
Cell-derived matrices for tissue engineering and regenerative medicine applications
Lindsay E.
Fitzpatrick
*a and
Todd C.
McDevitt
bc
aDepartment of Chemical Engineering, Queen's University, Kingston, Canada. E-mail: lindsay.fitzpatrick@queensu.ca
bThe Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology/Emory University, Atlanta, Georgia, USA
cThe Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia, USA
First published on 28th August 2014
Abstract
The development and application of decellularized extracellular matrices (ECM) has grown rapidly in the fields of cell biology, tissue engineering and regenerative medicine in recent years. Similar to decellularized tissues and whole organs, cell-derived matrices (CDM) represent bioactive, biocompatible materials consisting of a complex assembly of fibrillar proteins, matrix macromolecules and associated growth factors that often recapitulate, at least to some extent, the composition and organization of native ECM microenvironments. The unique ability to engineer CDM de novo based on cell source and culture methods makes them an attractive alternative to conventional allogeneic and xenogeneic tissue-derived matrices that are currently harvested from cadaveric sources, suffer from inherent heterogeneity, and have limited ability for customization. Although CDM have been investigated for a number of biomedical applications, including adhesive cell culture substrates, synthetic scaffold coatings, and tissue engineered products, such as heart valves and vascular grafts, the state of the field is still at a relatively nascent stage of development. In this review, we provide an overview of the various applications of CDM and discuss successes to date, current limitations and future directions.
 Lindsay E. Fitzpatrick | Lindsay Fitzpatrick is currently an assistant professor in the Department of Chemical Engineering at Queen's University. She graduated from McMaster University in 2006 with a B.Eng. in Chemical Engineering and Biosciences, and then completed her Ph.D. under the supervision of Michael Sefton at the University of Toronto in 2012. For her postdoctoral training, Lindsay worked with Todd McDevitt at the Georgia Institute of Technology. Her current research interests include innate immune responses to biomaterials and developing cell-derived materials for wound healing applications. |
 Todd C. McDevitt | Todd McDevitt is the Carol Ann and David D. Flanagan Professor in the Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory University, a Faculty Member in the Parker H. Petit Institute for Bioengineering and Bioscience, and Director of the Stem Cell Engineering Center at Georgia Tech. The McDevitt laboratory focuses on the application of microscale technologies to engineer stem cell fate and tissue morphogenesis, as well as provide scalable and robust approaches for stem cell bioprocessing and engineering of regenerative molecular therapies from stem cells. |
Introduction
The extracellular matrix (ECM) of tissues is a complex, highly organized assembly of macromolecules and incorporated signalling factors. In addition to providing tissue structure and a substrate for cell adhesion, the ECM also contributes cues for cell migration, proliferation, differentiation and survival, which are essential processes in development, homeostasis and tissue repair. The primary constituents of the ECM include fibrillar proteins (e.g. collagens, fibronectin, laminin), glycosaminoglycans (GAGs; e.g. heparan sulphate, chondroitin sulphate, hyaluronan), proteoglycans (e.g. decorin, versican, aggrecan) and matricellular proteins (e.g. osteopontin, thrombospondin). As well as contributing directly to the overall structural and mechanical properties of tissue, ECM components are also involved in the binding, sequestration and stabilization of signalling molecules incorporated within the matrix. The composition and distribution of specific matrix components varies with the type of tissue, and can also be altered by the tissue's developmental stage and/or pathological state.Biomaterials used in tissue engineering and regenerative medicine attempt to recapitulate the multifactorial aspects of ECM function, however synthetic materials and matrices formed from isolated biological materials, such as collagen, fibrin or hyaluronan, fail to achieve the molecular complexity and organization of native tissue matrices. This has motivated the use of native ECM itself as a biomaterial source. ECM derived from allogenic or xenogenic tissues, reviewed in-depth elsewhere,1–3 has emerged as an increasingly common source of natural biomaterials. Decellularized or devitalized skin, small intestinal submucosa, urinary bladder matrix, amniotic membrane, ligaments, blood vessels and heart valves are but a few examples of tissue ECM used in pre-clinical research and clinical therapies. The use of whole organ decellularization is also of interest for tissue engineering applications due to the preserved tissue architecture.4–7 However, naturally derived matrices are inherently limited in their capacity for manipulation beyond processing procedures and tissue source, and possess uncontrolled variability that may arise from the age, health and gender of individual sources.
ECM derived from cultured mammalian cells provides an alternative to native tissue-derived ECM. Cell-derived matrices (CDM) contain a complex yet organized mixture of macromolecules that can mimic aspects of native tissue microenvironments. In contrast to tissue-derived matrices, CDM have greater ability for customization by selecting the type(s) of cells used to generate the ECM, the culture system (e.g. 2D versus 3D culture; static versus perfusion), the application of external stimuli to modulate ECM production and also the ability to genetically modify the source cells to augment or silence the expression of target molecules. CDM can also be used to confer bioactivity to synthetic scaffolds through the deposition of matrix molecules on the scaffold surface. The trade-off with respect to tissue-derived matrices is that CDM typically have poorer mechanical properties, making them unsuitable for certain applications. However, mechanical preconditioning applied prior to decellularization has been shown to significantly improve the mechanical properties of CDM, particularly for cardiovascular applications.8,9
The purpose of this review is to explore the current uses of CDM in tissue engineering, stem cell therapies and other biomedical applications, and discuss their current successes, limitations and future directions. CDM have been used in a variety of applications, including as coatings for synthetic scaffolds, biomimetic microenvironments for stem cell differentiation, and decellularized tissue engineered heart valves and vascular grafts. While several results are promising, there remains a fair amount of variability in the reported effects CDM have in vitro and in vivo, likely due to many differences in the cells, methods and applications examined thus far. Throughout this review we will attempt to identify key factors, such as cell source(s), processing techniques and experimental designs, that may help to explain these inconsistencies and provide insights into the best practices for the derivation and application of CDM in biomedical engineering.
Overview of cell-derived matrices fabrication
Generating CDM involves three major considerations: cell source, culture method and processing method. The cell source is the primary determinant of the resulting CDM composition. Cells sourced from different tissues typically yield matrices that mimic the relative composition of the natural tissue matrix. Fibroblasts, a predominant cell type found in connective tissue, are well known for their ability to produce a collage-rich extracellular matrix, a characteristic that has been exploited in a number of different biomedical applications including vascular and dermal tissue engineering. Mesenchymal stem cells (MSCs) are also a common source of CDM due to their ability to deposit ECM that mimics various tissues (e.g. bone, cartilage, adipose) depending on culture conditions, and their prevalent use in tissue engineering applications. Recently, Lu et al. compared the composition of matrices derived from fibroblasts, chondrocytes and MSCs.10 All matrices stained positive for type I collagen, type III collagen, fibronectin, vitronectin, laminin and decorin. However, versican was only detected in chondrocyte CDM, and aggrecan was detected in both the MSC and chondrocyte matrix, but not the fibroblast CDM.10 Although this particular study did not quantify the relative amounts of individual matrix components, it does demonstrate the effect of cell source on matrix composition, and the relative complexity of CDM.Both primary cells and cell lines have been used to generate CDM. Primary cells harvested directly from tissues without passaging are typically considered to be an ideal cell source for tissue engineering and biomedical applications, as they closely resemble their native in vivo phenotype and are arguably more capable of generating a matrix that resembles the native microenvironments. An important caveat is that the native matrix can be generated and remodelled by multiple cell types, whereas primary cells are often sorted or isolated into a (quasi)-monoculture. Obtaining sufficient numbers of primary cells for a given application is often impossible or impractical; therefore primary cells are commonly expanded in vitro as finite cell lines. However, passaging of cells selects for the most rapidly dividing subpopulations and the in vitro environment can alter cell behaviour, resulting in a phenotypic drift from the native cell state. This is commonly observed for chondrocytes that undergo dedifferentiation and senesce,11 which has been shown to affect the quality of the deposited ECM.12 Alternatively, continuous (i.e. immortalized) cells lines can be used to generate CDM because they are easily cultured and can yield large numbers of homogeneous cells. However, continuous cell lines are often tumor-derived and can differ significantly from primary cell phenotypes. If mimicking a particular tissue microenvironment is important, a continuous cell line may not be an appropriate source. In contrast, if a particular molecular make-up is not critical, or a cell line produces a matrix with the desired molecular components (i.e. a tumor cell line-derived matrix that promotes angiogenesis), a continuous cell line may prove to be a simple solution. Genetic engineering of cell lines may also enable artificial depletion or enhancement of certain key molecular elements within a CDM.
A second important consideration that affects CDM production is the manner in which the cells are cultured. The most common methods for generating CDM are to culture cells for an extended period of time as adherent monolayers (2D) or on scaffold surfaces (coatings), as multicellular aggregates (3D), or within a degradable carrier until sufficient ECM has been deposited (Fig. 1). Additional culture considerations may also include mechanical pre-conditioning to improve the mechanical properties of the resulting material,8,9 or altering the environmental conditions (e.g. hypoxic conditioning13,14) to better model in vivo microenvironments (0.5–14% O2, depending on tissue vascularization,15 compared to 21% O2 in classic culture conditions).
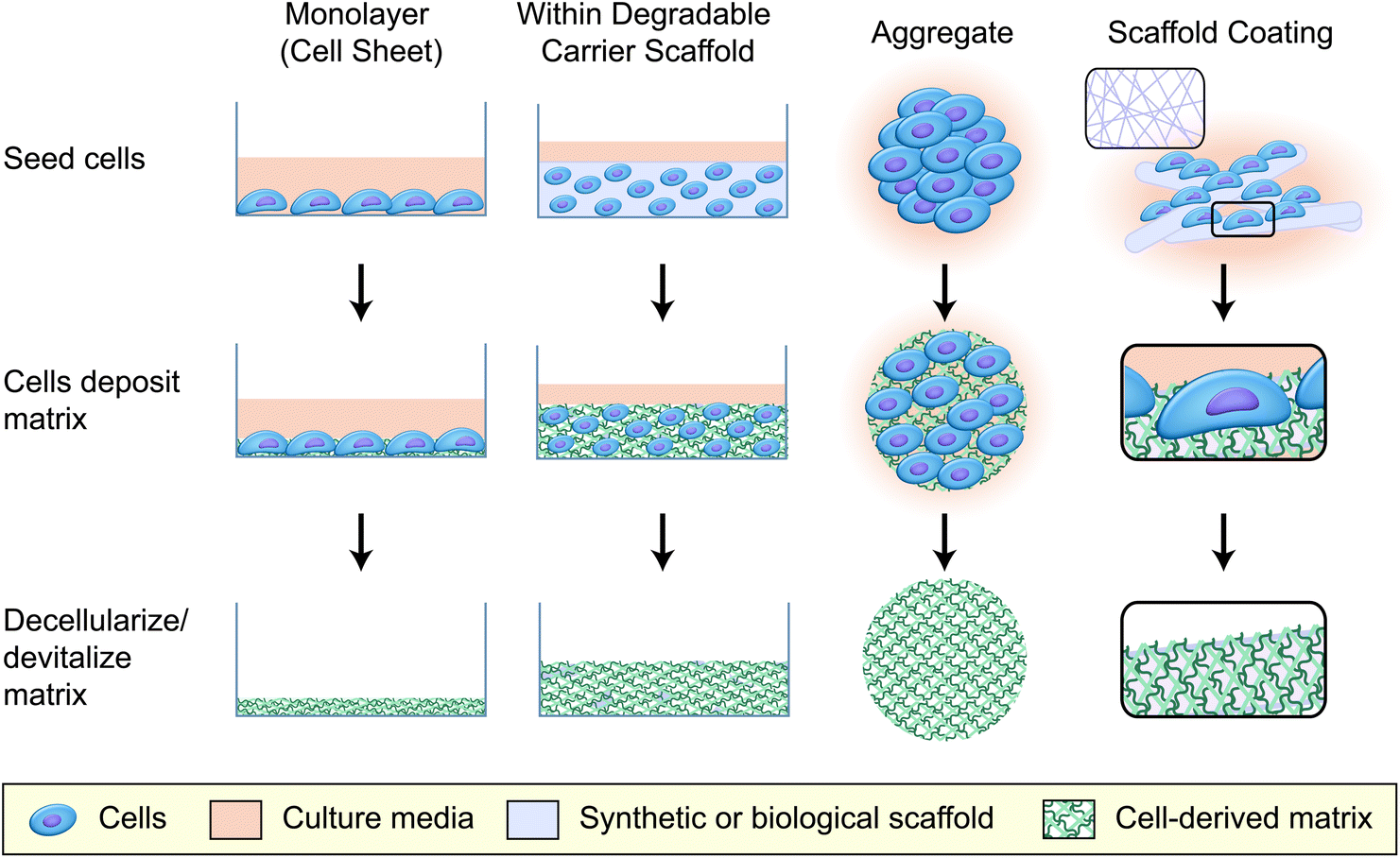 | ||
| Fig. 1 Common culture methods for generating cell-derived matrices (CDM). CDM can be generated using a variety of culture methods that offer flexibility and tunability to the desired applications. When grown in monolayers, adherent cells deposit a thin layer of matrix molecules upon the underlying surface. For thicker CDM constructs, cells can be embedded in a degradable carrier scaffold. Over time, the degradable scaffold is replaced by ECM deposited by the embedded cells, which yields a three-dimensional ECM construct following decellularization. Culturing cells as multicellular aggregates also produces three-dimensional matrices, without the need for a carrier scaffold. However, individual aggregates tend to be much smaller than the thick constructs obtained when using a carrier scaffold. Similar to monolayer cultures, cells can also be cultured on the surface of synthetic scaffolds. The deposited matrix molecules remain on the scaffold surface following decellularization, and can improve the bioactivity and biocompatibility of the synthetic material. | ||
Once a sufficient ECM has been deposited, the cellular component can be disrupted and removed from the ECM through a combination of chemical, physical and/or enzymatic processing methods (refer to Table 1 for examples of typical decellularization and devitalization protocols). The primary goal of decellularization is the removal of allogenic or xenogenic cellular antigens and other immunogenic components, such as DNA, in order to minimize the risk of adverse immunological responses.1 This objective must be balanced against preserving the molecular composition, bioactivity and structural integrity of the matrix itself, thus careful consideration and subsequent evaluation must be taken when selecting/developing a decellularization method. Decellularization techniques and the use of decellularized tissues and organs have been reviewed in depth elsewhere,1,16–18 therefore it will only be discussed briefly here. Chemical decellularization typically uses alkaline or acidic reagents (ammonium hydroxide, peracetic acid) and/or detergents (Triton X-100, SDS, CHAPS) to solubilize and disrupt cellular components and membranes. Chemical reagents are very efficient in removing cellular components, including DNA, however, chemical processing can also result in the loss of GAGs and disrupt ECM structure. Alternatively, physical methods such as lyophilisation and freeze–thaw cycling devitalize the cells but do not necessarily remove all cellular components from the matrix. Physical methods may enable retention of more matrix components, but can still result in alteration of ECM structure. Often, both chemical and physical methods are combined with DNase treatment to degrade any remnant DNA,16 which can induce sterile inflammatory responses in humans.19 However, there is some clinical evidence suggesting that in certain cases, CDM generated by allogenic cells do not elicit immune or inflammatory responses when prepared by simple dehydration without DNase treatment.20 Within the context of this review, the term cell-derived matrices (CDM) is used to describe any non-living cell-generated matrix, regardless of the specific processing method.
| Culture method | Application | Examples of common processing methods | DNase treatment | References |
|---|---|---|---|---|
| NH4OH, ammonium hydroxide (alkaline); CHAPS (zwitterionic detergent); EDTA, ethylenediaminetetraacetic acid (chelating agent); SDS, sodium dodecyl sulphate (ionic detergent); Sodium deoxycholate (ionic detergent); Triton X-100 (non-ionic detergent). | ||||
| Monolayer | Substrate for cell culture | 0.5% Triton X-100 and 20 mM NH4OH for 5 min at 37 °C | Yes | 14,29,33,39,60,63,103 |
| No | 38,59,62 | |||
| Cell sheet layering for tissue engineered blood vessels | Air dried then frozen at −80 °C | No | 20,41 | |
| Degradable Carrier Scaffold | Tissue engineered blood vessel | 8 mM CHAPS, 1 M NaCl and 25 mM EDTA for 1 hour at 37 °C; followed by 1.8 mM SDS, 1 M NaCl and 25 mM EDTA for 1 hour at 37 °C | No | 8,43 |
| Tissue engineered heart value | 1% SDS for 6–24 hours at RT; followed by 1% Triton X-100 for 30 min at RT | Yes | 46,51,54 | |
| 0.25% Triton X-100, 0.25% sodium deoxycholate, 0.02% EDTA overnight at 37 °C | Yes | 50,52 | ||
| Aggregate | Embryoid body matrices | 1% Triton X-100 for 30 min at RT | Yes | 89,90 |
| No | 92 | |||
| Lyophilized | No | 91,93 | ||
| Scaffold Coating | Scaffolds for bone repair and osteogenesis | Freeze–thaw cycling (3 cycles): liquid nitrogen for 10 min, then 37 °C water bath for 10 min | No | 21–23,26,27,31,33 |
Cell-derived matrices in skeletal tissue engineering
Orthopaedic tissue engineering and regenerative strategies aim to improve upon current clinical practices for repairing large bone defects and damaged cartilage. CDM are being investigated (i) as a scaffold coating to improve the biological activity and tissue integration of synthetic scaffolds for regenerative therapies and (ii) as alternative cell culture substrates for maintaining, expanding and differentiating stem cells, and primary chondrocytes and osteoblasts for future use in orthopaedic cell therapies.The effect of CDM on MSC osteogenic differentiation has been extensively studied in vitro.21–27 MSCs cultured on titanium (Ti) fibre meshes and poly(ε-caprolactone) (PCL) microfibre scaffolds coated with previously deposited osMSC-synthesized matrix exhibited enhanced osteogenic differentiation, characterized by increased alkaline phosphatase (ALP) activity and calcium deposition, relative to MSCs cultured on unmodified Ti and PCL scaffolds.21–24 Furthermore, MSCs cultured on Ti/ECM scaffolds upregulated expression of a subset of osteogenic genes (alkaline phosphatase, osteopontin, osteocalcin, osteonectin, osteomodulin and parathyroid hormone receptor) and decreased expression of aggrecan, a protein associated with chondrogenesis.27 The MSC-derived matrices also supported increased mineralization, even in the absence of dexamethasone, a common osteogenic additive. However, increased calcium deposition appeared to be primarily due to mineralization of the pre-existing matrix as the acellular construct exhibited only slightly less calcium deposition than PCL/ECM construct with MSCs.24 While the ECM/scaffolds did not appear to be able to induce osteogenic differentiation in naïve MSCs, they were capable of maintaining osteogenic differentiation of MSCs without dexamethasone if MSCs had already been primed towards the osteogenic lineage prior to being seeded on the scaffold.22–24 The ability to maintain osteogenic differentiation of primed MSCs has important implications for promoting osteogenesis in vivo in the absence of exogenous soluble factor stimulation.
Decellularized matrices derived from fibroblasts and pre-osteoblasts yielded similar MSC osteogenic differentiation on composite poly(lactic-co-glycolic acid; PLGA)/hydroxyapatite (HA)/β-tricalcium phosphate (β-TCP) scaffolds.28 The CDM-coated composite scaffolds contained fibronectin and collage type I, and stimulated increased ALP activity and mineralization when reseeded with MSCs in osteogenic media, compared to uncoated PLGA/HA/β-TCP scaffolds.28 Decaris et al. demonstrated that osteogenic CDM generated as 2D culture substrates could be coated onto a 3D scaffold, while retaining their capacity to modulate osteogenic differentiation.29,30 MSC-derived matrices deposited TCPS culture dishes were decellularized using Triton X-100, NH4OH and DNAse, then homogenized in acetic acid and deposited as a matrix solution on 3D macroporous poly(lactide-co-glycolide; PLG) scaffolds.29,30 The transferred matrix maintained the ability to enhance osteogenic differentiation of MSCs in the presence of β-glycerophosphate (β-GP) and dexamethasone, demonstrated by increased expression of osteogenic markers and ALP activity.29,30 The retention of bioactivity by the transferred CDM implies that composition of the matrix, and not necessarily the matrix structure, is sufficient to enhance a cellular response. However, as discussed further below, these positive in vitro outcomes did not accurately predict in vivo responses.
CDM for bone regeneration and osteogenesis have been evaluated in ectopic and orthotopic sites in vivo. Subcutaneous implants of polyesterurethane scaffolds coated with mineralized osMSC-derived ECM induced the ingrowth of host cells and the formation of a mineralized matrix that stained positive for bone sialoprotein (BSP), whereas no mineralization or BSP was found in the uncoated polymer scaffold explants.31 Similarly, Hong et al. demonstrated enhanced osteogenic differentiation of naïve MSCs implanted on PLGA/HA/β-TCP scaffolds coated with fibroblast-CDM using a subcutaneous ectopic bone formation model.28 Explanted matrix-coated scaffolds had greater collagen and calcium deposition and ALP expression than uncoated scaffolds.28 The increased osteogenic differentiation of implanted MSCs was attributed primarily to instructive cues present in the CDM, as the MSCs were not treated with soluble exogenous osteogenic factors prior to implantation. Rat femoral segmental defects treated with decellularized MSCs and amniotic fluid stem cells (AFSCs) matrices on collagen/PCL scaffolds increased the rate of bone bridging, although neither matrix affected the total volume of mineralized matrix in the defect over 12 weeks.32 Matrix deposited by either calvarial osteoblasts or fibroblasts was found to enhance the osteogenic properties of hydroxyapatite (HA) microparticles in critical-sized calvarial defects in rats. Defects treated with ECM-coated HA microparticles yielded multiple areas of new bone formation throughout the defect, similar to bone formation observed in response to HA microparticles mixed with Tissue Fleece (Baxter), a commercial equine type I collagen scaffold; while defects treated with uncoated HA microparticles had limited new bone formation that was restricted to the defect margins.33 Despite some of the positive individual results observed in these in vivo studies suggesting that CDM can have osteoinductive potential, the current lack of significant improvements over clinical standards, such as Tissue Fleece, indicate that CDM examined to date may only yield modest improvements in skeletal regeneration.
In contrast, several other studies examining the osteogenic properties of CDM failed to observe significant improvements in vivo. In an ectopic bone formation model, MSC-derived matrix coating did not improve the mineralization of PLG scaffolds seeded with MSCs, regardless of whether the MSCs had been previously been grown in osteogenic media.29 However, increased vascular ingrowth was observed in the MSC-containing CDM-coated scaffolds, relative to MSC-containing uncoated scaffolds.29 At 2 weeks, no human MSCs were detected within the coated and uncoated PLG scaffolds, suggesting implanted MSC death due to hypoxia or migration away from the implant site.29 The lack of cell survival and engraftment may have impaired ectopic bone formation and masked any potential effect of the CDM. Decellularized MSC-derived matrix also failed to induce bone formation on titanium scaffolds implanted intramuscularly for up to 56 days,26 although, again, the matrix did enhance vascularization of the scaffold. The increase in vascular infiltration of ECM-coated scaffolds without significant improvement of bone formation is an interesting observation in both studies, especially considering the increase in vessel density was observed with and without the inclusion of viable MSCs in the scaffolds.26,29 In both cases, the lack of bone formation may be attributed to reduced or altered osteoinductive properties of the matrices due to processing methods. In the first study, the matrix was transferred from a 2D culture plate and coated onto a scaffold. While previous in vitro studies indicated that the bioactivity of the matrix was preserved, it is possible that the homogenization in acetic acid affected the in vivo response. In the second study, the matrix was sterilized with ethylene oxide (EtO) following decellularization using freeze–thaw cycles since in vitro studies suggested that CDM retained its bioactivity following EtO sterilization.21 However, EtO sterilization of demineralized bone matrix is still debated due to evidence that the doses required to sterilize the products reduces their osteoinductivity, specifically when high temperatures are used.34
CDM rich in collagen and GAGs have also been used to enhance the chondrogenic differentiation of MSCs and synovium-derived stem cells (SDSCs) for cartilage regeneration. In vitro chondrogenesis of MSCs is typically induced by supplementation with transforming growth factor β (TGF-β), dexamethasone and ascorbate-2-phosphate.35,36 Differentiating MSCs on chondrocyte-derived matrices instead of uncoated PCL scaffolds moderately enhanced the GAG deposition and GAG synthetic activity (GAG/DNA per scaffold) in the presence of TGF-β125 and decreased the expression of collagen I in the absence of TGF-β1. However, the gene expression of aggrecan, collagen II and collagen I was not affected by the presence of the ECM in TGF-β1 treated cultures. Furthermore, this effect was TGF-β dependent, as the matrix alone was not sufficient to induce chondrogenesis.25 In the reverse scenario, in vitro culture of chondrocytes on MSC-derived matrices had increased collagen type II and aggrecan expression in the presence of chondrogenic media,37 but this improvement was only observed when the matrices were derived from MSCs cultured in a basic growth medium and not chondrogenic medium containing TGF-β3 and dexamethasone. Unfortunately, in vitro culture on either MSC-derived matrix did not rescue the phenotype of chondrocytes obtained from an osteoarthritic donor.37
SDSCs cultured on decellularized SDSC matrix (DSCM) had improved proliferation and attenuated oxidative stress-induced senescence during culture in vitro.14,38 SDSCs expanded on DSCM also had enhanced chondrogenic potential but impaired differentiation along adipogenic and osteogenic lineages, compared to SDSCs expanded on tissue culture polystyrene (TCPS).14,38 The benefit of culturing the SDSCs on DSCM was retained after the cells were removed from the matrix and cultured as pellets14,38 or injected into a cartilage defect in vivo.39 DSCM-expanded SDSC pellets underwent striking chondrogenic differentiation in response to TGF-β1 and TGF-β3 in vitro, characterized by increased size, intensified staining for collagen type II, sulphated GAGs and decreased collagen type I staining compared to TCPS-expanded SDSC pellets.39 Intra-articular injection of DSCM-expanded SDSCs improved the repair of partial-thickness cartilage defects in pigs.39 Upon explant at 3 months, the defect area and newly formed tissue in the group treated with DSCM-expanded cells stained more intensely for GAGs and collagen type II, and less collagen type I compared to defects treated with TCPS-expanded SDSCs.39
In a direct therapeutic application of CDM, Li et al. developed a membrane composed of layered decellularized chondrocyte-derived ECM to improve the outcome of bone marrow stimulation in a canine model.40 The thin, flexible membrane, composed primarily of collagen and sulphated GAGs, was applied to cover a cartilage defect and protect the bone marrow stem cell-containing blot clot that resulted from bone marrow stimulation. Coverage with the decellularized membrane resulted in more cartilage-like tissue formed within the defect by 18 weeks than uncovered defects.40
CDM have successfully been applied in in vitro osteogenic and chondrogenic differentiation schemes, but thus far they have had limited success in improving regenerative skeletal therapies in vivo. For the most part, the CDM in these applications were used as a coating for metallic and polymeric scaffolds. It is possible that the osteogenic or chondrogenic cues within the CDM lack the potency required to drive an in vivo response or that the bulk scaffold effects remain dominant in the in vivo environment. Another possibility is the lifespan of the CDM, which would likely be remodelled over the span of weeks, could be much shorter than the time scale required for bone or cartilage defect repair, thus limiting the impact of the transiently present CDM. In the case of cell-seeded scaffolds, retention of viable cells within the scaffold continues to be a limitation due to hypoxic cell death or migration of cells away from the scaffold29 that may interfere with properly evaluating CDM in vivo.
Cell-derived matrices in cardiovascular tissue engineering
In contrast to skeletal engineering, CDM in cardiovascular engineering applications tend to be used as regenerative therapies, where the CDM itself is used to replace damaged or diseased vessels and valves, rather than as a scaffold coating or cell culture substrate. The shortage of human allogenic heart valves and autogenic or allogenic vascular grafts for cardiovascular therapy has motivated the used of decellularized xenogenic and allogenic valves and vascular grafts. However, the limited availability of cadaveric allogenic tissues and potential risk(s) associated with xenogenic tissue sources has prompted significant research into the development of tissue engineered blood vessels (TEBVs) and tissue engineered heart valves (TEHVs).Tissue engineered small diameter vascular grafts have been a critical yet elusive goal of vascular tissue engineering for a long time. Although synthetic grafts function acceptably for large diameter vessels (>6 mm), there currently is no suitable alternative for small diameter autologous grafts. One method of fabrication for TEBVs is culturing fibroblasts41,42 or vascular smooth muscle cells8,43–45 in sheets, then wrapping the cell sheets around a mandrel to form a tubular structure. The engineered collagen-rich tissue is cultured for months, in some cases conditioned by pulsatile flow, until the tissue is mechanically robust enough for implantation. In some instances, endothelial cells are seeded on the luminal side of the vessel to improve hemocompatibility.8,41 While TEBVs are considered a success story for the field of tissue engineering with encouraging performances in pre-clinical44,46,47 and clinical studies,42,48 the long manufacturing times coupled with the regulatory challenges facing the use of living engineered tissues pose potential limitations on widespread clinical use. Such concerns motivated the development of potential “off-the-shelf” products through devitalization of TEBVs. Devitalization of vascular grafts has a clinical precedence with the use of cryopreserved allogenic grafts, which retain their mechanical functionality as arterial allografts but are nonviable at the time of implantation.49
In a 2011 case study, an autologous fibroblast-derived TEBV (Lifeline™, Cytograft) was devitalized by air drying then stored for 5 months at −80 °C prior to being thawed, lined with autologous endothelial cells and implanted as an atrioventricular shunt in a 72-year old male patient who required vascular access for hemodialysis.41 During the first 8 weeks, the graft remained patent with a stable 5 mm diameter, however no reports have been released to date regarding the performance of the graft following puncture for hemodialysis access. With this approach, the patient wait time to prepare the devitalized graft with a viable, autologous endothelium and conduct quality control testing was less then 2 weeks.41 In a follow-up case study, allogenic devitalized TEBVs prepared without the endothelial layer were implanted as brachial-axillary arteriovenous shunts for hemodialysis access in three patients.20 Grafts were used for hemodialysis access as early as 7 weeks post-implantation, and functioned without signs of aneurysms for up to 11 months; however, two of the three patients did require interventions to maintain graft patency. The authors noted that the decellularized graft, similar to synthetic grafts, did bleed slightly into the surrounding tissue at previous puncture sites whereas previously tested living, autologous TEBVs did not. An interesting observation was that lymphotoxic cross-reactivity and panel reactive antibody tests indicated that the allogenic grafts were well-tolerated and did not elicit an immune response despite the fact the grafts were simply dehydrated instead of decellularized using detergents and DNase treatments.20
Using a similar approach, Niklason and collaborators also developed a TEBV using human smooth muscle cells cultured on a tubular biodegradable polyglycolic acid (PGA) scaffold for 8 weeks, at which point the PGA scaffolds had mostly degraded and the constructs were decellularized.8In vitro mechanical testing indicated that the decellularized grafts possessed similar burst strength and suture retention as a human saphenous vein.8 Furthermore, the decellularized grafts had an 80% patency rate over six weeks in an arterial graft model in nude rats, despite lacking any endothelial lining prior to implantation.43 Histological analysis did reveal that host endothelial cells (von Willebrand factor positive) created a confluent endothelium, while host smooth muscle actin (SMA)-positive cells invaded the outer side of the graft. Taken together, these results demonstrate the potential of completely devitalized, non-endothelialized allogenic TEBVs to serve as a true “off-the-shelf” tissue engineered product. Furthermore, they demonstrate that CDM used in this application can be remodelled and repopulated by host cells, without generating a massive inflammatory response.
Similar studies have also been conducted with TEBVs developed using ovine dermal fibroblasts seeded in fibrin gels in tubular molds for 2 weeks, then conditioned under pulsatile flow for 3 weeks prior to decellularization.46 As expected from previous reports, the decellularization process had little effect on the mechanical properties of the TEBVs and the grafts remained patent, without dilation, over 24 weeks in an ovine femoral graft model.46 The grafts were completely re-endothelialized by 24 weeks, and exhibited a high degree of cellular infiltration with primary α-SMA positive cells. Interestingly, this immune-competent model also showed evidence of immune cell infiltration with CD45+CD3+ cells (T-lymphocytes) and CD45+CD11b+ (macrophages) cells present within the graft at 8 weeks, mainly in the acellular region. However, by 24 weeks very few CD45+ cells were observed within the graft, reflecting an acute inflammatory response associated with the remodelling and recellularization of the decellularized TEBVs rather than a chronic inflammatory or immunogenic response.
CDM have also been used to construct tissue engineered heart valves. In this application, human vascular fibroblasts,50 human neonatal dermal fibroblasts51 and ovine fibroblasts52–54 have been seeded in fibrin carriers on degradable synthetic templates and cultured 4–5 weeks to allow the deposition of CDM. The matrix derived from fibroblasts generally contained collagen (based upon hydroxyproline content) and GAGs, but not elastin, which is a major component of native valve matrix.50,52 Decellularization and long-term storage (18 months) did not significantly alter the mechanical properties or matrix composition of the valves.52 When implanted in baboons, human vascular fibroblast-derived valves performed well over the 8-week study, similar to decellularized human native heart valve.50 However, radial shortening of the leaflet led to impaired coaptation and regurgitation that persisted throughout the 8 weeks. Contraction of valve leaflets is a well-establish limitation of viable TEHVs, and had been attributed to the presence of contractile cells within the scaffold.52,54,55 The contraction of the decellularized leaflets in vivo indicate that the remodeling of the valve wall and leaflet matrix by invading host α-SMA positive cells and MAC-387 positive cells (i.e. macrophages), respectively, may be detrimental to the long-term functioning of the engineered tissue. Residual stresses from TEHV being cultured in a closed position have also been suggested as a contributing factor to valve retraction.50 In contrast, decellularized human native heart valves exhibited limited recellularization and maintained good coaptation without regurgitation over the duration of the study. Culturing the matrix producing fibroblasts with TGF-β1 to stimulate the deposition of elastin may help preserve the leaflet geometry.56
The use of CDM for tissue engineered heart valve and blood vessel replacement offers a complementary alternative to xenogenic products. The studies discussed in this section have demonstrated that CDM can be repopulated and remodelled by host cells, integrate with the host tissue and maintain mechanical integrity and functionality. While there are still challenges to overcome, “off-the-shelf” tissue engineered vascular grafts and heart valves have the potential to have significant impacts on clinical cardiovascular therapies in the near future.
Cell-derived matrices as models of stem cell niches and ex vivo tissue-mimetic microenvironments
A common limitation in expanding sensitive primary cells ex vivo is loss of the native phenotype, characterized by loss of function, senescence, dedifferentiation and/or spontaneous differentiation.57–61 One approach to mitigate culture-induced phenotypic changes is to recapitulate the tissue-specific microenvironment, in addition to the use of soluble factors. Recently, an in-depth evaluation of cell-derived ECM and the ability of MSC-derived ECM to support MSC and hematopoietic stem and progenitor cell (HSPC) expansion ex vivo was conducted by Prewitz et al.62 Matrices generated by mouse embryonic fibroblasts (MEFs), human umbilical vein endothelial cells (HUVECs) and human neonatal dermal fibroblasts were compared to matrices generated by human MSCs grown in osteogenic factors (osteoECM) or ascorbic acid (aaECM). The matrices were deposited on a fibronectin (FN) layer covalently bonded to the TCPS to prevent the delamination of the matrix, which is a common problem with matrices simply physisorbed onto surfaces. Once decellularized, all the matrices were characterized as having a complex fibrillar structure, with tethered GAG aggregates within the network. The MSC-ECM was thicker than the other matrices, and also had an elasticity that resembled native bone marrow (∼0.1–0.3 kPa), whereas the other matrices exhibited elasticities of approximately 1 kPa. Proteomic analysis revealed that both MSC-ECM had similar proteomic composition, including proteins associated with the bone marrow matrix such as collagen, decorin, laminins, nidogens, tenascin, thrombospondins, vitronectin and fibronectin. However, the aaECM contained significantly more sulphated GAGs and collagen than the osteoECM. aaECM also released greater amounts of growth factors (hepatocyte growth factor, HGF; fibroblast growth factor, FGF; interleukin-8, IL-8; vascular endothelial growth factor, VEGF) compared to osteoECM. Compared to TCPS and FN-coated TCPS, the MSC matrices supported higher proliferation rates, greater osteogenic and adipogenic differentiation efficiencies and increased secretion of growth factors (angiopoietin 1, Ang-1; stromal cell-derived factor 1, SDF-1; IL-8) in cultured MSCs, with aaECM outperforming osteoECM. The aaECM also supported greater expansion of CD34+ and CD34+CD133+ cells compared to the other surfaces, without adversely affecting the long-term engraftment of HSPCs. Overall, the improved expansion of both the MSC and HSPC populations were attributed to the ability of the aaECM to mimic the native bone marrow niche.MSCs have the multipotent ability to differentiate into several musculoskeletal cell types, but their low abundance in native tissue is thought to require ex vivo expansion to obtain clinically relevant cell numbers. Long-term expansion of MSC typically diminishes their differentiation potential and leads to cellular senescence. Consequently, researchers have attempted to recapitulate the microenvironment of the bone marrow niche using MSC and bone marrow cell (BMC)-derived matrices to prevent the loss of MSC differentiation and proliferative potential.58–60 BMC-derived matrix contained a composition similar to bone marrow, including the presence of collagen I and III, fibronectin, biglycan, decorin, perlecan and laminin.58 Expansion of MSCs on BMC-derived matrix promoted proliferation and reduced intracellular levels of reactive oxygen species, relative to expansion on TCPS. Multipotentiality was also retained in MSCs expanded on BMC-derived matrix, compared to TCPS. In vivo, CDM-expanded MSCs retained their potential for osteogenesis, while TCPS-expanded MSCs had reduced osteogenic potential after 7 passages.58 Similar improvements were reported for adult MSCs grown on fetal MSC-derived ECM and SDSCs grown on SDSC-derived matrix.38,63
Human and mouse embryonic stem cells (ESCs) are of great interest for regenerative medicine due to their capacity to differentiate into all somatic cell types.64–67 Traditionally, they have been cultured with serum on feeder layers of mitotically inactivated MEFs to maintained the ESCs in an undifferentiated, pluripotent state.64–66 However, inconsistent and undefined culture conditions produced by the feeder cells and the transmission risk of xenogenic pathogens to human ESCs (hESCs), among other concerns, motivated a move away from feeder-dependent cultures.68 Undifferentiated mouse ESCs (mESC) can be grown in feeder-free conditions on gelatin with supplementation of leukemia inhibitory factor (LIF), however, LIF is unable to maintain undifferentiated hESCs on gelatin-coated substrates. Instead, a feeder-free system for undifferentiated hESCs was established by culturing the cells in MEF conditioned media on Matrigel™, a cancer cell derived matrix.69,70 Although feeder-free, the Matrigel/MEF-conditioned media culture system does not address the primary concerns of xenogenic contamination and variability of culture conditions.71 Other serum-free, non-xenogenic culture systems have since been developed for the undifferentiated maintenance of hESCs. Of particular interest for this review, Klimanskaya and colleagues reported using decellularized ECM from MEFs to maintain hESCs in a feeder layer-free and serum-free culture.61 Mitotically inactivated MEFs were grown on gelatin-coated plates for 7 to 21 days, then decellularized with a sodium deoxycholate solution. The MEF-CDM was used to establish and maintain a hESC line (ACT-14), along with six other existing hESC lines.61 While the MEFs are of xenogeneic origin, the ability to decellularize and sterilize the matrix layer significantly reduces the risk of animal component contamination and pathogen transmission.
Other examples of ex vivo expansion of therapeutic cell populations include the use of CDM to reverse chondrocyte dedifferentiation and prevent loss of phenotype.11 Cha et al. evaluated matrices derived from fibroblasts, pre-osteoblasts and chondrocytes for their ability to rescue chondrocyte dedifferentiation in culture57 and each contained collagen type I, collagen type II, fibronectin and laminin. To evaluate the effect of different CDM on chondrocyte dedifferentiation, rat primary chondrocytes were cultured on TCPS for 4 passages to induce dedifferentiation, marked by loss of collagen type II and aggrecan expression. When transferred to the CDM and cultured for an additional 2 weeks, the passaged chondrocytes exhibited signs of redifferentiation, including increased GAG and collagen type II deposition and reduced collagen type I gene expression, although the gene expression levels of collagen type II and aggrecan were similar in all cultures.57 Pellet cultures of dedifferentiated chondrocytes expanded on CDM for 2 passages also contained more GAGs, stronger peripheral staining of collagen type II and were larger in size compared to pellets formed from TCPS-expanded chondrocytes.57 Similar studies conducted using SDSC-derived matrix with primary pig chondrocytes,11 and chondrocyte-derived matrix with primary bovine chondrocytes12,72 have also demonstrated delayed chondrocyte dedifferentiation when cultured on CDM substrate.
As noted earlier, an important consideration in the use of CDM is that the composition and quality of the matrix is dependent on the source cell population. For example, aging has been shown to have a profound effect on the quality of matrices that MSCs deposit.59 Matrix derived from BMCs isolated from young mice (3 month old) improved the proliferation and osteogenic potential of MSCs from aged mice (18 months old) to a level comparable with young MSCs, and reduced intracellular reactive oxygen species.59 In comparison, both young and aged MSCs grown on aged MSC-derived matrix had significant impairment of proliferation and osteogenesis. These results suggest that matrices derived from young MSCs can rejuvenate aged MSC populations and contributes to maintaining healthy MSC function. Confocal Raman microscopic analysis of the young and old MSC-derived matrices suggested that the old matrix contained more mineralized phosphate, while the young ECM appeared to contain more collagens, indicating that aging impacts the composition of MSC-derived matrix.59 This study has far reaching implications for MSC biology and regenerative medicine, as it suggests that culture on matrix derived from young MSCs may improve the quality and quantity of MSCs isolated from aged patients for regenerative cell therapies.
Just as disease states are often reflected by cell phenotypes, the native ECM of disease tissues and tumours can also have distinct features that may contribute to or result from a disease state.73 Studying the composition and properties of matrices derived from diseased cell populations may enable researchers to identify and better understand the relationship between cells and matrix in disease propagation, and enable the development of superior disease models for basic research. Reichert et al. used matrices produced by human osteoblasts to develop an in vitro model of bone metastasis, which recapitulated features of prostate cancer bone metastasis, including loss of epithelial phenotype.74 Also, the role of ECM in craniosynostosis (CS), the premature fusion of cranial sutures, was investigated using decellularized matrix produced by osteoblasts from patients with CS and healthy individuals.75 Previous studies had detected decreased expression of TGF-β, a protein involved in regulating bone resorption,76 in MSCs harvested from fused versus unfused sutures in mice.77 TGF-β had significantly reduced binding affinity for ECM secreted by CS osteoblasts, compared to osteoblasts from healthy individuals, indicating that differences in the ECM of patients with CS likely contribute to the pathogenesis of the disease.75
Embryonic stem cell-derived matrices
In most cases, CDM are used to recapitulate the microenvironment of adult tissues and the cells used to derive the matrix are sourced directly from the tissue of interest. However, there are instances, as noted above, when adult matrices may be limited in their ability to promote a desired behaviour, such as tissue regeneration. For example, scarring and impaired wound healing are significant clinical challenges related to traumatic and surgical wounds, and co-morbidities of diseases like diabetes. The main matrix components in skin are collagens, elastin, GAGs, laminin, nidogen and fibronectin,78 and matrices derived from fibroblasts, the primary cell type within the dermis, recapitulate some the main components in adult skin.10,57,79 Acellular dermal matrix allografts and skin substitutes, such as INTEGRA® Dermal Regeneration Template, are routinely used in the clinic to treat burns and chronic wounds.80–83 However, adult skin is also known to have reduced regenerative capability compared to fetal skin. At early stages of development, fetal skin can undergo scarless healing and is rich in matrix proteins, GAGs and growth factors that have are known to promote wound repair.84–86 Therefore, instead of recapitulating adult skin matrix, a matrix reflecting the protein composition of fetal tissue may provide a better therapeutic material for promoting scarless wound healing.Previous studies from our group have demonstrated that differentiating embryonic stem cells grown in multicellular aggregates, called embryoid bodies (EBs), express many of the matrix and matricellular proteins and morphogens that are expressed during early development and are also associated with improved wound healing.87 Gene expression analysis of spontaneously differentiating mESC EBs revealed expression of many ECM and matricellular proteins, including collagens (Col1a1, Col2a1, Col3a1, Col4a1-3, Col6a1 etc.), laminins (Lama1, Lamb1, Lamb3 etc.), fibronectin (Fn1), vitronectin (Vtn), osteopontin (Spp1), fibulin 1 (Fbln1), tenascin (Tnc) and sparc (Sparc).87 An interesting trend in the expression profile was that while many matrix proteins had constant or decreased expression initially, the expression for many matrix-associated proteins started to increase expression by day 10. Similarly, the EBs were also shown to express genes of many therapeutically relevant secreted morphogens, including bone morphogenic proteins (BMPs), FGFs, TGFs, platelet-derived growth factors (PDGFs) and VEGFs, some of which would be retained within the EB matrix.87 The presence of versican and hyaluronan within the EB matrix was independently confirmed in a separate study.88 The rich and complex nature of the EB matrix (EBM), particularly at later time points, led us to postulate that the EBM and its associated growth factors may provide a therapeutic platform for delivering embryonic-like matrices and paracrine factors to damaged tissues and organs, such as skin and bone. Successful decellularization of EBs has been achieved using both chemical and mechanical methods, matrix components are retained, and the EBM can support exogenous cell attachment and proliferation.89–91
Similar to the use of MSC-derived matrices to improve somatic stem cell expansion, Goh et al. proposed that ESC-derived ECM be used to recapitulate the embryonic microenvironment to improve the efficiency of current ESC lineage-specific differentiation protocols.92 Two distinct EBM were generated by culturing mESC EBs with or without retinoic acid (RA), an inducer of neural differentiation. Prior to decellularization, both RA-treated and untreated EBs contained collagen I, collagen IV, fibronectin and laminin, although staining for collagen I, collagen IV and laminin staining appeared to be more intense in the spontaneously differentiating EBs than the RA-EBM and the distribution pattern of fibronectin differed between the two groups. While the expression of these four proteins was retained following the decellularization process, the total protein content and sulphated GAG content was significantly reduced in both groups. Total collagen and sulphated GAG content remained higher in the EBM generated from spontaneously differentiating EBs (SPT-EBM) than RA-EBM, although the spatial distribution of ECM components was lost. When reseeded with mESC, both SPT-EBM and RA-EBM supported greater cell proliferation compared to the proliferation of ESCs cultured as native EBs. However, SPT-EBM increased expression of early differentiation markers Brachyury, FGF-5 and FGF-8 of mESC, whereas the gene expression of ESC cultured on RA-EBM more closely resembled that of native EBs.92 This study demonstrated that EB-derived matrices can support ESC proliferation and differentiation, and that the differentiation protocol used to obtain the EBM can influence the phenotype of reseeded ESCs. Furthermore, it suggests that the use of an appropriate ESC matrix may improve soluble factor-directed differentiation.
Sart et al. also examined the effect of culture conditions on the composition of ESC-derived ECM and whether any differences in ECM exerted an effect on the proliferation and differentiation of ESCs reseeded on the EBM.93 Mouse ESCs were cultured as EBs differentiating spontaneously or in the presence of retinoic acid (RA) or BMP-4 to induce ectodermal or mesodermal differentiation, respectively. The matrices derived from these different culture conditions all contained varying amounts of fibronectin, laminin, vitronectin, collagen IV and GAGs, with the highest fibronectin, vitronectin and collagen IV expression occurring in the matrices derived from BMP-4 and RA-treated EBs, compared to the spontaneously differentiating EBs. However, very few differences were observed when fresh ESCs were reseeded and grown on the different EBM for 4 days then replated on gelatin for 7 days, with the exception of ESCs grown on day 10 RA-EBM. However, the decreased proliferation and increased β-tubulin III (ectoderm), α-actinin (mesoderm) and FoxA2 expression (endoderm) observed with the day 10 RA-EBM was thought to be due to residual RA entrapped within the matrix, as the expression of α-actinin and FoxA2 was attenuated by a pan-RA receptor antagonist.93 Contrary to the results from the previous study, in this experiment the different EBM did not appear to have a direct effect on ESC differentiation. The differing outcomes of the two studies are not surprising, as there were fundamental experimental differences, including cell type, EB culture time, media formulations, dosing of RA, decellularization and reseeding methods, and culture of reseeded cells.92,93
Arguably, one of the strengths of ESC-derived matrices is the rich and complex expression profile of pluripotent cells. While the matrices retained both the matrix proteins and sequestered growth factors and signalling molecules, alternative approaches to deliver stem cell-secreted factors in more concentrated manners may yield greater potency and more significant results. Recently it was reported that hESC-secreted factors significantly improved the viability of human cortical neurons following exposure to a toxic form of Amyloid beta in an in vitro model of Alzheimer's disease.94 Moreover, hESC-conditioned media improved myoblast proliferation and the pro-myogenic proteins secreted by hESCs were found to bind heparin.94 The heparin-binding capacity of many of the morphogens secreted by ESCs may provide an efficient method for capturing stem cell morphogens for subsequent delivery.
Future directions
The use of cell derived matrices as bulk biomaterials, scaffold coatings and cell culture substrates has permeated the field of regenerative medicine. This review sought to highlight the current status of CDM across various applications and identify some of the successes and major challenges that remain. CDM offer promising alternative biomaterials as decellularization and sterilization techniques are optimized and appropriate cell sources and culture conditions are identified for producing more potent and instructive materials.As the use of CDM has become more established, one can foresee the potential for engineering matrices with desired properties using genetically engineered cells, small molecules or other soluble factors to induce overexpression of specific matrix components. An example would be inducing overexpression of elastin95 by fibroblasts generating the TEHV and TEBV matrix in an effort to reduce leaflet retraction or to improve the elasticity and resilience, respectively. As the cellular components of these transgenic cells would be removed prior to in vivo applications, the risk typically associated with transgenic, immortalized or tumorigenic cell lines would be eliminated.
In response to the damage current chemical, enzymatic and physical decellularization and devitalization techniques cause to the ECM, Martin and collaborators have proposed a novel method of decellularization that exploits programmed cell death.96 The authors propose to induce apoptosis by (i) delivery of a death receptor ligand, such as FasL,97,98 to activate the extrinsic apoptotic pathway; (ii) induction of mitochondrial apoptosis through lethal environmental conditions, such as hyper- or hypo-thermic temperatures,99,100 or nitric oxide;101 or (iii) genetically engineering cells to induce apoptotic pathways in response to a chemical inducer, such as the inducible caspase 9 suicide system.102 By inducing cell death through apoptotic pathways, the dying cells fragment into small apoptotic bodies, which in theory would be removed by perfusing or washing the ECM.96 There are a number of limitations and challenges that would need to be addressed to ensure complete decellularization and limit undesirable effects, such as the release of pro-inflammatory factors in response to the stimuli. However, decellularization by intentional activation of programmed cell death is an intriguing proposal that warrants further investigation.
Summary
Cell-derived matrices offer an alternative to tissue-derived matrices and have been used in a number of pre-clinical and clinical applications. Some advantages of CDM are accessibility of human cell sources, increased tunability of matrix properties and conformations (bulk material or scaffold coating) compared to tissue-derived matrices, and the ability to use a variety of somatic and stem cells to generate matrices with desirable properties. CDM have yielded promising results in generating off-the-shelf tissue engineered vascular grafts and heart valves, and novel cell culture substrates mimicking specific niche microenvironments. CDM-scaffold coatings have also shown promise in in vitro studies of osteo- and chondrogenesis, however there are apparent challenges in transferring the technology to in vivo orthopaedic applications. The future of CDM in regenerative medicine is exciting, with the potential for using genetically engineered cells and continued optimization of decellularization techniques.Abbreviations
| Aa | Ascorbic acid |
| AFSC | Amniotic fluid stem cell |
| ALP | Alkaline phosphatase |
| ANG-1 | Angiopoietin 1 |
| β-GP | β-Glycerophosphate |
| β-TCP | β-Tricalcium |
| BMC | Bone marrow cell |
| BMP | Bone morphogenic protein |
| BSP | Bone sialoprotein |
| CDM | Cell-derived matrix |
| CS | Craniosynostosis |
| DSCM | Decellularized SDSC matrix |
| EB | Embryoid bodies |
| EBM | Embryoid body matrix |
| ECM | Extracellular matrix |
| ESC | Embryonic stem cells |
| FGF | Fibroblast growth factor |
| FN | Fibronectin |
| GAG | Glycosaminoglycan |
| HA | Hydroxyapatite |
| HGF | Hepatic growth factor |
| HSPC | Hematopoietic stem and progenitor cell |
| IL-8 | Interleukin 8 |
| LIF | Leukemia inhibitory factor |
| MEF | Mouse embryonic fibroblast |
| MSC | Mesenchymal stem cells |
| PCL | Poly(ε-caprolactone) |
| PDGF | Platelet derived growth factor |
| PGA | Polyglycolic acid |
| PLG | Poly(lactide-co-glycolide) |
| PLGA | Poly(lactic-co-glycolic acid) |
| RA | Retanoic acid |
| SDF-1 | Stromal derived factor 1 |
| SDS | Sodium dodecyl sulfate |
| SDSC | Synovium-derived stem cells |
| SMA | Smooth muscle actin |
| TCPS | Tissue culture polystyrene |
| TEBV | Tissue engineered blood vessel |
| TEHV | Tissue engineered heart valve |
| TGF | Transforming growth factor |
| Ti | Titanium |
| VEGF | Vascular endothelial growth factor |
Acknowledgements
We dedicate this work to Dr Michael V. Sefton for his support, mentorship and leadership to the fields of biomaterials and tissue engineering. All of those fortunate enough to have interacted with Michael and witness his influence on the science and careers of many will forever appreciate his meaningful and lasting impacts.The authors appreciate financial support from the NIH (TR01 AR062006).
References
- S. F. Badylak, D. O. Freytes and T. W. Gilbert, Acta Biomater., 2009, 5, 1–13 CrossRef CAS PubMed.
- K. E. M. Benders, P. R. van Weeren, S. F. Badylak, D. l. B. F. Saris, W. J. A. Dhert and J. Malda, Trends Biotechnol., 2013, 31, 169–176 CrossRef CAS PubMed.
- J. J. Song and H. C. Ott, Trends Mol. Med., 2011, 17, 424–432 CrossRef CAS PubMed.
- H. C. Ott, T. S. Matthiesen, S. K. Goh, L. D. Black, S. M. Kren, T. I. Netoff and D. A. Taylor, Nat. Med., 2008, 14, 213–221 CrossRef CAS PubMed.
- T. H. Petersen, E. A. Calle, L. Zhao, E. J. Lee, L. Gui, M. B. Raredon, K. Gavrilov, T. Yi, Z. W. Zhuang, C. Breuer, E. Herzog and L. E. Niklason, Science, 2010, 329, 538–541 CrossRef CAS PubMed.
- H. C. Ott, B. Clippinger, C. Conrad, C. Schuetz, I. Pomerantseva, L. Ikonomou, D. Kotton and J. P. Vacanti, Nat. Med., 2010, 16, 927–933 CrossRef CAS PubMed.
- B. E. Uygun, A. Soto-Gutierrez, H. Yagi, M. L. Izamis, M. A. Guzzardi, C. Shulman, J. Milwid, N. Kobayashi, A. Tilles, F. Berthiaume, M. Hertl, Y. Nahmias, M. L. Yarmush and K. Uygun, Nat. Med., 2010, 16, 814–820 CrossRef CAS PubMed.
- C. Quint, Y. Kondo, R. J. Manson, J. H. Lawson, A. Dardik and L. E. Niklason, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9214–9219 CrossRef CAS PubMed.
- Z. H. Syedain, L. A. Meier, J. W. Bjork, A. Lee and R. T. Tranquillo, Biomaterials, 2011, 32, 714–722 CrossRef CAS PubMed.
- H. Lu, T. Hoshiba, N. Kawazoe and G. Chen, Biomaterials, 2011, 32, 2489–2499 CrossRef CAS PubMed.
- M. Pei and F. He, J. Cell. Physiol., 2012, 227, 2163–2174 CrossRef CAS PubMed.
- T. Hoshiba, T. Yamada, H. Lu, N. Kawazoe and G. Chen, J. Biomed. Mater. Res., Part A, 2012, 100, 694–702 CrossRef PubMed.
- M. Pei, M. Shoukry, J. Li, S. D. Daffner, J. C. France and S. E. Emery, Spine, 2012, 37, 1538–1547 CrossRef PubMed.
- M. Pei, Y. Zhang, J. Li and D. Chen, Stem. Cells Dev., 2013, 22, 889–900 CrossRef CAS PubMed.
- Z. Ivanovic, J. Cell. Physiol., 2009, 219, 271–275 CrossRef CAS PubMed.
- T. W. Gilbert, T. L. Sellaro and S. F. Badylak, Biomaterials, 2006, 27, 3675–3683 CAS.
- T. J. Keane and S. F. Badylak, The host response to allogeneic and xenogeneic biological scaffold materials, J. Tissue Eng. Regen. Med., 2014 DOI:10.1002/term.1874.
- M. L. Wong and L. G. Griffiths, Acta Biomater., 2014, 10, 1806–1816 CrossRef CAS PubMed.
- M. H. Zheng, J. Chen, Y. Kirilak, C. Willers, J. Xu and D. Wood, J. Biomed. Mater. Res., Part B, 2005, 73, 61–67 CrossRef CAS PubMed.
- W. Wystrychowski, T. N. McAllister, K. Zagalski, N. Dusserre, L. Cierpka and N. L'Heureux, J. Vasc. Surg., 2013 DOI:10.1016/j.jvs.2013.08.018.
- N. Datta, Q. P. Pham, U. Sharma, V. I. Sikavitsas, J. A. Jansen and A. G. Mikos, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2488–2493 CrossRef CAS PubMed.
- N. h. Datta, H. L. Holtorf, V. I. Sikavitsas, J. A. Jansen and A. G. Mikos, Biomaterials, 2005, 26, 971–977 CrossRef CAS PubMed.
- J. Liao, X. Guo, D. Nelson, F. K. Kasper and A. G. Mikos, Acta Biomater., 2010, 6, 2386–2393 CrossRef CAS PubMed.
- R. A. Thibault, L. Scott Baggett, A. G. Mikos and F. K. Kasper, Tissue Eng., Part A, 2010, 16, 431–440 CrossRef CAS PubMed.
- J. Liao, X. Guo, K. J. Grande-Allen, F. K. Kasper and A. G. Mikos, Biomaterials, 2010, 31, 8911–8920 CrossRef CAS PubMed.
- Q. P. Pham, F. K. Kasper, A. S. Mistry, U. Sharma, A. W. Yasko, J. A. Jansen and A. G. Mikos, J. Biomed. Mater. Res., Part A, 2009, 88, 295–303 CrossRef PubMed.
- Q. P. Pham, F. Kurtis Kasper, L. Scott Baggett, R. M. Raphael, J. A. Jansen and A. G. Mikos, Biomaterials, 2008, 29, 2729–2739 CrossRef CAS PubMed.
- Y. J. Hong, S. E. Bae, S. H. Do, I. H. Kim, D. K. Han and K. Park, Macromol. Res., 2011, 19, 1090–1096 CrossRef CAS PubMed.
- M. L. Decaris, B. Y. Binder, M. A. Soicher, A. Bhat and J. K. Leach, Tissue Eng., Part A, 2012, 18, 2148–2157 CrossRef CAS PubMed.
- M. L. Decaris, A. Mojadedi, A. Bhat and J. K. Leach, Acta Biomater., 2012, 8, 744–752 CrossRef CAS PubMed.
- N. Sadr, B. E. Pippenger, A. Scherberich, D. Wendt, S. Mantero, I. Martin and A. Papadimitropoulos, Biomaterials, 2012, 33, 5085–5093 CrossRef CAS PubMed.
- E. R. Deutsch and R. E. Guldberg, J. Mater. Chem., 2010, 20, 8942–8951 RSC.
- G. Tour, M. Wendel and I. Tcacencu, Tissue Eng., Part A, 2011, 17, 127–137 CrossRef CAS PubMed.
- J. Glowacki, Cell Tissue Bank, 2005, 6, 3–12 CrossRef PubMed.
- A. M. Mackay, S. C. Beck, J. M. Murphy, F. P. Barry, C. O. Chichester and M. F. Pittenger, Tissue Eng., 1998, 4, 415–428 CrossRef CAS.
- J. U. Yoo, T. S. Barthel, K. Nishimura, L. Solchaga, A. I. Caplan, V. M. Goldberg and B. Johnstone, J. Bone Joint Surg. Am., 1998, 80, 1745–1757 CAS.
- S. Thakkar, C. A. Ghebes, M. Ahmed, C. Kelder, C. A. van Blitterswijk, D. Saris, H. A. M. Fernandes and L. Moroni, Biofabrication, 2013, 5, 025003 CrossRef PubMed.
- F. He, X. Chen and M. Pei, Tissue Eng., Part A, 2009, 15, 3809–3821 CrossRef CAS PubMed.
- M. Pei, F. He, J. Li, J. E. Tidwell, A. C. Jones and E. B. McDonough, Tissue Eng., Part A, 2013, 19, 1144–1154 CrossRef CAS PubMed.
- T. Z. Li, C. Z. Jin, B. H. Choi, M. S. Kim, Y. J. Kim, S. R. Park, J. H. Yoon and B.-H. Min, Adv. Funct. Mater., 2012, 22, 4292–4300 CrossRef CAS.
- W. Wystrychowski, L. Cierpka, K. Zagalski, S. Garrido, N. Dusserre, S. Radochonski, T. N. McAllister and N. L'Heureux, J. Vasc. Access, 2011, 12, 67–70 CrossRef.
- T. N. McAllister, M. Maruszewski, S. A. Garrido, W. Wystrychowski, N. Dusserre, A. Marini, K. Zagalski, A. Fiorillo, H. Avila, X. Manglano, J. Antonelli, A. Kocher, M. Zembala, L. Cierpka, L. M. de la Fuente and N. L'Heureux, Lancet, 2009, 373, 1440–1446 CrossRef.
- C. Quint, M. Arief, A. Muto, A. Dardik and L. E. Niklason, J. Vasc. Surg., 2012, 55, 790–798 CrossRef PubMed.
- S. L. M. Dahl, A. P. Kypson, J. H. Lawson, J. L. Blum, J. T. Strader, Y. Li, R. J. Manson, W. E. Tente, L. DiBernardo, M. T. Hensley, R. Carter, T. P. Williams, H. L. Prichard, M. S. Dey, K. G. Begelman and L. E. Niklason, Sci. Transl. Med., 2011, 3, 68ra69 Search PubMed.
- S. L. M. Dahl, J. Koh, V. Prabhakar and L. E. Niklason, Cell Transplant., 2003, 12, 659–666 Search PubMed.
- Z. H. Syedain, L. A. Meier, M. T. Lahti, S. L. Johnson and R. T. Tranquillo, Tissue Eng., Part A, 2014, 20(11–12), 1726–1734 CrossRef CAS PubMed.
- N. L'Heureux, N. Dusserre, G. Konig, B. Victor, P. Keire, T. N. Wight, N. A. Chronos, A. E. Kyles, C. R. Gregory, G. Hoyt, R. C. Robbins and T. N. McAllister, Nat. Med., 2006, 12, 361–365 CrossRef PubMed.
- N. L'Heureux, T. N. McAllister and L. M. de la Fuente, N. Engl. J. Med., 2007, 357, 1451–1453 CrossRef PubMed.
- S. E. Langerak, M. Groenink, E. E. van der Waals, C. Wassenaar, E. Vanbavel, M. C. van Baal and J. A. Spaan, Transpl. Int., 2001, 14, 248–255 CrossRef CAS PubMed.
- B. Weber, P. E. Dijkman, J. Scherman, B. Sanders, M. Y. Emmert, J. Grunenfelder, R. Verbeek, M. Bracher, M. Black, T. Franz, J. Kortsmit, P. Modregger, S. Peter, M. Stampanoni, J. Robert, D. Kehl, M. van Doeselaar, M. Schweiger, C. E. Brokopp, T. Walchli, V. Falk, P. Zilla, A. Driessen-Mol, F. P. T. Baaijens and S. P. Hoerstrup, Biomaterials, 2013, 34, 7269–7280 CrossRef PubMed.
- Z. H. Syedain, A. R. Bradee, S. Kren, D. A. Taylor and R. T. Tranquillo, Tissue Eng., Part A, 2013, 19, 759–769 CrossRef CAS PubMed.
- P. E. Dijkman, A. Driessen-Mol, L. Frese, S. P. Hoerstrup and F. P. T. Baaijens, Biomaterials, 2012, 33, 4545–4554 CrossRef CAS PubMed.
- D. van Geemen, A. Driessen-Mol, L. G. M. Grootzwagers, R. S. Soekhradj-Soechit, P. W. Riem Vis, F. P. T. Baaijens and C. V. C. Bouten, Regen. Med., 2012, 7, 59–70 CrossRef CAS PubMed.
- Z. H. Syedain, L. A. Meier, J. M. Reimer and R. T. Tranquillo, Ann. Biomed. Eng., 2013, 41(12), 2645–2654 CrossRef PubMed.
- T. C. Flanagan, J. S. Sachweh, J. Frese, H. Schnoring, N. Gronloh, S. Koch, R. H. Tolba, T. Schmitz-Rode and S. Jockenhoevel, Tissue Eng., Part A, 2009, 15, 2965–2976 CrossRef CAS PubMed.
- Z. H. Syedain and R. T. Tranquillo, J. Biomech., 2011, 44, 848–855 CrossRef PubMed.
- M. H. Cha, S. H. Do, G. R. Park, P. Du, K.-C. Han, D. K. Han and K. Park, Tissue Eng., Part A, 2013, 19, 978–988 CrossRef CAS PubMed.
- Y. Lai, Y. Sun, C. M. Skinner, E. L. Son, Z. Lu, R. S. Tuan, R. L. Jilka, J. Ling and X.-D. Chen, Stem Cells Dev., 2010, 19, 1095–1107 CrossRef CAS PubMed.
- Y. Sun, W. Li, Z. Lu, R. Chen, J. Ling, Q. Ran, R. L. Jilka and X.-D. Chen, FASEB J., 2011, 25, 1474–1485 CrossRef CAS PubMed.
- X.-D. Chen, V. Dusevich, J. Q. Feng, S. C. Manolagas and R. L. Jilka, J. Bone Miner. Res., 2007, 22, 1943–1956 CrossRef CAS PubMed.
- I. Klimanskaya, Y. Chung, L. Meisner, J. Johnson, M. D. West and R. Lanza, Lancet, 2005, 365, 1636–1641 CrossRef CAS.
- M. C. Prewitz, F. P. Seib, M. von Bonin, J. Friedrichs, A. Stissel, C. Niehage, K. Muller, K. Anastassiadis, C. Waskow, B. Hoflack, M. Bornhauser and C. Werner, Nat. Methods, 2013, 10, 788–794 CrossRef CAS PubMed.
- C. P. Ng, A. R. Sharif, D. E. Heath, J. W. Chow, C. B. Zhang, M. B. Chan-Park, P. T. Hammond, J. K. Chan and L. G. Griffith, Biomaterials, 2014, 35, 4046–4057 CrossRef CAS PubMed.
- J. A. Thomson, J. Itskovitz-Eldor, S. S. Shapiro, M. A. Waknitz, J. J. Swiergiel, V. S. Marshall and J. M. Jones, Science, 1998, 282, 1145–1147 CrossRef CAS.
- M. J. Evans and M. H. Kaufman, Nature, 1981, 292, 154–156 CrossRef CAS.
- G. R. Martin, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 7634–7638 CrossRef CAS.
- J. Itskovitz-Eldor, M. Schuldiner, D. Karsenti, A. Eden, O. Yanuka, M. Amit, H. Soreq and N. Benvenisty, Mol. Med., 2000, 6, 88–95 CAS.
- T. C. McDevitt and S. P. Palecek, Curr. Opin. Biotechnol., 2008, 19, 527–533 CrossRef CAS PubMed.
- C. Xu, M. S. Inokuma, J. Denham, K. Golds, P. Kundu, J. D. Gold and M. K. Carpenter, Nat. Biotechnol., 2001, 19, 971–974 CrossRef CAS PubMed.
- H. K. Kleinman, M. L. McGarvey, L. A. Liotta, P. G. Robey, K. Tryggvason and G. R. Martin, Biochemistry, 1982, 21, 6188–6193 CrossRef CAS.
- L. G. Villa-Diaz, A. M. Ross, J. Lahann and P. H. Krebsbach, Stem Cells, 2013, 31, 1–7 CrossRef CAS PubMed.
- T. Hoshiba, H. Lu, N. Kawazoe, T. Yamada and G. Chen, Biotechnol. Prog., 2013, 29, 1331–1336 CrossRef CAS PubMed.
- P. Carinci, E. Becchetti and M. Bodo, Int. J. Dev. Biol., 2000, 44, 715–723 CAS.
- J. C. Reichert, V. M. C. Quent, L. J. Burke, S. H. Stansfield, J. A. Clements and D. W. Hutmacher, Biomaterials, 2010, 31, 7928–7936 CrossRef CAS PubMed.
- A. Bhat, S. A. Boyadjiev, C. W. Senders and J. K. Leach, PLoS One, 2011, 6, e25990 CAS.
- Y. Tang, X. Wu, W. Lei, L. Pang, C. Wan, Z. Shi, L. Zhao, T. R. Nagy, X. Peng, J. Hu, X. Feng, W. Van Hul, M. Wan and X. Cao, Nat. Med., 2009, 15, 757–765 CrossRef CAS PubMed.
- Y. Xu, P. Malladi, M. Chiou and M. T. Longaker, Plast. Reconstr. Surg., 2007, 119, 819–829 CrossRef CAS PubMed.
- J. Uitto, D. R. Olsen and M. J. Fazio, J. Invest. Dermatol., 1989, 92, 61S–77S CrossRef CAS.
- P. A. Soucy and L. H. Romer, Matrix Biol., 2009, 28, 273–283 CrossRef CAS PubMed.
- R. S. Kirsner, G. Bohn, V. R. Driver, J. L. Mills Sr., L. B. Nanney, M. L. Williams and S. C. Wu, Lancet, 2012, 380(9846), 977–985 CrossRef.
- A. Reyzelman, R. T. Crews, J. C. Moore, L. Moore, J. S. Mukker, S. Offutt, A. Tallis, W. B. Turner, D. Vayser, C. Winters and D. G. Armstrong, Int. Wound J., 2009, 6, 196–208 CrossRef PubMed.
- N. Moiemen, J. Yarrow, E. Hodgson, J. Constantinides, E. Chipp, H. Oakley, E. Shale and M. Freeth, Plast. Reconstr. Surg., 2011, 127, 1149–1154 CrossRef CAS PubMed.
- S. A. Rehim, M. Singhal and K. C. Chung, Hand Clin., 2014, 30, 239–252 CrossRef PubMed , vii.
- A. S. Colwell, M. T. Longaker and H. P. Lorenz, Adv. Biochem. Eng. Biotechnol., 2005, 93, 83–100 CrossRef CAS.
- M. W. Ferguson and S. O'Kane, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 839–850 CrossRef CAS PubMed.
- B. J. Larson, M. T. Longaker and H. P. Lorenz, Plast. Reconstr. Surg., 2010, 126, 1172–1180 CrossRef CAS PubMed.
- R. Nair, A. V. Ngangan, M. L. Kemp and T. C. McDevitt, PLoS One, 2012, 7, e42580 CAS.
- S. Shukla, R. Nair, M. W. Rolle, K. R. Braun, C. K. Chan, P. Y. Johnson, T. N. Wight and T. C. McDevitt, J. Histochem. Cytochem., 2010, 58, 345–358 CrossRef CAS PubMed.
- R. Nair, A. V. Ngangan and T. C. McDevitt, J. Biomater. Sci., Polym. Ed., 2008, 19, 801–819 CrossRef CAS PubMed.
- R. Nair, S. Shukla and T. C. McDevitt, J. Biomed. Mater. Res., Part A, 2008, 87, 1075–1085 CrossRef PubMed.
- A. V. Ngangan and T. C. McDevitt, Biomaterials, 2009, 30, 1143–1149 CrossRef CAS PubMed.
- S.-K. Goh, P. Olsen and I. Banerjee, PLoS One, 2013, 8, e61856 CAS.
- S. Sart, T. Ma and Y. Li, Tissue Eng., Part A, 2014, 20, 54–66 CrossRef CAS PubMed.
- H. Yousef, M. J. Conboy, J. Li, M. Zeiderman, T. Vazin, C. Schlesinger, D. V. Schaffer and I. M. Conboy, Aging, 2013, 5, 357–372 CAS.
- S. H. Li, Z. Sun, L. Guo, M. Han, M. F. Wood, N. Ghosh, I. A. Vitkin, R. D. Weisel and R. K. Li, J. Cell. Mol. Med., 2012, 16, 2429–2439 CrossRef CAS PubMed.
- P. E. Bourgine, B. E. Pippenger, A. Todorov, L. Tchang and I. Martin, Biomaterials, 2013, 34, 6099–6108 CrossRef CAS PubMed.
- M. Rodrigues, H. Blair, L. Stockdale, L. Griffith and A. Wells, Stem Cells, 2013, 31, 104–116 CrossRef CAS PubMed.
- M. Yamaoka, S. Yamaguchi, T. Suzuki, M. Okuyama, J. Nitobe, N. Nakamura, Y. Mitsui and H. Tomoike, J. Mol. Cell. Cardiol., 2000, 32, 881–889 CrossRef CAS PubMed.
- E. H. Nijhuis, A. A. Poot, J. Feijen and I. Vermes, Int. J. Hyperthermia., 2006, 22, 687–698 CrossRef CAS.
- U. Rauen, B. Polzar, H. Stephan, H. G. Mannherz and H. de Groot, FASEB J., 1999, 13, 155–168 CAS.
- C. M. Snyder, E. H. Shroff, J. Liu and N. S. Chandel, PLoS One, 2009, 4, e7059 Search PubMed.
- P. Bourgine, C. Le Magnen, S. Pigeot, J. Geurts, A. Scherberich and I. Martin, Stem Cell Res., 2014, 12, 584–598 CrossRef CAS PubMed.
- G. R. Park, J. G. Lee, H. J. Chun, D. K. Han and K. Park, Macromol. Res., 2012, 20, 868–874 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |