
Reducible, dibromomaleimide-linked polymers for gene delivery†
James-Kevin Y.
Tan
*,
Jennifer L.
Choi
,
Hua
Wei
,
Joan G.
Schellinger
and
Suzie H.
Pun
University of Washington, Department of Bioengineering and Molecular Engineering and Sciences Institute, William H. Foege Building, Box 355061, 3720 15th Ave NE, Seattle, WA 98195, USA. E-mail: spun@uw.edu; Fax: +1 206 543 3488; Tel: +1 206 685 3488
First published on 12th August 2014
Abstract
Polycations have been successfully used as gene transfer vehicles both in vitro and in vivo; however, their cytotoxicity has been associated with increasing molecular weight. Polymers that can be rapidly degraded after internalization are typically better tolerated by mammalian cells compared to their non-degradable counterparts. Here, we report the use of a dibromomaleimide-alkyne (DBM-alkyne) linking agent to reversibly bridge cationic polymer segments for gene delivery and to provide site-specific functionalization by azide–alkyne cycloaddition chemistry. A panel of reducible and non-reducible, statistical copolymers of (2-dimethylamino)ethyl methacrylate (DMAEMA) and oligo(ethylene glycol)methyl ether methacrylate (OEGMA) were synthesized and evaluated. When complexed with plasmid DNA, the reducible and non-reducible polymers had comparable DNA condensation properties, sizes, and transfection efficiencies. When comparing cytotoxicity, the DBM-linked, reducible polymers were significantly less toxic than the non-reducible polymers. To demonstrate polymer functionalization by click chemistry, the DBM-linked polymers were tagged with an azide-fluorophore and were used to monitor cellular uptake. Overall, this polymer system introduces the use of a reversible linker, DBM-alkyne, to the area of gene delivery and allows for facile, orthogonal, and site-specific functionalization of gene delivery vehicles.
Introduction
Gene-based drugs typically require a carrier to facilitate delivery into cell nuclei, where they are transcribed and later translated into proteins that can elicit a therapeutic effect. While virally-derived carriers have been the preferred vectors in clinical trials, a number of safety concerns have arisen due to their ability to alter the genome (retrovirus and lentivirus) and activate the immune system.1–3 While non-viral vectors generally have lower transfection efficiencies compared to viral vectors, they offer potential safety advantages. A class of materials that is highly investigated for non-viral gene transfer is cationic polymers, which condense DNA through electrostatic interactions, protect cargo from early elimination, and can be readily functionalized with molecular targeting agents.4–7 One of the most effective and commonly used cationic polymers is polyethylenimine (PEI); however, it is non-degradable and has a high density of positive amino groups that contribute to its cytotoxicity.8In vivo, PEI aggregates and accumulates in the lungs and liver upon systemic administration due to strong electrostatic interactions with cell membranes, proteins, and the extracellular matrix.5,6 Several other cationic materials such as poly[L-lysine],9,10 chitosan,11–13 poly[(N-2-hydroxypropyl)methacrylamide] (pHPMA),14–16 and poly[(2-dimethylamino)ethyl methacrylate] (pDMAEMA)17–20 have also been investigated and are able to achieve transfection efficiencies comparable to that of PEI but with the advantage of improved cell viability. Nevertheless, cationic polymers such as pDMAEMA still have inherent cytotoxicity due to their non-degradable nature.20,21 To ameliorate these effects, degradable linkages have been incorporated into cationic polymers so they can dissociate into smaller, less toxic fragments.3,20,22,23 Two commonly used chemistries for synthesizing degradable polymers are reducible disulfide bonds and hydrolyzable ester bonds.20,21,24 Several degradable polymers containing ethylene imine units have been reported with comparable gene delivery efficiency to that of PEI but with reduced cytotoxicity.14,22,25,26Bromomaleimide-functional groups have recently been developed for their ability to reversibly react with thiols. For example, dibromomaleimide (DBM) has two bromines that can be substituted with free thiols to form two thioether bonds.27 Upon the addition of an excess of thiols, the thioether bonds can be readily cleaved to regenerate the original, thiolated product. Since the cell cytoplasm contains reducing agents such as glutathione, intracellular bromomaleimide-linked conjugates can potentially be cleaved in vivo. In fact, these thioether bonds have been shown to be cleaved within 1 hour in the cytoplasm of mammalian cells.28 Another advantage of DBM is that it has a third point of attachment off the maleimide group. This has allowed for a very site-specific and quantity-controlled conjugation of various moieties ranging from fluorophores to polymers.27–29
Herein, we describe synthesis of cleavable gene delivery vehicles that utilize DBM as a reversible bridging agent for cationic polymer segments as well as a moiety for site-specific functionalization. A panel of reducible, statistical copolymers of DMAEMA and oligo(ethylene glycol)methyl ether methacrylate (OEGMA) were synthesized by reacting thiol-terminated polymers with a DBM-alkyne derivative. Polymer fragmentation in reducing environments was confirmed. While polymer reducibility did not significantly affect gene transfer ability, the reducible polymers were better tolerated by mammalian cells than their non-reducible analogs. We further demonstrated site-specific modification of polymers by click conjugation of an azide-functionalized fluorophore to the alkyne site. Fluorophore-labeled polymers were used to monitor polyplex uptake by flow cytometry. Overall, the DBM-alkyne derivative offers a facile way to reversibly connect different polymers for gene delivery while the alkyne allows for the site-specific functionalization of polymers.
Materials and methods
Materials
Reagents for solid phase peptide synthesis were purchased from Apptec (Louisville, KY) except 5-azido pentanoic acid which was purchased from Bachem (Torrance, CA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Cell culture reagents were purchased from Cellgro/Mediatech (Fisher Scientific, Pittsburgh PA). The pCMV-Luc2 plasmid was isolated using the Qiagen Plasmid Giga Kit (Hilden, Germany). The bicinchoninic acid (BCA) protein quantification assay kit was purchased from Thermo Fischer Scientific (Waltham, MA) while the luciferase expression quantification kit and MTS assay kit were obtained from Promega (Madison, WI).Polymer synthesis and characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 hexane–ethyl acetate) to yield the product as an off-white powder (0.38 g, 82%). The product identity and purity was confirmed by GC-MS on a Hewlett-Packard (Palo Alto, CA) 5971A GC-MSD.
:1 methanol–300 mM acetate buffer pH 4.4 on a Shodex (Kawasaki, Japan) SB-804 HQ column connected to a Shimadzu (Kyoto, Japan) LC-20AD liquid chromatography pump and Wyatt (Sana Barbara, CA) MiniDawn Treos and Optilab rEX system. Number and weight average molecular weight (Mn and Mw) and dn/dc values were calculated using ASTRA software (Wyatt).
:1 (v/v) DMF–0.05 mM PBS to a final concentration of 0.02 M. The solution was stirred for 30 minutes before the addition of DBM-alkyne (1.5 mg, 5.0 μmol). After 2 hours of stirring, the solution was dialyzed to remove DMF and salts and subsequently lyophilized to yield an orange solid gel. Polymers were characterized by aqueous SEC as mentioned previously.
:0.5 hexane–ethyl acetate) to yield the product as an off-white powder (0.38 g, 82%). The product identity and purity was confirmed by GC-MS on a Hewlett-Packard (Palo Alto, CA) 5971A GC-MSD.
:1 methanol–300 mM acetate buffer pH 4.4 on a Shodex (Kawasaki, Japan) SB-804 HQ column connected to a Shimadzu (Kyoto, Japan) LC-20AD liquid chromatography pump and Wyatt (Sana Barbara, CA) MiniDawn Treos and Optilab rEX system. Number and weight average molecular weight (Mn and Mw) and dn/dc values were calculated using ASTRA software (Wyatt).
:1 (v/v) DMF–0.05 mM PBS to a final concentration of 0.02 M. The solution was stirred for 30 minutes before the addition of DBM-alkyne (1.5 mg, 5.0 μmol). After 2 hours of stirring, the solution was dialyzed to remove DMF and salts and subsequently lyophilized to yield an orange solid gel. Polymers were characterized by aqueous SEC as mentioned previously.
Polyplex characterization
:100 and incubated at room temperature for 1 hour.
Polyplexes were prepared at an N/P = 10 by complexing YOYO-labeled plasmid with reducible and non-reducible polymers as previously mentioned. Polyplexes were pre-treated with 1 mM glutathione for 8 hours and then treated with 5 μg mL−1 heparin sulfate for 1 hour. The fluorescence (ex: 491 nm, ex: 509 nm) of each well was normalized to its respective DNA only control.
In vitro studies
Statistical analysis
All statistical analyses were performed using a two-tailed Student's t-test with unequal variance.Results and discussion
Polymer synthesis and characterization
Three cationic, reducible polymers were synthesized by substituting a DBM-alkyne molecule with thiolated copolymers of DMAEMA and OEGMA (Scheme 1a). DBM-alkyne was synthesized as previously reported (Fig. S1 & S2†).27 A double-headed ATRP initiator with an internal disulfide was prepared by N,N′-dicyclohexylcarbodiimide coupling between 2-hydroxyethyl disulfide and 2-bromo-2-methylpropionic acid (Fig. S3†). The double-headed initiator was used to prepare statistical copolymers of DMAEMA and OEGMA with low (∼10 kDa), medium (∼20 kDa), and high (∼40 kDa) target molecular weights (Table 1). The DMAEMA monomer provides cationic charge for DNA condensation while the OEGMA affords colloidal stability as reported previously for similar systems.20,21,31 The resulting p(DMAEMA-s-OEGMA) materials possessed near-target molecular weights and compositions close to monomer feed ratios (5:1 DMAEMA to OEGMA) (Fig. S4†). These parent p(DMAEMA-s-OEGMA) polymers were reduced with TCEP to generate thiol-terminated fragments that were substituted onto DBM-alkyne. Under SEC-MALLS analysis, TCEP reduction caused a delayed elution compared to the elution of the parent polymers due to the reduction of the disulfide bond and decreased molecular weight (Fig. 1). DBM-alkyne was added to the thiolated polymers in the presence of TCEP to prevent disulfide formation and to yield the DBM-substituted polymers. The SEC traces of the DBM-substituted polymers were shifted to the earlier elution times, as expected for the increased molecular weight due to polymer substitution (Fig. 1 and Table 1). The reaction was rapid and highly efficient as only equimolar amounts of parent polymer and DBM-alkyne were needed. Excess TCEP ensured the complete reduction of parent p(DMAEMA-s-OEGMA) and did not reduce the newly formed thioether bonds. Furthermore, the substituted polymers showed a new increase in UV absorbance and exhibited fluorescent properties [data not shown]. These findings are consistent with reports of other substituted dibromomaleimides.27,29,30,32 As non-reducible controls, polymers of similar molecular weights were polymerized with a conventional ATRP initiator, α-bromoisobutyrate (Scheme 1b). The non-reducible polymers had molecular weights similar to those of the substituted polymers and monomer incorporations close to the initial feed ratio (5:1 DMAEMA to OEGMA) (Table 1 and Fig. S4†).
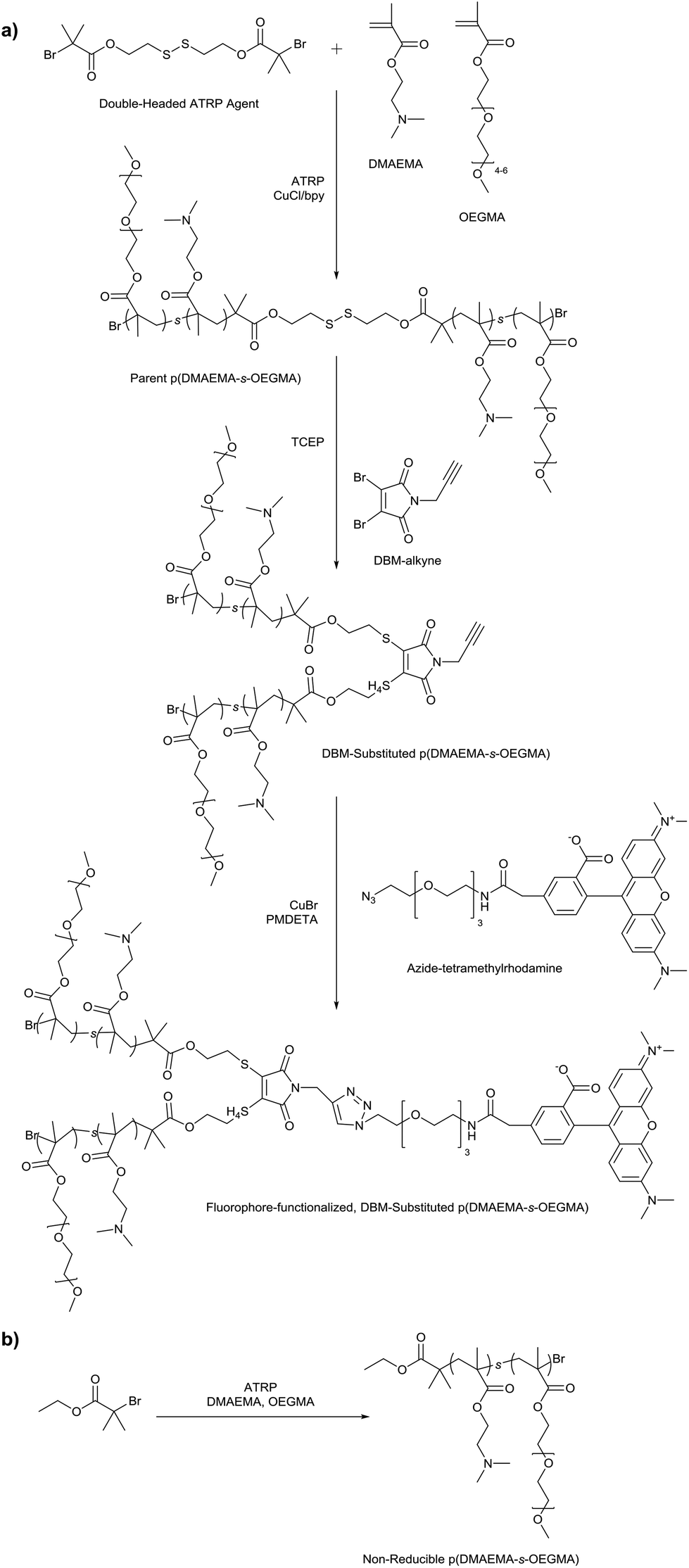 | ||
| Scheme 1 (a) Synthesis of fluorophore-functionalized, DBM-substituted p(DMAEMA-s-OEGMA); (b) Synthesis of non-reducible p(DMAEMA-s-OEGMA). | ||
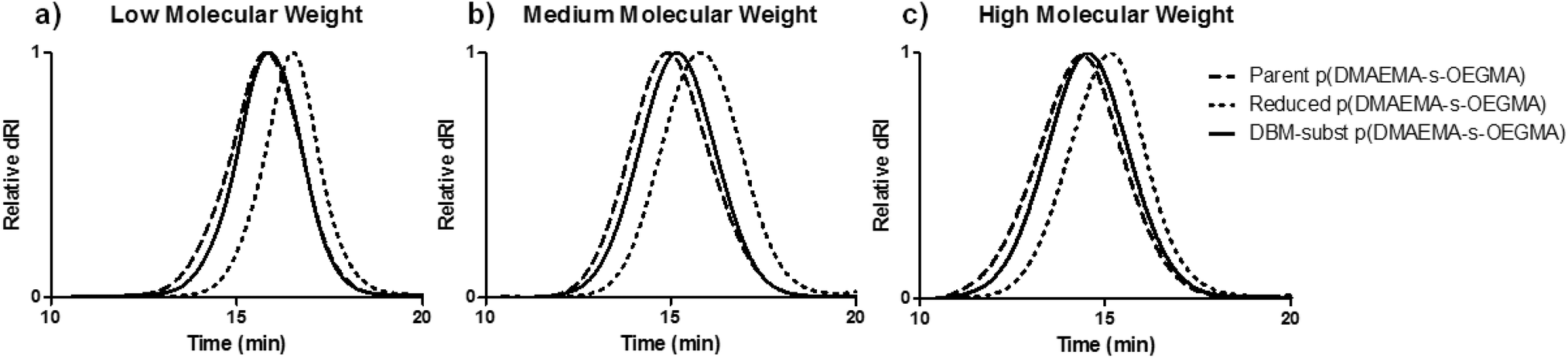 | ||
| Fig. 1 SEC traces of (a) low, (b) medium, and (c) high molecular weight parent, reduced, and DBM-substituted DMAEMA and OEGMA copolymers. Parent polymers were reduced with TCEP (right-shifted trace) and resulting thiolated polymer fragments were substituted to DBM-alkyne (overlapping trace with parent polymer). | ||
| Type of polymer | Determined Mn (kDa)a | M w/Mna | Determined DMAEMA:OEGMA ratiob |
|
|---|---|---|---|---|
| a Determined by SEC-MALLS. b Determined by 1H NMR. | ||||
| Parent p(DMAEMA-s-OEGMA) | Low | 13080 |
1.31 | 5.11 |
| Medium | 21010 |
1.42 | 5.41 | |
| High | 40090 |
1.55 | 6.23 | |
| DBM-substituted p(DMAEMA-s-OEGMA) | Low | 15440 |
1.24 | 5.11 |
| Medium | 23810 |
1.38 | 5.41 | |
| High | 35770 |
1.49 | 6.23 | |
| Non-reducible p(DMAEMA-s-OEGMA) | Low | 10170 |
1.29 | 4.61 |
| Medium | 19490 |
1.31 | 4.91 | |
| High | 42220 |
1.85 | 4.17 | |
Larger molecular weight pDMAEMA has been correlated with higher cytotoxicity.33 Unlike the slow hydrolysis rate of DMAEMA esters,21 the DBM-substituted polymers should be more rapidly reduced after intracellular uptake of the polyplexes in order to facilitate cargo release and reduce cytotoxicity. To demonstrate polymer reduction under physiological conditions, DBM-substituted polymers were incubated in a cytoplasmic mimic buffer27 containing 1 mM glutathione reducing agent for 2 hours and analyzed by SEC (Fig. 2a). Substituted polymers with glutathione had a delayed elution compared to the peak of the DBM-substituted polymers, demonstrating that the thioether bonds in the copolymer were cleaved by glutathione. The reducibility of this polymer system is consistent with other DBM systems and suggests that the material can be cleaved in the reducing environment of the cell cytoplasm.27,28,34 Non-reducible polymers were also incubated with glutathione and analyzed by SEC (Fig. 2b). No peak shift was seen in the SEC traces for the non-reducible polymers.
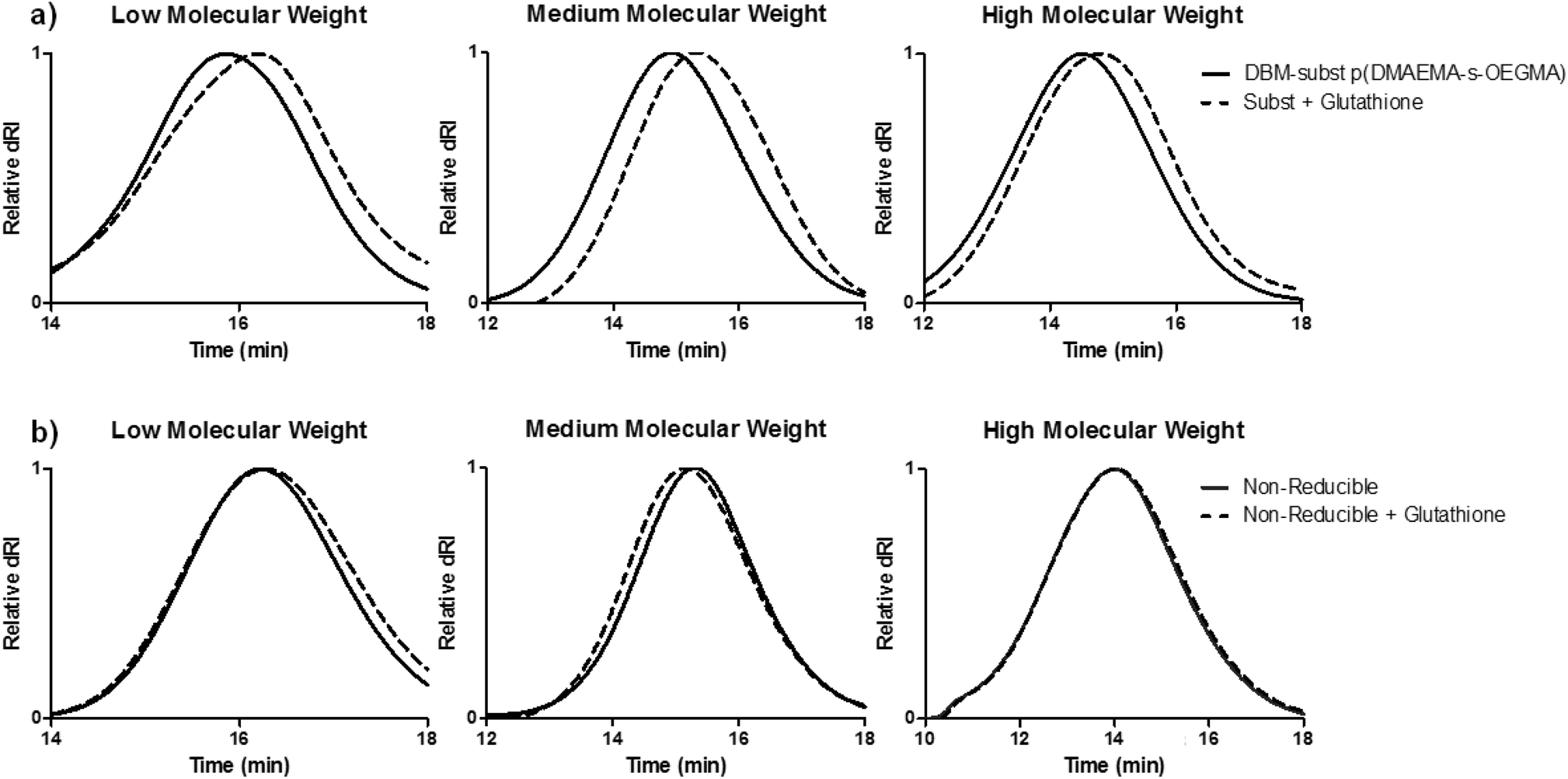 | ||
| Fig. 2 SEC traces of (a) DBM-substituted and (b) non-reducible DMAEMA and OEGMA copolymers with glutathione. The copolymers were incubated in a cell cytoplasm mimic buffer containing 20 mM HEPES, 100 mM KCl, 1 mM MgCl2, 1 mM EDTA, and 1 mM glutathione, pH 7.4. Incubation with glutathione reduced the DBM-substituted polymers and caused a rightward peak shift, signifying a decrease in MW. | ||
Characterization of polyplexes
Polyplexes were formed by mixing the polymers with plasmid DNA at various amine to phosphate (N/P) ratios. The ability of the polyplexes to complex DNA was assessed by a gel retardation assay (Fig. S5†). Complete complexation of DNA was observed starting at an N/P = 2 for reducible and non-reducible copolymers, suggesting that DBM-alkyne and the reducible thioethers did not affect DNA condensation. Polyplexes formed at N/P = 5 and 10 were characterized by DLS in either ddH2O or PBS containing physiological salt concentrations (Fig. 3). In water, reducible polyplexes showed comparable polyplex size (150–225 nm) to non-reducible polyplexes (175–260 nm), suggesting that DBM-alkyne and the reducible thioethers did not affect polyplex formation and size. In physiological salt concentrations, relatively good stability is observed as particles remained the same size or only slightly increased in size. Homopolymers of DMAEMA tend to form large complexes and aggregates with DNA (∼1 μm); thus, the incorporation of OEGMA was able to stabilize polyplexes in physiological salt concentrations.17,19 The uncharged, hydrophilic properties of OEGMA provided steric hindrance and an extra hydration layer that conferred increased stabilization as reported for other DMAEMA and OEGMA copolymers.17–19,35,36 Zeta potential measurements (Fig. S6†) indicate that the net surface charges for both reducible and non-reducible copolymers at N/P = 5 and 10 are all positive (∼20–40 mV in water, ∼5–20 mV in PBS). A positive charge is necessary for polyplex-mediated gene delivery since the excess positive charge interacts with the negatively charged cell membrane for enhanced uptake and gene delivery.37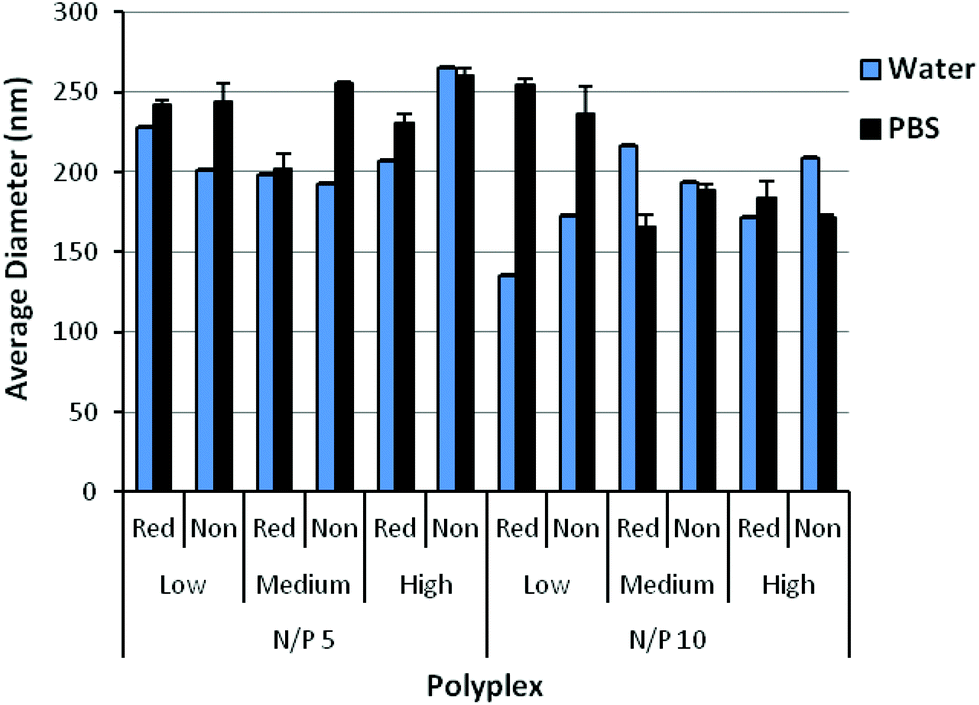 | ||
| Fig. 3 Average diameter of reducible and non-reducible polymer polyplexes measured by DLS. Polyplexes were formed in water and allowed to form for 10 minutes. Afterwards, polyplexes were diluted with water or 150 mM PBS and measured by DLS. Data is presented as mean ± SD, n = 3. | ||
Polyplex unpackaging in non-reducible and reducible conditions was assessed by incubating YOYO-1-labeled DNA polyplexes with heparin sulfate and glutathione (Fig. S7†). In condensed complexes, the YOYO-1 fluorescence is self-quenching; however, as the polyplex unpackages, the fluorescence is recovered. When comparing the heparin only versus heparin and glutathione conditions, the reducible medium MW and high MW polyplexes unpackaged more after exposure to a reducing environment. These results suggest that cleavable thioether bonds of the reducible, DBM-linked polymers were able to mediate greater DNA unpackaging in the presence of glutathione.
In vitro transfection and cytotoxicity
To investigate the effect of cleavable linkages on transfection efficiency, polymers were complexed with the luciferase reporter plasmid at N/P = 5 and 10 and exposed to HeLa cells. Branched polyethylenimine (bPEI, 25k) at an N/P = 5 was used as a standard for comparison. Transfection efficiency of both reducible and non-reducible polymers improved with increasing molecular weight as seen with other DMAEMA and OEGMA copolymers and other cationic systems (Fig. 4a).36 The transfection efficiencies of the polyplexes were comparable with only 5 of the 12 reducible and analogous non-reducible polyplex sets showing statistically significant differences in transfection efficiency (P < 0.05), as marked by an (*) in Fig. 4a. The reducible polymers had higher transfection efficiency for two of the statistically significant sets while the non-reducible polymers had higher transfection efficiency for the other three statistically significant sets. This suggests that polymer reducibility did not greatly affect transfection efficiency.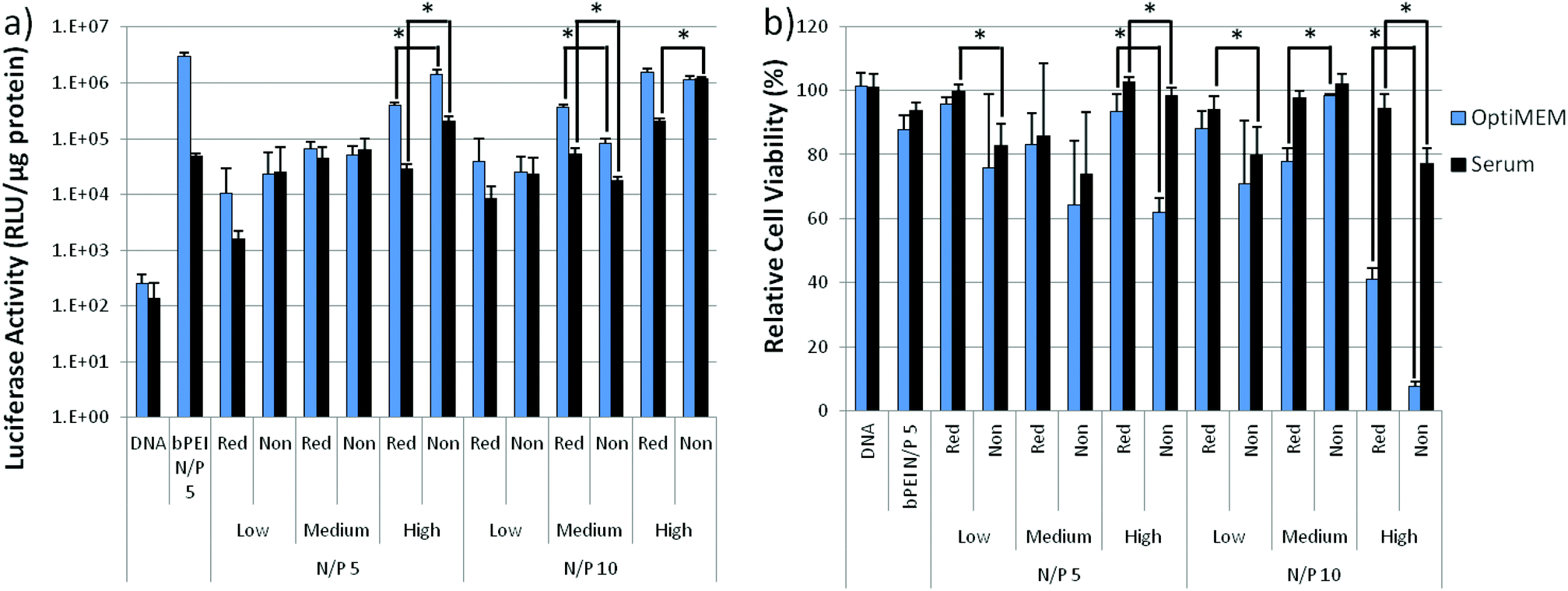 | ||
| Fig. 4 (a) Luciferase plasmid transfection efficiency and (b) cytotoxicity of reducible DBM-alkyne and non-reducible low, medium, and high MW polyplexes at N/P ratios = 5 and 10. Naked DNA and bPEI (25 kD) controls are included for comparison. Data is presented as mean ± SD, n = 4. Statistically significant (P < 0.05) differences are indicated with a (*). | ||
The cytotoxicity of the polymers was assessed by measuring the total protein content in cell lysates after transfection as compared to untreated cells (Fig. 4b). Larger molecular weight copolymers were more cytotoxic than smaller copolymers as is reported for other systems.8 Overall, the reducible, DBM-substituted polymers had higher cell viabilities than their non-reducible counterparts in both OptiMEM™ and serum. Seven of the 12 analogous polymer sets showed statistically significant (P < 0.05) differences in cytotoxicity, as marked with an (*). The reducible polymers had better viability for six of the statistically significant sets, suggesting that the reducible polyplexes were less cytotoxic than the non-reducible polyplexes. The reducible nature of the DBM-substituted polyplexes allowed the large size of positive charge to more readily disassemble into smaller, less toxic pieces as reported for other reducible systems.20,22,25
IC50 study
While the reducible polyplexes were less toxic than the non-reducible polyplexes, there is a lack of direct measurement of polymer toxicity without DNA. An MTS assay with HeLa cells was conducted to determine half maximal inhibitory concentration (IC50) values of polymers alone without DNA (Table 2). The IC50 values were based on DMAEMA monomer concentration to account for differences in molecular weight, amount of DMAEMA incorporation, and because the cytotoxicity of the polymers is heavily based on the cationic charge. The reducible polymers had higher IC50 values than their non-reducible counterparts, consistent with the lower toxicity seen with reducible polyplexes in Fig. 4b. Statistically significant (P < 0.05) differences are marked with an (*). The IC50 decreased with increasing molecular weight which is also consistent with the trend that larger molecular weight polymers are less tolerated by cells.8| Type of polymer | Low molecular weight | Medium molecular weight | High molecular weight |
|---|---|---|---|
| *indicates reducible polymers were less toxic than their counterpart and statistically significant (P < 0.05). | |||
| DBM-substituted p(DMAEMA-s-OEGMA) | 119 μM | 31.7 μM* | 5.97 μM* |
| Non-reducible p(DMAEMA-s-OEGMA) | 96.0 μM | 26.2 μM | 3.51 μM |
Cleavage of the reducible high and medium MW polymers results in polymers similar in size to the non-reducible medium and low MW polymers, respectively. Therefore, similar IC50 values might be expected for these pairs of polymers; however, the IC50 value of the reducible polymer is lower in each case. The higher cytotoxicity from the reducible polymer compared to the lower molecular non-reducible polymer may be due to the kinetics and location of the dithiomaleimide reduction. The expected location for reduction of DBM-substituted polymers is in the cytoplasm and another 2 hours is needed for complete reduction of thioether bonds once inside the cell cytoplasm.28 In addition, there is a higher concentration of reduced polymer than the non-reducible polymer since the reduction of each DBM-substituted polymer yields two polymers. Nonetheless, the reducible polymers are less toxic than their non-reducible counterparts. Together with the glutathione reduction study (Fig. 2), these results suggest that the DBM-substituted polymers are better tolerated by mammalian cells compared to their non-reducible counterparts due to fragmentation triggered after cellular internalization.
Polyplex uptake by flow cytometry
The reducible, DBM-substituted polymers may be easily modified at the alkyne functional group by using azide–alkyne cycloaddition. Most cationic polymers are functionalized by reaction with their primary amines; however, this can be undesirable in gene transfer applications since amine reactions may interfere with nucleic acid packaging. Therefore, the DBM-alkyne provides a location for site-specific functionalization without affecting polymer charge. As a proof-of-concept, we functionalized the DBM-substituted polymers with azide-modified rhodamine fluorophores (Scheme 1a) and used flow cytometry to monitor cellular uptake of the fluorescently-labeled polyplexes. Fluorophore alone, un-functionalized polyplexes, un-functionalized polyplexes with free fluorophore, and fluorophore-functionalized polyplexes were incubated with cells and polymer uptake was analyzed by flow cytometry (Fig. 5). Statistically significant (P < 0.05) differences are marked with an (*). Cells treated with fluorophore-labeled polyplexes exhibited the highest levels of mean fluorescence intensity compared to the fluorophore only, polymer only, and polyplex with free fluorophore groups. Thus, the fluorophore-labeled polymers were able to monitor the uptake of the polyplexes.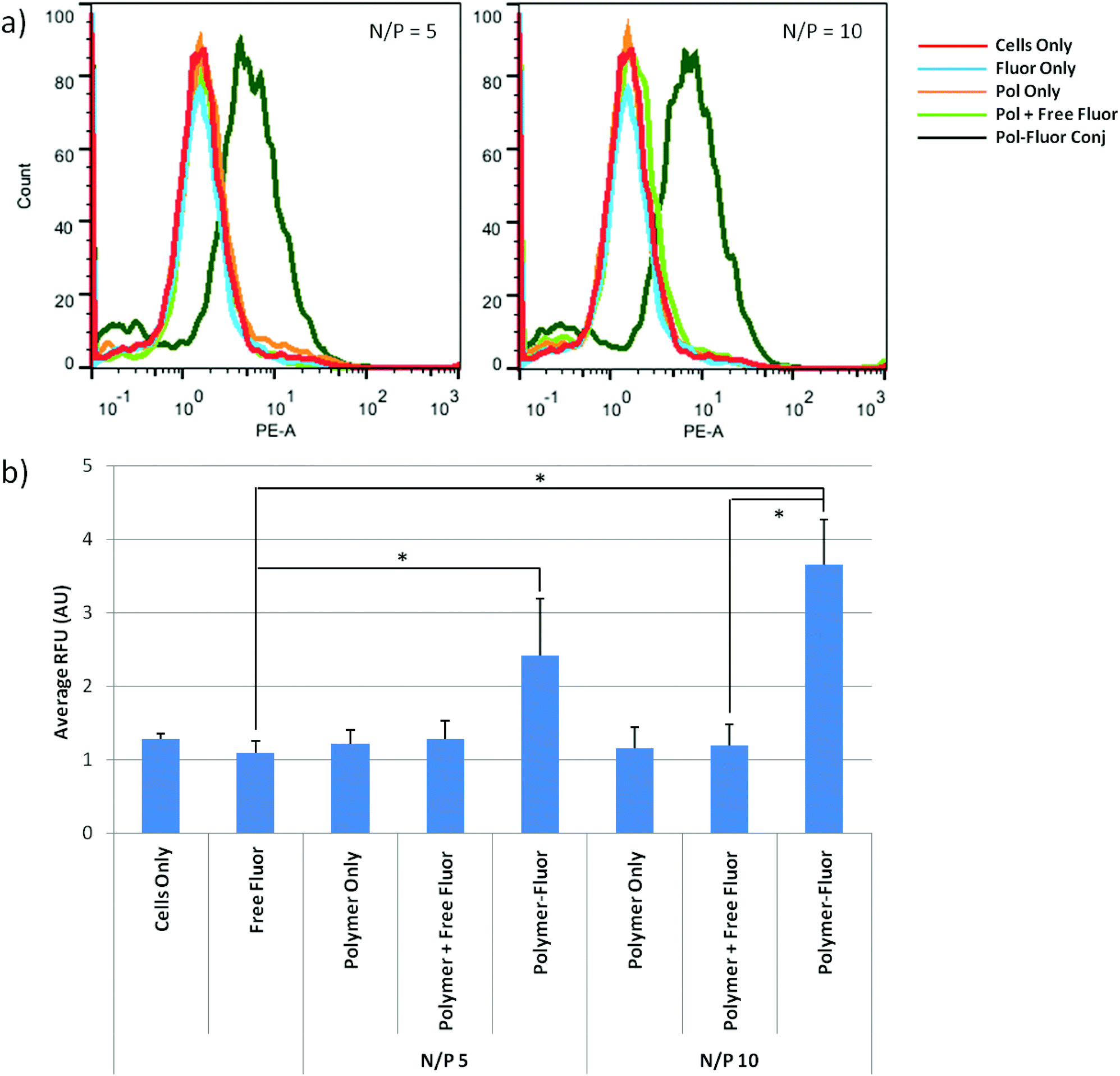 | ||
| Fig. 5 (a) Histograms representing the data and (b) average fluorescence as assessed by flow cytometry. Uptake of fluorophore, non-functionalized polyplexes, polyplexes plus free fluorophore, and fluorophore-functionalized polyplexes was monitored in HeLa cells. Data is presented as mean ± SD, n = 4. Statistically significant (P < 0.05) differences are indicated with a (*). | ||
In addition to imaging agents, these DBM-linked conjugates can be functionalized with other moieties such as peptides and antibodies. The DBM-alkyne derivative provides a site-specific and quantity-controlled method of conjugation that is less promiscuous than other conjugation methods such as amine chemistry. Another possible application is to use the alkyne handle to immobilize polymers or polyplexes to azide-functionalized surfaces. Polymers or drug carriers can then be easily released from surfaces by a reducing agent such as glutathione.
Conclusion
A panel of reducible and non-reducible DMAEMA and OEGMA copolymers was synthesized by ATRP. The double-headed polymers were reduced and substituted onto a DBM-alkyne derivative. All polymers condensed plasmid DNA and were stable in water and physiological salt concentrations. The reducibility afforded by DBM did not have a significant effect on transfection efficiency; however, it did confer much reduced cytotoxicity since the polymer can dissociate in the reducing environment of the cell cytoplasm. In addition to lowering cytotoxicity, DBM-alkyne provided a site-specific and quantity-controlled place of attachment. A fluorophore was clicked onto the DBM-linked polymers and these polyplexes were able to monitor cell uptake. The site-specific and quantity-controlled functionalization of DBM may allow for more controlled conjugation of biomolecules such as peptides and antibodies.Acknowledgements
This work was supported by NIH 2R01 NS064404. JKYT is supported by an NSF Graduate Research Fellowship (2011128558). We would also like to thank David S. Chu for his expertise and guidance and Binhan Pham for his assistance.Notes and references
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed.
- T. Niidome and L. Huang, Gene Ther., 2002, 9, 1647–1652 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- B. Abdallah, A. Hassan and C. Benoist, Hum. Gene Ther., 1996, 7, 1947–1954 CrossRef CAS PubMed.
- M. Jäger, S. Schubert, S. Ochrimenko, D. Fischer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 4755–4767 RSC.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS.
- J. G. Schellinger, J. A. Pahang, R. N. Johnson, D. S. H. Chu, D. L. Sellers, D. O. Maris, A. J. Convertine, P. S. Stayton, P. J. Horner and S. H. Pun, Biomaterials, 2013, 34, 2318–2326 CrossRef CAS PubMed.
- D. S. H. Chu, J. G. Schellinger, M. J. Bocek, R. N. Johnson and S. H. Pun, Biomaterials, 2013, 34, 9632–9637 CrossRef CAS PubMed.
- K. Wong, G. Sun, X. Zhang, H. Dai, Y. Liu, C. He and K. W. Leong, Bioconjugate Chem., 2006, 17, 152–158 CrossRef CAS PubMed.
- P. Erbacher, S. Zou and T. Bettinger, Pharm. Res., 1998, 15 Search PubMed.
- M. Köping-Höggård, K. M. Vårum, M. Issa, S. Danielsen, B. E. Christensen, B. T. Stokke and P. Artursson, Gene Ther., 2004, 11, 1441–1452 CrossRef PubMed.
- H. Wei, J. A. Pahang and S. H. Pun, Biomacromolecules, 2013, 14, 275–284 CrossRef CAS PubMed.
- D. S. H. Chu, R. N. Johnson and S. H. Pun, J. Controlled Release, 2012, 157, 445–454 CrossRef CAS PubMed.
- R. N. Johnson, R. S. Burke, A. J. Convertine, A. S. Hoffman, P. S. Stayton and S. H. Pun, Biomacromolecules, 2010, 3007–3013 CrossRef CAS PubMed.
- U. Rungsardthong, M. Deshpande, L. Bailey, M. Vamvakaki, S. P. Armes, M. C. Garnett and S. Stolnik, J. Controlled Release, 2001, 73, 359–380 CrossRef CAS.
- S. Uzgün, O. Akdemir, G. Hasenpusch, C. Maucksch, M. M. Golas, B. Sander, H. Stark, R. Imker, J.-F. Lutz and C. Rudolph, Biomacromolecules, 2010, 11, 39–50 CrossRef PubMed.
- M. C. Deshpande, M. C. Davies, M. C. Garnett, P. M. Williams, D. Armitage, L. Bailey, M. Vamvakaki, S. P. Armes and S. Stolnik, J. Controlled Release, 2004, 97, 143–156 CrossRef CAS PubMed.
- Y.-Z. You, D. S. Manickam, Q.-H. Zhou and D. Oupický, J. Controlled Release, 2007, 122, 217–225 CrossRef CAS PubMed.
- Y. Zhang, M. Zheng, T. Kissel and S. Agarwal, Biomacromolecules, 2012, 13, 313–322 CrossRef CAS PubMed.
- M. A. Gosselin, W. Guo and R. J. Lee, Bioconjugate Chem., 2001, 12, 989–994 CrossRef CAS PubMed.
- L. V. Christensen, C.-W. Chang, W. J. Kim, S. W. Kim, Z. Zhong, C. Lin, J. F. J. Engbersen and J. Feijen, Bioconjugate Chem., 2006, 17, 1233–1240 CrossRef CAS PubMed.
- R. S. Burke and S. H. Pun, Bioconjugate Chem., 2010, 21, 140–150 CrossRef CAS PubMed.
- H. Wei, J. G. Schellinger, D. S. H. Chu and S. H. Pun, J. Am. Chem. Soc., 2012, 134, 16554–16557 CrossRef CAS PubMed.
- Q. Peng, Z. Zhong and R. Zhuo, Bioconjugate Chem., 2008, 19, 499–506 CrossRef CAS PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- P. Moody, M. E. B. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS PubMed.
- Y. Cui, Y. Yan, Y. Chen and Z. Wang, Macromol. Chem. Phys., 2013, 214, 470–477 CrossRef CAS.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed.
- P. van de Wetering, J. Y. Cherng, H. Talsma, D. J. Crommelin and W. E. Hennink, J. Controlled Release, 1998, 53, 145–153 CrossRef CAS.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed.
- J. M. Layman, S. M. Ramirez, M. D. Green and T. E. Long, Biomacromolecules, 2009, 10, 1244–1252 CrossRef CAS PubMed.
- C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC.
- F. J. Verbaan, C. Oussoren, C. J. Snel, D. J. Crommelin, W. E. Hennink and G. Storm, J. Gene Med., 2004, 6, 64–75 CrossRef CAS PubMed.
- S. Venkataraman, W. L. Ong, Z. Y. Ong, S. C. Joachim Loo, P. L. R. Ee and Y. Y. Yang, Biomaterials, 2011, 32, 2369–2378 CrossRef CAS PubMed.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00240g |
| This journal is © The Royal Society of Chemistry 2015 |