement of the B block with the E block. Further, the influence of surfactant on the final conformation of the architecture is demonstrated. Adapted with permission from Chambon et al.84 copyright 2012 of the American Chemical Society. | ||
4.1 Delivery applications
An important aspect in the design of 3D assemblies for medical applications is the biocompatibility of its components. Variations in the preparation conditions (i.e. cross-linking density, UV irradiation time, solvent) during polymer vesicle formation determine their cytotoxic behaviour as shown for poly(ethylene glycol)-b-poly(diethylaminomethacrylate-stat-poly-3,4-dimethylmaleinimidobutylmethacrylate) (PEG-b-PDEAMA-s-PDMIBM) and poly(ethylene glycol)-b-poly(diethylaminomethacrylate-stat-poly-3,4-dimethylmaleinimidoethylmethacrylate) (PEG-b-PDEAMA-s-PDMIEM) poylmersomes.99 Here, long UV irradiation times and the presence of phosphate buffer during the vesicle preparation might induce formation of toxic byproducts.
4.2 Nanoreactors, artificial organelles and synthetic cells
Recently, polymer vesicles have been proposed as compartments for reactions ranging from nanoreactors101 to artificial organelles92,102,103 and cell mimics104 (Fig. 2). For applications as nanoreactors, the vesicles need to be highly stable for prolonged time periods, as opposed to their application as conventional drug delivery systems, where their cargo is released. In this respect, the vesicle membrane needs to be permeable to substrates/products of reactions occurring in the inner cavity. Permeability of the membrane can be generated by different approaches: a specific chemical nature of amphiphilic copolymers105–107 or chemical modification of the membrane after vesicle formation108,109 or insertion of channel proteins inside synthetic membrane.101,110–113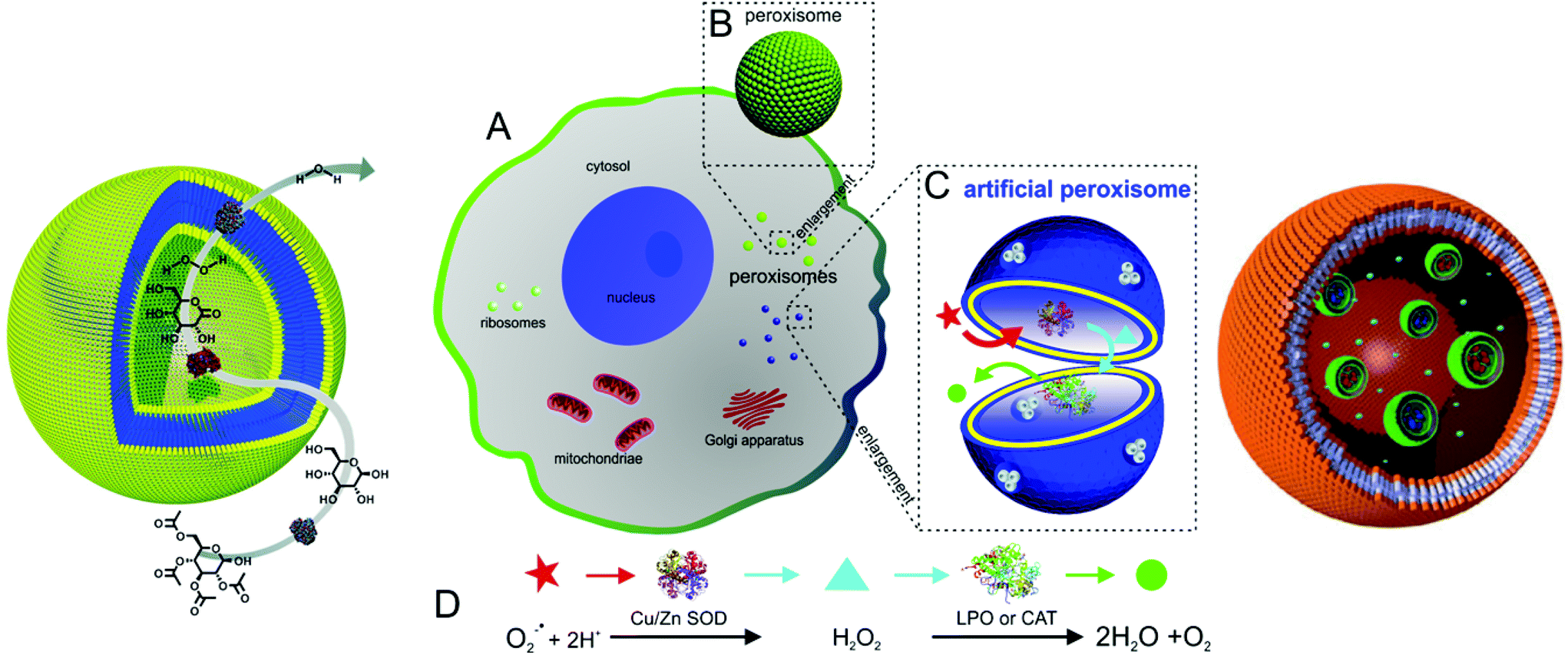 | ||
| Fig. 2 Typical applications of polymersomes as a nanoreactors (left, reproduced with permission from ref. 158), as an artificial peroxisome (centre, adapted with permission from ref. 92, copyright 2013 American Chemical Society) and as a cell mimic (right, adapted with permission from ref. 104). | ||
Several model systems have shown the potential of vesicular nanoreactors. In addition to single enzyme type nanoreactors,103,113,114 entrapped combinations of enzymes participating in reactions have been described.115,116 Haemoglobin was encapsulated in vesicles assembled from poly-(2-methyloxazoline)-b-poly(dimethylsiloxane)-b-poly(2-methyl-oxazoline) (PMOXA-b-PDMS-b-PMOXA) triblock copolymers, where it fulfilled a dual role. Haemoglobin detoxified peroxynitrites present in the vesicular environment and stores oxygen.117 Nanoreactors with triggered activity were introduced by encapsulation of Rose Bengal–bovine serum albumin conjugate (RB–BSA) inside polymersomes composed of PMOXA-b-PDMS-b-PMOXA. This membrane is permeable to oxygen, enabling the nanoreactors to release reactive oxygen species (ROS) “on demand”, thereby promoting cellular toxicity at predetermined time and location.118 Moreover, thymidine phosphorylase encapsulated in polymersomes of the same triblock copolymer maintained stability in blood serum for several days with only a slight decrease in enzyme activity over time.119 Interestingly, an increase of catalytic activity after enzyme encapsulation was reported for trypsin in polystyrene-b-poly(acrylic acid) (PS-b-PAA) block copolymer (BCP) vesicles.120 Successful nanoreactors were also formed with trypanosomavivax nucleoside hydrolase in PMOXA-b-PDMS-b-PMOXA vesicles, which can be applied in enzyme replacement therapy121 and by entrapment of β-lactamase in similar vesicles used for hydrolysis of antibiotics.101 A three-enzyme cascade reaction system has been developed by the encapsulation of glucose oxidase (GOx) in the inner cavity, insertion of Candida Antarctica lipase B (CalB) in the bilayer membrane and immobilisation of horseradish peroxidise (HRP) on the surface of polystyrene-b-poly(l-isocyanoalanine(2-thiophen-3-yl-ethyl)amide) (PS-b-PIAT) copolymer vesicles.115
Recently, research activities extended to the formation of artificial organelles, which represent nanoreactors that are uptaken and active inside cells. The first example of an artificial organelle mimicking natural peroxisomes was realised by co-encapsulation of antioxidant enzymes (Cu/Zn SOD, and lactoperoxidase, LPO, or catalase, CAT) in polymersomes (Fig. 2, centre).92 These enzymes are the main proteins inside peroxisomes and indeed, upon uptake in HeLa cells, the enzymes acted in tandem inside the artificial peroxisomes and allowed simultaneous detection and detoxification of reactive oxygen species (ROS). Moreover, an approach to mimic eukaryotic cells was recently described. Here, a polymersome-in-polymersome system is used to perform cascade reactions across multiple compartments (Fig. 2, right). First, individual enzymes such as Candida antarctica lipase B (CalB), alcalase and alcohol dehydrogenase (ADH) were encapsulated in polystyrene-b-poly(3-(isocyano-lalanyl-aminoethyl)thiophene) (PS-b-PIAT) polymersomes with sizes ranging from 180 to 300 nm.104 Subsequently, these vesicles were encapsulated in micrometer sized polybutadiene-b-poly(ethylene oxide) (PB-b-PEO) polymersomes together with free enzymes and reagents. This design provides a protected environment for the enzymes. The reactions are initiated once reactants and products diffuse across the inner polymersomes membrane. An initial profluorescent substrate undergoes a multi-step, enzyme-catalysed reaction to yield resorufin as a final, fluorescent product.
Further research and optimisation of artificial organelles and polymersome-in-polymersome systems is needed, particularly towards improvements of the encapsulation efficiency (of biomacromolecules and also small polymersomes), cell uptake and addressing specific pathologic conditions. An additional challenge is the enhancement of mechanical stability of polymersomes after internalisation in cells. The current examples show a promising and elegant way to assemble complex systems and gain a deeper understanding of the involved biological processes. Nevertheless, there are still a several issues to be addressed until advanced mimics of cells can be obtained. The ability of polymer vesicles to self-replicate for example or their capability to mimic processes that can be switched on and off – like in the living cell – are some interesting challenges to be investigated in the future.122
4.3 Special vesicular architectures
5. Nanoparticles
Polymer and peptide nanoparticles are solid, in most cases spherically shaped objects with different morphologies. Most commonly multi-compartment micelle (MCM) architectures and polyplexes are reported, which are formed by direct interaction of the payload with the carrier material (see Scheme 5). These self-assembled structures are very promising candidates to serve as carriers not only for small molecules but also for (bio-) macromolecules. MCMs differ from other nano-scale delivery systems, they possess similar volume fractions for entrapment of hydrophilic and hydrophobic payloads. Furthermore, high stability of the nanoparticles is achieved owing to their solid character. Integrated release mechanisms, stealth coatings and specific targeting moieties have been included in the design towards smart and functional nanoparticles based carriers. In contrast to the previously discussed architectures, which form regardless of their cargo, the driving force for polyplex formation is the physical interaction between payload and carrier material. This direct interaction of guest molecules with the carrier polymer/peptide typically occurs either via charge compensation or by entrapment using hydrophobic interactions during nanoparticle formation. These nanoparticles can be unimolecular or they can be composed of a combination of different molecular species. Besides spontaneous degradation, the release of the payloads is achieved via stimuli responsiveness of nanoparticles, provided they have the appropriate chemical nature. Delivered biomacromolecules include enzymes, antigens, proteins, and nucleic acids, which are usually condensed using polycationic molecules. Moreover, positive ratios of cations to the anionic phosphate in nucleic acids (N/P-ratio) favour the adsorption to mammalian cell membranes and internalisation is accelerated due to ionic interactions with negatively charged cell membranes.13 | ||
| Scheme 5 Schematic view of a polyplex and a multicompartment micellar assembly, both carrying condensed nucleic acids (purple). On the left, a random coiled structure derived from properties and interactions of both carrier and nucleic acid payload is shown, whereas the architecture on the right consists of the well-defined arrangement of micelles driven by the amphiphilicity of their carrier material. | ||
Functional nanoparticles can be assembled from synthetic polymers and/or peptides. Often, amphiphilic or charged macromolecules are used to form nanoparticles via self-assembly. The peptides consist of sequences built from naturally occurring amino acids and the predominant synthetic polymer is the FDA approved poly(lactic-co-glycolic) acid (PLGA) copolymer. Owing to its biocompatibility and biodegradability, PLGA is replacing polyethylenimine (PEI), which is cytotoxic at high dosage. In addition, biodegradable polymers composed of are poly(β-amino ester), poly(α-(4-aminobutyl)-L-glycolic acid (PAGA) and polyphosphoester are used for generation of nanoparticles.133 An elegant approach to induce stimuli-responsiveness is to use polymers and peptides containing reductively cleavable groups or pH-sensitive linkers.
However, to date nanoparticles have not been applied to act as nanoreactors, because the intrinsic solid state of nanoparticles does not allow proteins or enzymes to act freely inside. Hence, nanoparticles are specific 3D assemblies serving only for transport and release of the entrapped biomacromolecules.
5.1 Delivery applications
Besides, nucleic acid cargo can also be delivered and released from polymeric nanoparticles. Linear cationic click polymers containing disulfide bonds, secondary amine groups and amide-triazole moieties were reported to deliver and release pDNA in a reductive environment, if the DNA had been condensed with PEI first.151 Moreover, efficient delivery of nucleic acid payload to mesenchymal stem cells, which is of high interest in regenerative medicine, was recently reported.71,152 In this study, a pH-responsive diblock copolymer composed of poly(dimethylaminoethyl methacrylate) (PDMAEMA), poly(butyl methacrylate) (PBMA) and poly(propylacrylic acid) (PAA) was used to form nanoparticles of siRNA and the copolymer. In addition, successful dual gene knockdown in ovarian tumours in vivo is described for shRNA condensed with PLLys/PLGA nanoparticles.153 The use of nuclear localisation signals (NLS) is important for nuclear delivery of DNA. This was observed for PEI/PLGA nanoparticles with covalently attached NF-κB (an NLS), which significantly increased the particle delivery.154
In addition to purely peptidic and purely polymeric systems, polymer–protein conjugates can also form nanoparticles and deliver pDNA, as shown for example with BSA-poly(dimethylamino) ethyl methacrylate (PDMA) based nanoparticles.155
To date, a variety of nanoparticles have been developed and proved their functionality in vitro. However, only few of them also showed applicability in vivo.146,148,151,153,156,157 Despite favourable effects on internalisation, positively charged nanoparticles tend to agglomerate upon contact with serum proteins, leading to accumulation in thin capillaries and ultimately cause blockages of the blood stream. Further, a balance has to be kept between the several desirable requirements for drug delivery systems and the need to reduce the complexity of assembled nanoparticles to avoid possible sources of error in the statistical preparation process.
6. Conclusions
In this review, the four most prominent architectures that can be used to deliver functional biomacromolecules to cells are discussed. All carriers are composed of natural or synthetic polymers, giving rise to a large pool of different functionalities and their combinations. These carriers have been employed to transport a variety of different biomacromolecules, ranging from enzymes to proteins and nucleic acids. An overview of the guest molecules discussed in this review and their corresponding carrier architectures is presented in Table 2. This data collection shows that nanoparticles and dendrimers are preferentially used to transport and deliver nucleic acids, while vesicles in particular are predominantly used to carry enzymes. In addition to ‘simple’ carriers, we also elucidated the application of these 3D architectures as containers for reactions. This strategy involves the encapsulation/insertion of active compounds (enzymes, proteins, mimics) that are free to act in situ, protected by the architecture of the 3D assembly. From model nanoreactors to artificial organelles, these systems combine the functionality of biomacromolecules with the molecular properties of 3D assemblies, such as stability, membrane flexibility and permeability or flexibility. However, the internalisation of nanocarriers into cells requires more and detailed investigations. This often occurs via endocytotic pathways, and the detailed mechanisms are not yet fully understood. Further hurdles come up during approval process of self-assembled systems through public health authorities, because the accumulation in the body and long-term toxicity is difficult to estimate for completely new 3D assemblies. Nevertheless, several exciting and innovative systems have been developed and investigated with respect to their behaviour in cells or in living organisms. These results are very promising and underline that research activities are going towards the right direction, but they also outline the potential for improvements. We hope that this compilation and the remaining challenges to be overcome for each system aid the reader with the selection of an appropriate polymeric carrier for their desired system.| Biomacromolecular guest | Carrier architectures |
|---|---|
| siRNA | Dendrimer,24,40,41,159 micelle,74–76 polymeric vesicle,87 multicompartment micelle,142 polyplex nanoparticle8,44,152,156 |
| DNA | Dendrimer,30,32,33,42,43,45,51,52,55 polymeric vesicle,91,100 peptidic vesicle,131 peptidic nanoparticle,143–145,147,148 polymeric polyplex nanoparticle151,154,157,160,161 |
| Alcalase | Polymeric vesicle104 |
| Alcohol dehydrogenase | Polymeric vesicle104 |
| Antigenic proteins | Polymeric nanoparticle8 |
| Beta-galactosidase | Peptidic nanoparticle135,136 |
| Beta-lactamase | Polymeric vesicle101 |
| Bovine serum albumin | Polymeric vesicle,93,94,96,130 nanoparticle11,12 |
| BSA–Rose Bengal conjugate | Polymeric vesicle118 |
| Candida antarctica lipase B | Polymeric vesicle158 |
| Catalase | Polymeric vesicle162 |
| Cu/Zn superoxide dismutase | Polymeric vesicle,162 polymeric nanoparticles9 |
| Cytochrome C | Polymeric vesicle93,94,96 |
| Haemoglobin | Micelles,61 polymeric vesicles117 |
| Horseradish peroxidase | Polymeric vesicle158 |
| Glucose oxidase | Polymeric vesicles115,158 |
| Immunoglobulin G | Polymeric vesicle,95 peptide vesicle130 |
| Influenza hemagglutinin | Polymeric nanoparticles9 |
| Insulin | Micelle,67 peptide vesicles130 |
| Laccase | Linear-dendritic copolymer59 |
| Lactoperoxidase | Polymeric vesicles92 |
| Lysozyme | Dendrimer,29 polymeric vesicles,93,94 peptide vesicles130 |
| Myoglobin | PICsomes125 |
| Ovalbumin | Micelles,68,69 polymeric vesicles93,94,97 polymeric nanoparticles141 |
| Thymidine phosphorylase | Polymeric vesicles119 |
| Trypanosomavivax nucleoside hydrolase | Polymeric vesicles121 |
| Trypsin | Polymeric vesicles120 |
Acknowledgements
Support by the Swiss National Science Foundation, the Swiss Nanoscience Institute and the Gebert Rüf Stiftung is gratefully acknowledged. Further, Ruth Pfalzberger is thanked for help with the preparation of images.Notes and references
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- H. D. Maynard, Nat. Chem., 2013, 5, 557–558 CrossRef CAS PubMed.
- Y. Nakayama, Acc. Chem. Res., 2012, 45, 994–1004 CrossRef CAS PubMed.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- C. Y. Ang, S. Y. Tan and Y. Zhao, Org. Biomol. Chem., 2014, 12, 4776–4806 CAS.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- X. Xu, K. Xie, X.-Q. Zhang, E. M. Pridgen, G. Y. Park, D. S. Cui, J. Shi, J. Wu, P. W. Kantoff, S. J. Lippard, R. Langer, G. C. Walker and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18638–18643 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41, 5599–5612 RSC.
- J. Wu, N. Kamaly, J. Shi, L. Zhao, Z. Xiao, G. Hollett, R. John, S. Ray, X. Xu, X. Zhang, P. W. Kantoff and O. C. Farokhzad, Angew. Chem., Int. Ed., 2014 DOI:10.1002/anie.201404766.
- J. Wu and C.-C. Chu, J. Mater. Chem. B, 2013, 1, 353–360 RSC.
- S. Hong, P. R. Leroueil, E. K. Janus, J. L. Peters, M.-M. Kober, M. T. Islam, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Bioconjugate Chem., 2006, 17, 728–734 CrossRef CAS PubMed.
- J. Sebestik, P. Niederhafner and J. Jezek, Amino Acids, 2010, 40, 301–370 CrossRef PubMed.
- C. G. Palivan, O. Fischer-Onaca, M. Delcea, F. Itel and W. Meier, Chem. Soc. Rev., 2012, 41, 2800 RSC.
- P. Tanner, S. Egli, V. Balasubramanian, O. Onaca, C. G. Palivan and W. Meier, FEBS Lett., 2011, 585, 1699–1706 CrossRef CAS PubMed.
- P. Hodge, Nature, 1993, 362, 18–19 CrossRef.
- Y. Segawa, T. Higashihara and M. Ueda, Polym. Chem., 2013, 4, 1746–1759 RSC.
- J. Khandare, M. Calderón, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- C. Wörner and R. Mülhaupt, Angew. Chem., Int. Ed., 1993, 32, 1306–1308 CrossRef PubMed.
- E. de Brabander van den Berg and E. W. Meijer, Angew. Chem., Int. Ed., 1993, 32, 1308–1311 CrossRef PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- W. Fischer, M. Calderón, A. Schulz, I. Andreou, M. Weber and R. Haag, Bioconjugate Chem., 2010, 21, 1744–1752 CrossRef CAS PubMed.
- J. F. Jansen, E. M. de Brabander-van den Berg and E. W. Meijer, Science, 1994, 266, 1226–1229 CAS.
- J. F. Jansen, E. W. Meijer and E. M. de Brabander-van den Berg, J. Am. Chem. Soc., 1995, 117, 4417–4418 CrossRef CAS.
- J. Zhu and X. Shi, J. Mater. Chem. B, 2013, 1, 4199–4211 RSC.
- C. S. Popeney, M. C. Lukowiak, C. Böttcher, B. Schade, P. Welker, D. Mangoldt, G. Gunkel, Z. Guan and R. Haag, ACS Macro Lett., 2012, 1, 564–567 CrossRef CAS.
- X. Zhang, J. Zhao, Y. Wen, C. Zhu, J. Yang and F. Yao, Carbohydr. Polym., 2013, 98, 1326–1334 CrossRef CAS PubMed.
- M. Byrne, D. Victory, A. Hibbitts, M. Lanigan, A. Heise and S.-A. Cryan, Biomater. Sci., 2013, 1, 1223–1234 RSC.
- Z. Kadlecova, L. Baldi, D. Hacker, F. M. Wurm and H.-A. Klok, Biomacromolecules, 2012, 13, 3127–3137 CrossRef CAS PubMed.
- A. Mann, R. Richa and M. Ganguli, J. Controlled Release, 2008, 125, 252–262 CrossRef CAS PubMed.
- A. U. Bielinska, J. F. Kukowska-Latallo and J. R. Baker, Biochim. Biophys. Acta, 1997, 1353, 180–190 CrossRef CAS.
- S. V. Lyulin, I. Vattulainen and A. A. Gurtovenko, Macromolecules, 2008, 41, 4961–4968 CrossRef CAS.
- V. A. Kabanov, A. B. Zezin, V. B. Rogacheva, Z. G. Gulyaeva, M. F. Zansochova, J. G. H. Joosten and J. Brackman, Macromolecules, 1999, 32, 1904–1909 CrossRef CAS.
- S. C. De Smedt, J. Demeester and W. E. Hennink, Pharm. Res., 2000, 17, 113–126 CrossRef CAS.
- J. Haensler and F. C. Szoka Jr., Bioconjugate Chem., 1993, 4, 372–379 CrossRef CAS.
- W. Fischer, M. Calderón and R. Haag, in Topics in Current Chemistry, Springer, Berlin Heidelberg, 2010, vol. 296, pp. 95–129 Search PubMed.
- J. F. Kukowska-Latallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897–4902 CrossRef CAS.
- J. Reyes-Reveles, R. Sedaghat-Herati, D. R. Gilley, A. M. Schaeffer, K. C. Ghosh, T. D. Greene, H. E. Gann, W. A. Dowler, S. Kramer, J. M. Dean and R. K. Delong, Biomacromolecules, 2013, 14, 4108–4115 CrossRef CAS PubMed.
- O. Taratula, O. B. Garbuzenko, P. Kirkpatrick, I. Pandya, R. Savla, V. P. Pozharov, H. He and T. Minko, J. Controlled Release, 2009, 140, 284–293 CrossRef CAS PubMed.
- K. Kono, H. Akiyama, T. Takahashi, T. Takagishi and A. Harada, Bioconjugate Chem., 2005, 16, 208–214 CrossRef CAS PubMed.
- J. S. Choi, K. Nam, J.-Y. Park, J.-B. Kim, J.-K. Lee and J.-S. Park, J. Controlled Release, 2004, 99, 445–456 CrossRef CAS PubMed.
- H. Zeng, H. C. Little, T. N. Tiambeng, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2013, 135, 4962–4965 CrossRef CAS PubMed.
- M. Yamagata, T. Kawano, K. Shiba, T. Mori, Y. Katayama and T. Niidome, Bioorg. Med. Chem., 2007, 15, 526–532 CrossRef CAS PubMed.
- H. G. Abdelhady, Y.-L. Lin, H. Sun and M. E. H. ElSayed, PLoS One, 2013, 8, e61710 CAS.
- M. A. Gosselin, W. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989–994 CrossRef CAS PubMed.
- Y. J. Kwon, Acc. Chem. Res., 2012, 45, 1077–1088 CrossRef CAS PubMed.
- H. H. Chen, Y.-P. Ho, X. Jiang, H.-Q. Mao, T.-H. Wang and K. W. Leong, Mol. Ther., 2008, 16, 324–332 CrossRef CAS PubMed.
- G. M. Ryan, L. M. Kaminskas, B. D. Kelly, D. J. Owen, M. P. McIntosh and C. J. H. Porter, Mol. Pharmaceutics, 2013, 10, 2986–2995 CAS.
- K. C. Wood, S. M. Azarin, W. Arap, R. Pasqualini, R. Langer and P. T. Hammond, Bioconjugate Chem., 2008, 19, 403–405 CrossRef CAS PubMed.
- D. Pandita, J. L. Santos, J. Rodrigues, A. P. Pêgo, P. L. Granja and H. Tomás, Biomacromolecules, 2011, 12, 472–481 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- T. Okuda, T. Niidome and H. Aoyagi, J. Controlled Release, 2004, 98, 325–332 CrossRef CAS PubMed.
- J.-H. S. Kuo, M.-J. Liou and H.-C. Chiu, Mol. Pharmaceutics, 2010, 7, 805–814 CAS.
- A. D. Wong, M. A. DeWit and E. R. Gillies, Adv. Drug Delivery Rev., 2012, 64, 1031–1045 CrossRef CAS PubMed.
- J. L. Santos, E. Oramas, A. P. Pêgo, P. L. Granja and H. Tomás, J. Controlled Release, 2009, 134, 141–148 CrossRef CAS PubMed.
- L. Wang, Q. Yang, G. Ma, W. Lin, Z. Wang, M. Huang and S. Chen, J. Mater. Chem. B, 2013, 1, 4259–4266 RSC.
- I. Gitsov, J. Hamzik, J. Ryan, A. Simonyan, J. P. Nakas, S. Omori, A. Krastanov, T. Cohen and S. W. Tanenbaum, Biomacromolecules, 2008, 9, 804–811 CrossRef CAS PubMed.
- X. Huang, Y. Xiao and M. Lang, Carbohydr. Polym., 2012, 87, 790–798 CrossRef CAS PubMed.
- Y. Li, K. Xiao, W. Zhu, W. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2013, 1–16 Search PubMed.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2012, 38, 503–535 CrossRef PubMed.
- A. Kowalczuk, R. Trzcinska, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2013, 39, 43–86 CrossRef PubMed.
- P. Cotanda, A. Lu, J. P. Patterson, N. Petzetakis and R. K. O'Reilly, Macromolecules, 2012, 45, 2377–2384 CrossRef CAS.
- P. Cotanda and R. K. O'Reilly, Chem. Commun., 2012, 48, 10280–10282 RSC.
- A. Lu and R. K. O'Reilly, Curr. Opin. Biotechnol., 2013, 24, 639–645 CrossRef CAS PubMed.
- B. Wang, R. Ma, G. Liu, Y. Li, X. Liu, Y. An and L. Shi, Langmuir, 2009, 25, 12522–12528 CrossRef CAS PubMed.
- J. K. Eby, K. Y. Dane, C. P. O'Neil, S. Hirosue, M. A. Swartz and J. A. Hubbell, Acta Biomater., 2012, 8, 3210–3217 CrossRef CAS PubMed.
- Z. Luo, P. Li, J. Deng, N. Gao, Y. Zhang, H. Pan, L. Liu, C. Wang, L. Cai and Y. Ma, J. Controlled Release, 2013, 170, 259–267 CrossRef CAS PubMed.
- B. Li, T. Li, G. Chen, X. Li, L. Yan, Z. Xie, X. Jing and Y. Huang, Macromol. Biosci., 2013, 13, 893–902 CrossRef CAS PubMed.
- Y. Lee and K. Kataoka, in Advances in Polymer Science, Springer, Berlin, Heidelberg, 2012, vol. 249, pp. 95–134 Search PubMed.
- H. Parhiz, W. T. Shier and M. Ramezani, Int. J. Pharm., 2013, 457, 237–259 CrossRef CAS PubMed.
- M. Ahmed and R. Narain, Prog. Polym. Sci., 2013, 38, 767–790 CrossRef CAS PubMed.
- M. Omedes Pujol, D. J. L. Coleman, C. D. Allen, O. Heidenreich and D. A. Fulton, J. Controlled Release, 2013, 172, 939–945 CrossRef CAS PubMed.
- M. C. Palanca-Wessels, A. J. Convertine, R. Cutler-Strom, G. C. Booth, F. Lee, G. Y. Berguig, P. S. Stayton and O. W. Press, Mol. Ther., 2011, 19, 1529–1537 CrossRef CAS PubMed.
- B. B. Lundy, A. Convertine, M. Miteva and P. S. Stayton, Bioconjugate Chem., 2013, 24, 398–407 CrossRef CAS PubMed.
- A. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yang, P. L. R. Ee, P. T. Hammond and Y.-Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228–1246 CrossRef CAS PubMed.
- B. M. Discher, Science, 1999, 284, 1143–1146 CrossRef CAS.
- D. E. Discher, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- S.-H. Kim, J. Nam, J. W. Kim, D.-H. Kim, S.-H. Han and D. A. Weitz, Lab Chip, 2013, 13, 1351–1356 RSC.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- F. Caruso and K. Ariga, Modern Techniques for Nano- and Microreactors/-reactions, Springer, Berlin, Heidelberg, 2010 Search PubMed.
- Y. Du, W. Chen, M. Zheng, F. Meng and Z. Zhong, Biomaterials, 2012, 33, 7291–7299 CrossRef CAS PubMed.
- H.-O. Kim, E. Kim, Y. An, J. Choi, E. Jang, E. B. Choi, A. Kukreja, M.-H. Kim, B. Kang, D.-J. Kim, J.-S. Suh, Y.-M. Huh and S. Haam, Macromol. Biosci., 2013, 13, 745–754 CrossRef CAS PubMed.
- J.-F. Le Meins, C. Schatz, S. Lecommandoux and O. Sandre, Mater. Today, 2013, 16, 397–402 CrossRef CAS PubMed.
- V. Pata and N. Dan, Biophys. J., 2003, 85, 2111–2118 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS PubMed.
- T. Matini, N. Francini, A. Battocchio, S. G. Spain, G. Mantovani, M. J. Vicent, J. Sanchis, E. Gallon, F. Mastrotto, S. Salmaso, P. Caliceti and C. Alexander, Polym. Chem., 2014, 5, 1626–1636 RSC.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS PubMed.
- G. Liu, S. Ma, S. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2010, 31, 7575–7585 CrossRef CAS PubMed.
- R. Cheng, F. Meng, S. Ma, H. Xu, H. Liu, X. Jing and Z. Zhong, J. Mater. Chem., 2011, 21, 19013–19020 RSC.
- I. Canton, M. Massignani, N. Patikarnmonthon, L. Chierico, J. Robertson, S. A. Renshaw, N. J. Warren, J. P. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, FASEB J., 2013, 27, 98–108 CrossRef CAS PubMed.
- J. Zhang, L. Wu, F. Meng, Z. Wang, C. Deng, H. Liu and Z. Zhong, Langmuir, 2012, 28, 2056–2065 CrossRef CAS PubMed.
- A. Stano, E. A. Scott, K. Y. Dane, M. A. Swartz and J. A. Hubbell, Biomaterials, 2013, 34, 4339–4346 CrossRef CAS PubMed.
- R. Tang, C. S. Kim, D. J. Solfiell, S. Rana, R. Mout, E. M. Velázquez-Delgado, A. Chompoosor, Y. Jeong, B. Yan, Z.-J. Zhu, C. Kim, J. A. Hardy and V. M. Rotello, ACS Nano, 2013, 7, 6667–6673 CrossRef CAS PubMed.
- J. Gaitzsch, I. Canton, D. Appelhans, G. Battaglia and B. Voit, Biomacromolecules, 2012, 13, 4188–4195 CAS.
- Z. Huang, W. Teng, L. Liu, L. Wang, Q. Wang and Y. Dong, Nanotechnology, 2013, 24, 265104 CrossRef PubMed.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434 RSC.
- N. Ben-Haim, P. Broz, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2008, 8, 1368–1373 CrossRef CAS PubMed.
- S. F. M. van Dongen, W. P. R. Verdurmen, R. J. R. W. Peters, R. J. M. Nolte, R. Brock and J. C. M. van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213–7216 CrossRef PubMed.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2013, 53, 146–150 CrossRef PubMed.
- A. Napoli, M. J. Boerakker, N. Tirelli, R. J. M. Nolte, N. A. J. M. Sommerdijk and J. A. Hubbell, Langmuir, 2004, 20, 3487–3491 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS PubMed.
- A. Carlsen, N. Glaser, J.-F. Le Meins and S. Lecommandoux, Langmuir, 2011, 27, 4884–4890 CrossRef CAS PubMed.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, J. Am. Chem. Soc., 2013, 135, 9204–9212 CrossRef CAS PubMed.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- M. Sauer and W. Meier, Aust. J. Chem., 2001, 54, 149–151 CrossRef CAS.
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5064–5068 CrossRef CAS PubMed.
- M. Nallani, S. Benito, O. Onaca, A. Graff, M. Lindemann, M. Winterhalter, W. Meier and U. Schwaneberg, J. Biotechnol., 2006, 123, 50–59 CrossRef CAS PubMed.
- M. Grzelakowski, O. Onaca, P. Rigler, M. Kumar and W. Meier, Small, 2009, 5, 2545–2548 CrossRef CAS PubMed.
- K. Langowska, C. G. Palivan and W. Meier, Chem. Commun., 2012, 49, 128–130 RSC.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2008, 15, 1107–1114 CrossRef PubMed.
- P. Tanner, O. Onaca, V. Balasubramanian, W. Meier and C. G. Palivan, Chem. – Eur. J., 2011, 17, 4552–4560 CrossRef CAS PubMed.
- D. Dobrunz, A. C. Toma, P. Tanner, T. Pfohl and C. G. Palivan, Langmuir, 2012, 28, 15889–15899 CrossRef CAS PubMed.
- P. Baumann, V. Balasubramanian, O. Onaca-Fischer, A. Sienkiewicz and C. G. Palivan, Nanoscale, 2012, 5, 217–224 RSC.
- C. De Vocht, A. Ranquin, R. Willaert, J. A. Van Ginderachter, T. Vanhaecke, V. Rogiers, W. Versées, P. Van Gelder and J. Steyaert, J. Controlled Release, 2009, 137, 246–254 CrossRef CAS PubMed.
- Q. Chen, H. Schönherr and G. J. Vancso, Small, 2009, 5, 1436–1445 CrossRef CAS PubMed.
- A. Ranquin, W. Versées, W. Meier, J. Steyaert and P. Van Gelder, Nano Lett., 2005, 5, 2220–2224 CrossRef CAS PubMed.
- R. E. Roodbeen and J. C. M. van Hest, BioEssays, 2009, 31, 1299–1308 CrossRef CAS PubMed.
- A. Koide, A. Kishimura, K. Osada, W.-D. Jang, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2006, 128, 5988–5989 CrossRef CAS PubMed.
- A. Kishimura, Polym. J., 2013, 45, 892–897 CrossRef CAS.
- A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085–6088 CrossRef CAS PubMed.
- B. Tian, X. Tao, T. Ren, Y. Weng, X. Lin, Y. Zhang and X. Tang, J. Mater. Chem., 2012, 22, 17404 RSC.
- L. Zhao, N. Li, K. Wang, C. Shi, L. Zhang and Y. Luan, Biomaterials, 2013, 1–18 Search PubMed.
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed.
- S. Kimura, D.-H. Kim, J. Sugiyama and Y. Imanishi, Langmuir, 1999, 15, 4461–4463 CrossRef CAS.
- A. Mishra, J. J. Panda, A. Basu and V. S. Chauhan, Langmuir, 2008, 24, 4571–4576 CrossRef CAS PubMed.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 119, 2483–2486 CrossRef PubMed.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2006, 6, 52–57 CrossRef PubMed.
- J. Luten, C. F. van Nostrum, S. C. De Smedt and W. E. Hennink, J. Controlled Release, 2008, 126, 97–110 CrossRef CAS PubMed.
- C. Weber, C. Coester, J. Kreuter and K. Langer, Int. J. Pharm., 2000, 194, 91–102 CrossRef CAS.
- L. H. Estrada, S. Chu and J. A. Champion, J. Pharm. Sci., 2014, 103, 1863–1871 CrossRef CAS PubMed.
- H. J. Lee, H. H. Park, J. A. Kim, J. H. Park, J. Ryu, J. Choi, J. Lee, W. J. Rhee and T. H. Park, Biomaterials, 2014, 35, 1696–1704 CrossRef CAS PubMed.
- Z. Zhang, S. Tongchusak, Y. Mizukami, Y. J. Kang, T. Ioji, M. Touma, B. Reinhold, D. B. Keskin, E. L. Reinherz and T. Sasada, Biomaterials, 2011, 32, 3666–3678 CrossRef CAS PubMed.
- T. Yoshikawa, N. Okada, A. Oda, K. Matsuo, K. Matsuo, Y. Mukai, Y. Yoshioka, T. Akagi, M. Akashi and S. Nakagawa, Biochem. Biophys. Res. Commun., 2008, 366, 408–413 CrossRef CAS PubMed.
- T. Jung, W. Kamm, A. Breitenbach, K. D. Hungerer, E. Hundt and T. Kissel, Pharm. Res., 2001, 18, 352–360 CAS.
- A. Vila, A. Sánchez, M. Tobío, P. Calvo and M. J. Alonso, J. Controlled Release, 2002, 78, 15–24 CrossRef CAS.
- S. L. Demento, W. Cui, J. M. Criscione, E. Stern, J. Tulipan, S. M. Kaech and T. M. Fahmy, Biomaterials, 2012, 33, 4957–4964 CrossRef CAS PubMed.
- D. de Bruyn Ouboter, T. Schuster, V. Shanker, M. Heim and W. Meier, J. Biomed. Mater. Res., 2013, 102, 1155–1163 CrossRef PubMed.
- Rajpal, A. Mann, R. Khanduri, R. J. Naik and M. Ganguli, J. Controlled Release, 2012, 157, 260–271 CrossRef CAS PubMed.
- X. D. Guo, N. Wiradharma, S. Q. Liu, L. J. Zhang, M. Khan, Y. Qian and Y.-Y. Yang, Biomaterials, 2012, 33, 6284–6291 CrossRef CAS PubMed.
- J.-X. Chen, X.-D. Xu, S. Yang, J. Yang, R.-X. Zhuo and X.-Z. Zhang, Macromol. Biosci., 2012, 13, 84–92 CrossRef PubMed.
- C. Föst, F. Duwe, M. Hellriegel, S. Schweyer, G. Emons and C. Gründker, Oncol. Rep., 2011, 25, 1481–1487 Search PubMed.
- Q. Tang, B. Cao, H. Wu and G. Cheng, Langmuir, 2012, 28, 16126–16132 CrossRef CAS PubMed.
- J. Gu, X. Wang, X. Jiang, Y. Chen, L. Chen, X. Fang and X. Sha, Biomaterials, 2012, 33, 644–658 CrossRef CAS PubMed.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, Cell. Mol. Life Sci., 2005, 62, 1839–1849 CrossRef CAS PubMed.
- Y. Lee and K. Kataoka, in Advances in Polymer Science, Springer, Berlin Heidelberg, 2012, vol. 249, pp. 95–134 Search PubMed.
- Y. Gao, L. Chen, Z. Zhang, Y. Chen and Y. Li, Biomaterials, 2011, 32, 1738–1747 CrossRef CAS PubMed.
- D. S. W. Benoit and M. E. Boutin, Biomacromolecules, 2012, 13, 3841–3849 CrossRef CAS PubMed.
- L. Zou, X. Song, T. Yi, S. Li, H. Deng, X. Chen, Z. Li, Y. Bai, Q. Zhong, Y. Wei and X. Zhao, Cancer Gene Ther., 2013, 20, 242–250 CrossRef CAS PubMed.
- T. Kanazawa, Y. Takashima, M. Murakoshi, Y. Nakai and H. Okada, Int. J. Pharm., 2009, 379, 187–195 CrossRef CAS PubMed.
- J. Zhang, Y. Lei, A. Dhaliwal, Q. K. Ng, J. Du, M. Yan, Y. Lu and T. Segura, Biomacromolecules, 2011, 12, 1006–1014 CrossRef CAS PubMed.
- D. S. Wilson, G. Dalmasso, L. Wang, S. V. Sitaraman, D. Merlin and N. Murthy, Nat. Mater., 2010, 9, 923–928 CrossRef CAS PubMed.
- S.-W. Kang, H.-W. Lim, S.-W. Seo, O. Jeon, M. Lee and B.-S. Kim, Biomaterials, 2008, 29, 1109–1117 CrossRef CAS PubMed.
- D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS PubMed.
- P. Ofek, W. Fischer, M. Calderon, R. Haag and R. Satchi-Fainaro, FASEB J., 2010, 24, 3122–3134 CrossRef CAS PubMed.
- Y. Patil and J. Panyam, Int. J. Pharm., 2009, 367, 195–203 CrossRef CAS PubMed.
- M. Sedlak, C. Konak and J. Dybal, Open Conf. Proc. J., 2010, 1, 87–90 CAS.
- I. Louzao and J. C. M. van Hest, Biomacromolecules, 2013, 14, 2364–2372 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |