
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comparison of one-dimensional and microscale two-dimensional liquid chromatographic approaches coupled to high resolution mass spectrometry for the analysis of complex samples†
Juri
Leonhardt
ab,
Thorsten
Teutenberg
*a,
Jochen
Tuerk
ad,
Michael P.
Schlüsener
c,
Thomas A.
Ternes
c and
Torsten C.
Schmidt
bd
aInstitut für Energie- und Umwelttechnik e. V., IUTA (Institute of Energy and Environmental Technology), Bliersheimer Str. 58-60, 47229 Duisburg, Germany. E-mail: teutenberg@iuta.de; Fax: +49 2065 418 211; Tel: +49 2065 418 179
bInstrumental Analytical Chemistry, University of Duisburg-Essen, Universitätsstr. 5, 45141 Essen, Germany
cBundesanstalt für Gewässerkunde, BfG (Federal Institute of Hydrology), Am Mainzer Tor 1, 56068 Koblenz, Germany
dCentre for Water and Environmental Research, ZWU, University of Duisburg-Essen, Universitätsstr. 2, 45141 Essen, Germany
First published on 28th July 2015
Abstract
The interest in two-dimensional liquid chromatography separations is growing every year together with the number of open questions on the benefits of multidimensional systems in comparison to one-dimensional liquid chromatography. In order to solve some of these open questions this work presents a comparison of one-dimensional and microscale two-dimensional liquid chromatography coupled to high resolution mass spectrometry for targeted analysis in wastewater. The comparison is based on the evaluation of a reference standard mixture containing 99 compounds and a real wastewater sample. For the evaluation and compound identification three different criteria were chosen. At first, a deviation of ±5 ppm from the exact mass was defined as acceptable to include the compound for further evaluation. To eliminate false positive results, a maximum retention time deviation of less than 2.5% for each compound of the reference standard and the compounds detected in the wastewater sample was defined for a positive identification as a second criterion for 1D-LC and the second dimension of 2D-LC. In the third step, fragment information from MS/MS experiments was used for further identification of compounds in wastewater. Additionally, the influence of a higher mass accuracy of 1 ppm on the number of identified compounds in comparison to a mass accuracy of 5 ppm was investigated. The results showed that the number of identified compounds was higher by a factor of three in the wastewater sample when using the microscale 2D-LC approach. Moreover, a higher reliability for compound identification is obtained when using retention time and MS/MS information as identification criteria instead of only applying high mass accuracy of 1 or 5 ppm.
1. Introduction
One-dimensional liquid chromatography (1D-LC) coupled to mass spectrometric detection is a powerful tool for the analysis of complex environmental samples that might contain several thousands of different components.1,2 However, the analysis of such complex samples with one-dimensional liquid chromatography has limitations in terms of peak capacity.3 Alternative analysis techniques with higher peak capacity are therefore deemed necessary to resolve as many compounds as possible. In that regard, two-dimensional liquid chromatography (2D-LC), which is well established in several analytical fields including proteomic and genomic research,4 might also be a powerful tool for the comprehensive analysis of environmental samples.In general two main variations of 2D-LC are applied. The first one is the heart-cut or selective two-dimensional technique (LC-LC, sLC × LC), which allows to cut one5 or a few selected6,7 fractions of the effluent of the first dimension that are then transferred to a second column. The second technique is the comprehensive 2D-LC (LC × LC),5,8,9 where the whole eluate of the first dimension column (D1) is transferred in small fractions to the second dimension column (D2). Such multidimensional approaches offer the possibility to separate complex samples on stationary phases with different selectivity.
In contrast to 1D-LC, 2D-LC is often associated with very long analysis times of several hours or even days if offline LC × LC is applied.10 Moreover, a more complex system configuration is needed if online LC × LC is used. A further disadvantage is the dilution introduced by the modulation between the first and second-dimension of an online LC × LC system. Furthermore, very fast cycle times of less than 1 min are necessary for the second dimension separation which often results in high flow rates up to 5 mL min−1.11–13 Such high flow rates are not compatible with electrospray ionization (ESI) MS detection. Therefore, flow splitters are used to minimize the solvent load that is introduced into the ESI source.14
In order to provide a splitless hyphenation with mass spectrometry, a microscale online LC × LC system was developed in a previous work.15 That system was based on nano-LC in first and micro-LC in second dimension. The flow rate for the second dimension separation was adjusted to 40 μL min−1, which is compatible to ESI-MS. For miniaturized systems, however, the injection volume needs to be reduced, which also reduces the absolute mass injected onto the first dimension column. In this view, it must be critically asked whether a two-dimensional separation with a higher peak capacity is generally favorable for the analysis of complex samples. Although a higher peak capacity might be obtained for a two-dimensional separation, the total number of detected peaks may be lower when compared to a one-dimensional separation. The reason is that the mass spectrometer, which separates the analytes according to their mass-over-charge ratio, is capable of detecting and distinguishing peaks that are not totally resolved chromatographically.
In this context, a comparison between one-dimensional and microscale comprehensive two-dimensional liquid chromatography coupled to high resolution mass spectrometry was performed. First of all, a reference standard mix containing 99 target compounds was analyzed to obtain the retention time and additional MS/MS information. In the second step, a native wastewater sample was analyzed on the basis of a suspected target screening. A small part of the data set was already used to demonstrate the capability of the miniaturized 2D-LC system in a previous work.15
Three different criteria for analyte identification were defined for both approaches. First of all, the accurate mass and a mass accuracy of less than ±5 ppm were chosen. As second and third criteria retention time deviations and MS/MS information were defined and applied to increase the reliability of analyte identification. For each step, the total number of detected peaks was compared. To our knowledge this is the first comprehensive comparison of 1D- and 2D-separations for complex environmental samples.
2. Experimental section
2.1. Solvents and additives
Ultra-pure water (J. T. Baker, LC/MS reagent grade) was purchased from Mallinckrodt Baker (Griesheim, Germany). Acetonitrile and methanol were both LC-MS Optigrade from Promochem (LGC Standards, Wesel, Germany). All LC eluents were acidified by adding 0.1% formic acid (FA) by volume (puriss. p. a., ∼98%, Sigma-Aldrich, Schnelldorf, Germany).2.2. Multi-component reference mix and wastewater sample
Overall 99 substances were included in the reference mix to obtain information for a suspected target screening of a wastewater sample. This mixture contained 3 corrosion inhibitors, 4 metabolites of sulfonamide antibiotics, 6 mycotoxins, 72 pharmaceuticals (antibiotics, cytostatics, psychotropics and contrast media) as well as 14 pesticides. A detailed list of all components is provided in ESI Table S-1.† The resulting multi-component mix containing 1 μg mL−1 of each target analyte was prepared in acidified (0.1% FA) water–acetonitrile (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v).
:5, v/v).
To investigate the applicability of the comparison, a real wastewater sample (200 mL) was taken after the first sedimentation step of a municipal wastewater treatment plant. The sample preparation steps are given in ESI.†
Prior to injection, the multi-component reference mix as well as the wastewater sample were filtered through a 0.2 μm CHROMAFIL RC 20/25 disposable syringe filter (Macherey-Nagel, Düren, Germany).
2.3. 1D-HPLC instrument
The conventional 1D-LC separations were performed on an Agilent 1260 HPLC system (Agilent Technologies, Waldbronn, Germany). This instrument was equipped with an Agilent 1260 pump (Model Number G1311B), an autosampler (Model Number G1367E) and column oven (Model Number G1316A).The separation was carried out on a Luna C18(2) column (150 mm × 2.0 mm i.d., 3 μm particles, Phenomenex, Aschaffenburg, Germany). The injection volume was 20 μL and corresponds to 6% of the column void volume. The flow rate was 200 μL min−1 and the oven temperature was set to 30 °C. The mobile phase consisted of acidified (0.1% FA) water (eluent A) and acetonitrile (eluent B). A solvent gradient was applied according to the following program: 3 min hold at 2% B, in 15 min 2–98% B, 6 min hold at 98% B, in 0.5 min 98–2% B, re-equilibration for 5.5 min (∼4 column void volumes).
2.4. 2D-HPLC instrument
The separations with on-line comprehensive 2D-LC were performed on an Eksigent NanoLC-Ultra 2D pump system (Sciex, Dublin, CA). This HPLC system contained a column oven compartment with two integrated ten port two-position valves and two binary-gradient pneumatic pumps which are able to generate a maximum backpressure of 680 bar (10 kpsi). The system was controlled by the Eksigent software version 3.12.1. The modification of this system to work in comprehensive mode is described by Haun et al.15For the first dimension (D1) separation a commercially available Hypercarb column (50 mm × 0.1 mm i.d., 5 μm particles, Thermo Fisher Scientific, Dreieich, Germany) was used. This stationary phase contained porous graphitic carbon (PGC) and was selected because of its high retentivity towards polar compounds.16 The flow rate was adjusted to 200 nL min−1 and the oven temperature was set to 60 °C. The mobile phase consisted of acidified (0.1% FA) water (% A) and methanol (% B). The injection volume on the D1 column was 1.57 μL. A solvent gradient was applied according to the following program: 8 min hold at 1% B, in 45 min 1–99% B, 35 min hold at 99% B, in 5 min 99–1% B, re-equilibration for 16 min (∼11 column void volumes).
For the second dimension (D2) separation a superficially porous 2.6 μm SunShell C18 particle (ChromaNik Technologies, Osaka, Japan) packed by Grace Davison (Worms, Germany) into a 50 mm × 0.3 mm i.d. hardware, was used. The flow rate was 40 μL min−1 and the oven temperature was set to 60 °C. The mobile phase consisted of acidified (0.1% FA) water (% A) and acetonitrile (% B). A solvent gradient was applied according to the following program: 3–97% B in 0.5 min, 0.1 min hold at 97% B, in 0.1 min 97–3% B, re-equilibration at 3% B for 0.3 min (∼6 column void volumes). The complete gradient cycle time took 1 min and was usually repeated without flow-stop until the end of the D1 program. The transfer volume on the D2 column was 300 nL. Additional information on the choice of the inner diameter of the columns can be found in ESI.†
Different solvents for the first and second dimension were used to increase the selectivity of the phase system. Acetonitrile was used in the second dimension because it has a much lower viscosity maximum than methanol. This is an important prerequisite in order to increase the flowrate as much as possible to achieve a very fast cycle time of 60 seconds in the second dimension. A slightly elevated temperature of 60 °C was used to reduce the viscosity maximum when a solvent gradient is applied. This is also very important because the column was operated near the maximum pressure of the pumps around 680 bars. During one analysis 109 gradient runs were performed. At a temperature of 60 °C the column is run nearly at constant pressure which greatly reduces the risk of a rapid column degradation. Increasing the temperature above 60 °C will decrease the column lifetime and also poses a risk to the modulation valves which are only specified to a maximum temperature of 60 °C.
2.5. MS instrumentation
For the mass spectrometric detection a Sciex (Darmstadt, Germany) hybrid HRMS system (TripleTOF 5600) with a DuoSpray ion source and a TurboIonSpray probe for ESI experiments was used. For the 1D-LC experiments with flow rates of 200 μL min−1, the standard probe was used. To minimize the dead volume and to avoid severe band broadening after the second dimension column of the 2D-LC, the standard emitter tip (i.d. 130 μm) of the source was replaced by an emitter with an i.d. of 50 μm. MS data acquisition was controlled with Sciex Analyst TF 1.5.1 and the data were analysed using Sciex PeakView 1.2.0.3 and MultiQuant 2.1.1742.0. The data acquired by the 2D-LC approach had to be manually evaluated due to the lack of commercially available 2D software packages.2.6. MS parameters
A suspected target screening approach with information dependent acquisition (IDA)17,18 was performed to obtain additional structural information. With this combination it is possible to detect a broad m/z range and afterwards to generate one or more product ion spectra of the most abundant precursor ions. The cycle time of such an IDA experiment depends on the selected m/z range and the defined number of precursor ions with a constant dwell time. The important MS parameters are listed in Table S-2.† All measurements were performed in positive electrospray ionization (ESI +) mode.Ions 214.090 Da (n-butyl benzenesulfonamide, plasticizer) and 221.190 Da (butylated hydroxytoluene, antioxidant) are both well-known contaminants in the field of LC-MS and were excluded for all IDA experiments. Dynamic background subtraction was enabled.
3. Results and discussion
3.1. Comparison of a reference standard mix and a wastewater sample
For the comparison of the 1D- and 2D-LC approach, a multi-component reference mix and a native wastewater sample were analysed with both approaches using high resolution mass spectrometry. The resulting total ion current (TIC) chromatograms are shown in Fig. 1.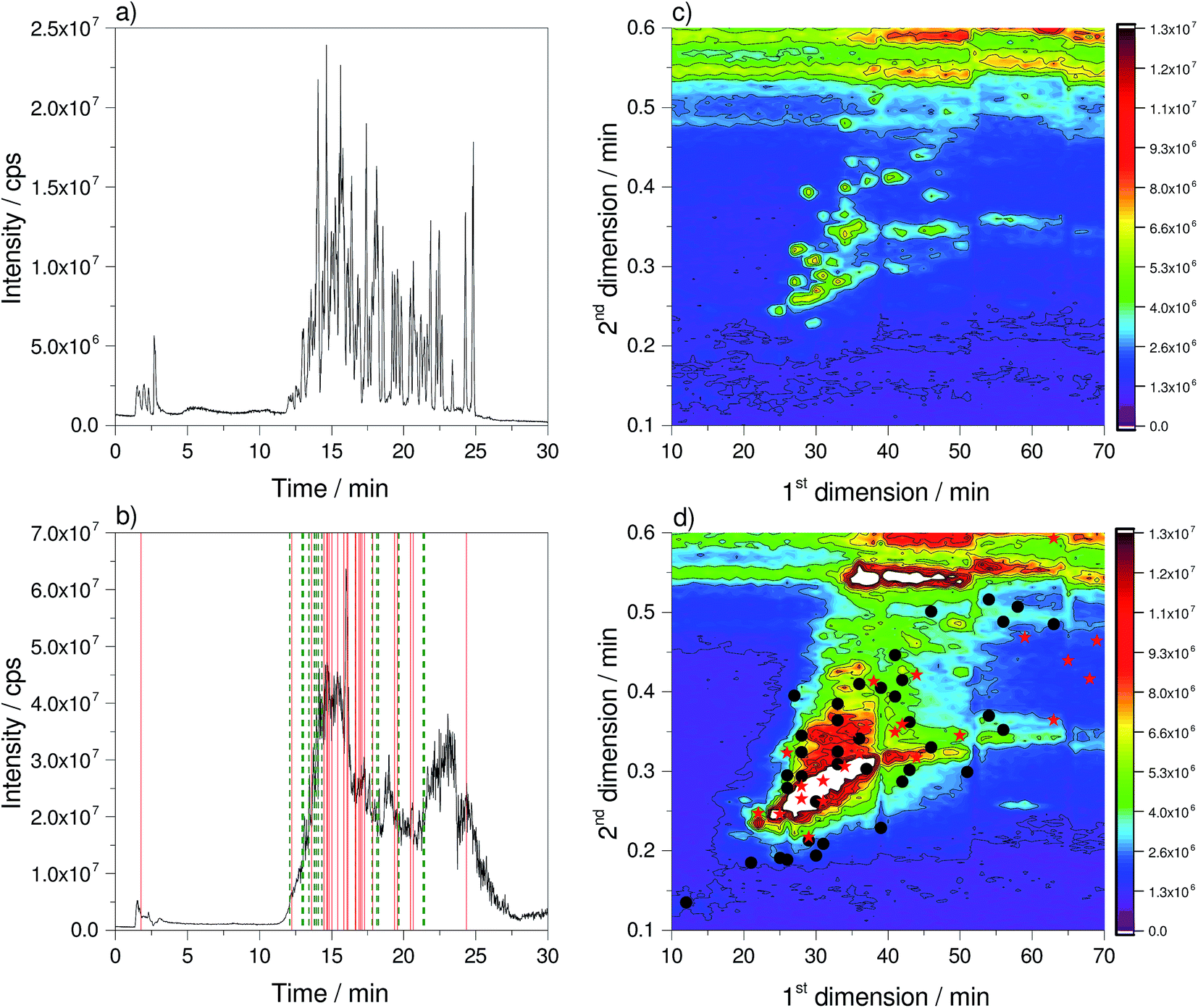 | ||
| Fig. 1 Total ion current chromatograms of a standard mixture and a native real wastewater sample. (a) Standard mixture using 1D-HPLC-MS; (b) wastewater sample using 1D-HPLC-MS (red solid lines = detected target compounds without MS/MS spectra; green dashed lines = detected target compounds with MS/MS spectra); (c) standard mixture using 2D-nLC × μLC-MS; (d) wastewater sample using 2D-nLC × μLC-MS (red stars = detected target compounds without MS/MS spectra; black dots = detected target compounds with MS/MS spectra). 2D-LC plots are zoomed in and redrawn with permission from Haun J., Leonhardt J., Portner C., Hetzel T., Tuerk J., Teutenberg T., Schmidt T. C., (2013), Anal. Chem., 85(21), 10083–10090. Copyright 2013 American Chemical Society. | ||
While for the standard mixture of the 1D separation distinct peaks can be seen in the TIC, a single merged peak is obtained for the wastewater sample (Fig. 1a and b). Here, the peaks cannot be resolved chromatographically, therefore mass spectrometry is required to obtain a separation in the m/z dimension. The observed single merged peak is a result of a co-elution of hundreds of compounds and underlines the complexity of the sample. It also clearly demonstrates that one-dimensional LC approaches will not provide the necessary resolution. The 2D-plots (Fig. 1c and d) show that the peaks are distributed over the chromatographic space. Nonetheless, there are regions with low and high peak density. The horizontal lines, which spread over the whole chromatographic run in D1 at a retention time in D2 of 0.5 and 0.6 min, resulted from the high content of the organic modifier at the end of the solvent gradient and the fast re-equilibration. In contrast to these signals that can be attributed to solvent effects, there is a horizontal signal at 0.34 min during the solvent gradient in D2, which occurs in fractions 30 to 70 in the reference standard as well as the wastewater sample. In this area, ions with m/z 648.3925, 670.3753 and 692.3902 could be observed and were assigned to Triton X detergents.19 Because of the fact that these ions are also present in the reference standard, they might be introduced by a contamination of the solvent reservoirs. Although the elution profile in D1 is very broad and covers 40 fractions, the retention time in D2 is constant. This underlines that although the elution strength of the transfer solvent continuously increases during the gradient run in D1, this has no effect on the retention on the D2 column. This is an important result because it underlines that the retention time obtained on the D2 column does not depend on the fraction of the organic modifier transferred from the D1 column to the D2 column. The additional solid and dashed lines as well as stars and dots that are also highlighted in Fig. 1 will be discussed later in Section 3.6.
3.2. Analyte identification by 5 ppm criterion in reference standard
On the basis of the molecular formula an exact monoisotopic mass and therefore the molecular formula of the protonated molecule [M + H]+ can be calculated. For the identification of detected signals with a high resolution mass spectrometer, a maximum deviation of ±5 ppm is often used as a criterion for compound identification in targeted analysis.20 A difference of less than 5 ppm between calculated and accurately measured m/z values is then taken as the major criterion for the presence of an analyte with the assumed sum formula. Applying this criterion it was possible to detect all chosen 99 components both with the 1D- and 2D-LC approach (for more information see Table S-3†).3.3. Modulation and the associated loss of intensity
The modulation process of the 2D-LC approach reduces the absolute peak intensities obtained after the second dimension separation. This negative effect is the result of the dilution when a one-dimensional peak is sampled in a number of fractions. For many compounds, at least five fractions per peak were obtained. By combining the effect of the lower injection volume (20 μL for 1D-LC and 1.57 μL for 2D-LC) which is applied onto the D1 column and the modulation, the peak area of a 2D-F2 signal in Fig. 2a is reduced by a factor of 86. Theoretically, the same factor would be expected for the differences in signal intensity between 1D-LC and 2D-LC. Interestingly, the absolute signal intensity is only reduced by a factor of 10 for the 2D-LC approach in comparison to the 1D-LC approach as is shown exemplarily for carbamazepine in Fig. 2a.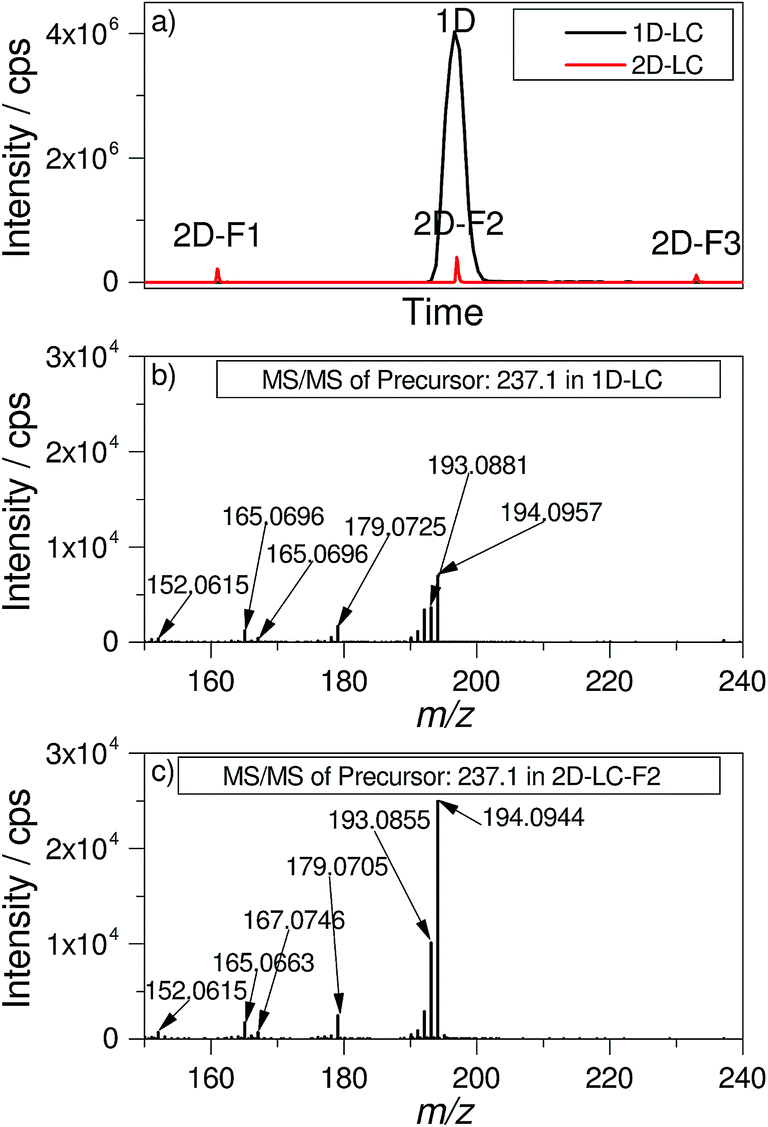 | ||
| Fig. 2 (a) Comparison of the absolute signal intensity of carbamazepine (XIC m/z 237.1022) for 1D-LC and 2D-LC approaches as an overlay. F1 to F3 in 2D-LC are three fractions that contain the corresponding target. Intensity: 1D-LC = 4026237 cps, 2D-LC-F2 = 399951 cps; signal-to-noise ratio: 1D-LC = 596, 2D-LC-F2 = 391; factor intensity: 10.1; factor signal-to-noise: 1.5; (b) MS/MS-spectrum of the precursor-ion m/z 237.1 in 1D-LC; (c) MS/MS-spectrum of the precursor-ion m/z 237.1 in 2D-LC fraction 2. | ||
Furthermore, the sensitivity and the detection limit do not necessarily depend on the absolute intensity, but on the signal-to-noise ratio (S/N-ratio). Due to the minimized solvent input into the ESI MS source, a lower dispersion by extra column volume and higher chromatographic efficiency of the 2D-LC system, the noise is substantially lower than for 1D-LC. The S/N-ratio is only 1.5 times smaller for carbamazepine in 2D-LC compared with 1D-LC. A similar observation was made for other target analytes (data not shown here).
A further visual inspection of the MS/MS spectra in Fig. 2b and c shows nearly the same pattern and a slightly higher intensity for the 2D-LC spectrum. This example underlines that the influence of the modulation and the strongly reduced injection volume on the identification of targets is very low.
3.4. Influence of mass accuracy on number of identified analytes
A suspected target screening was applied to the wastewater sample to reveal the number of detected compounds in both approaches. When using the 5 ppm criterion, 48 positive hits were obtained for the 1D-LC approach, while 65 positive hits were found for the 2D-LC approach.However, analyte identification which is only based on the accurate mass can lead to a high number of false positive results when a suspected target screening is applied. For the comparison of the 1D-LC and 2D-LC approaches a deviation of ±5 ppm was chosen. For a general unknown screening, often a higher mass accuracy is required21 to reduce the number of possible molecular formula which can be assigned to the molecular mass. Therefore, the question arises whether a smaller deviation of 1 ppm will lead to the elimination of false positive results. Fig. 3 shows the total number of detected compounds if the deviation is reduced from 5 ppm to 1 ppm in steps of 1 ppm.
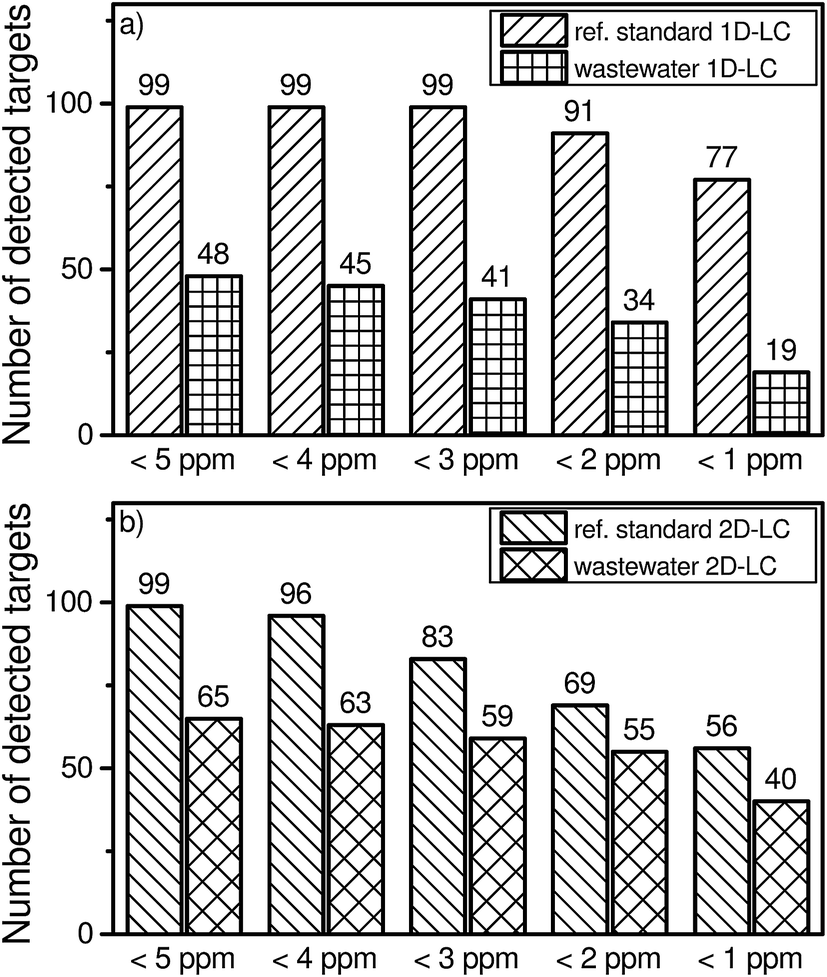 | ||
| Fig. 3 Number of detected targets versus a variable mass accuracy of 5 ppm to 1 ppm in multi-component standard and a real wastewater sample in the 1D-LC (a) and 2D-LC (b) approach. | ||
By reducing the mass accuracy from 5 ppm to 3 ppm for 1D-LC, the total number of detected targets of the reference standard remains constant at 99. If a deviation of 1 ppm is chosen, 78% of all targeted compounds can still be identified. For the wastewater sample, a higher mass accuracy leads to a more pronounced effect on the absolute number of excluded compounds. For a deviation of 1 ppm, the number of identified compounds is reduced by 60% when compared with a deviation of 5 ppm.
By reducing the mass accuracy from 5 ppm to 1 ppm for 2D-LC, the total number of detected targets of the reference standard decreases continuously. If a deviation of 1 ppm is chosen, 57% of all compounds can still be identified. For the wastewater sample and a higher mass accuracy of 1 ppm the number of identified compounds is reduced by 38% in comparison to a deviation of 5 ppm.
The results underline that already a small deviation of the accuracy of the mass spectrometer can lead to a preliminary exclusion of contained compounds in the sample or reference standard by using a too strong criterion. In this case 22 targets of the reference standard and 29 possible hits of the wastewater sample could be lost in the 1D-LC approach. In the 2D-LC approach, 43 targets of the reference standard and 25 possible hits of the wastewater sample might not be assigned. To avoid the exclusion of such a high number of targets, a mass accuracy of ±5 ppm was therefore chosen.
3.5. Retention time stability
To use the retention time of the analyte as an additional identification criterion and to exclude false positive hits, the retention time stability needs to be evaluated. A margin of ±2.5% was used for the deviation of the retention time measured in the sample from the retention time measured in the reference standard.To evaluate the retention time stability as an additional criterion for analyte identification, all detected targets that fulfilled the ±5 ppm for standard and sample and ±2.5% retention time stability criteria were plotted against each other in Fig. 4 for both approaches.
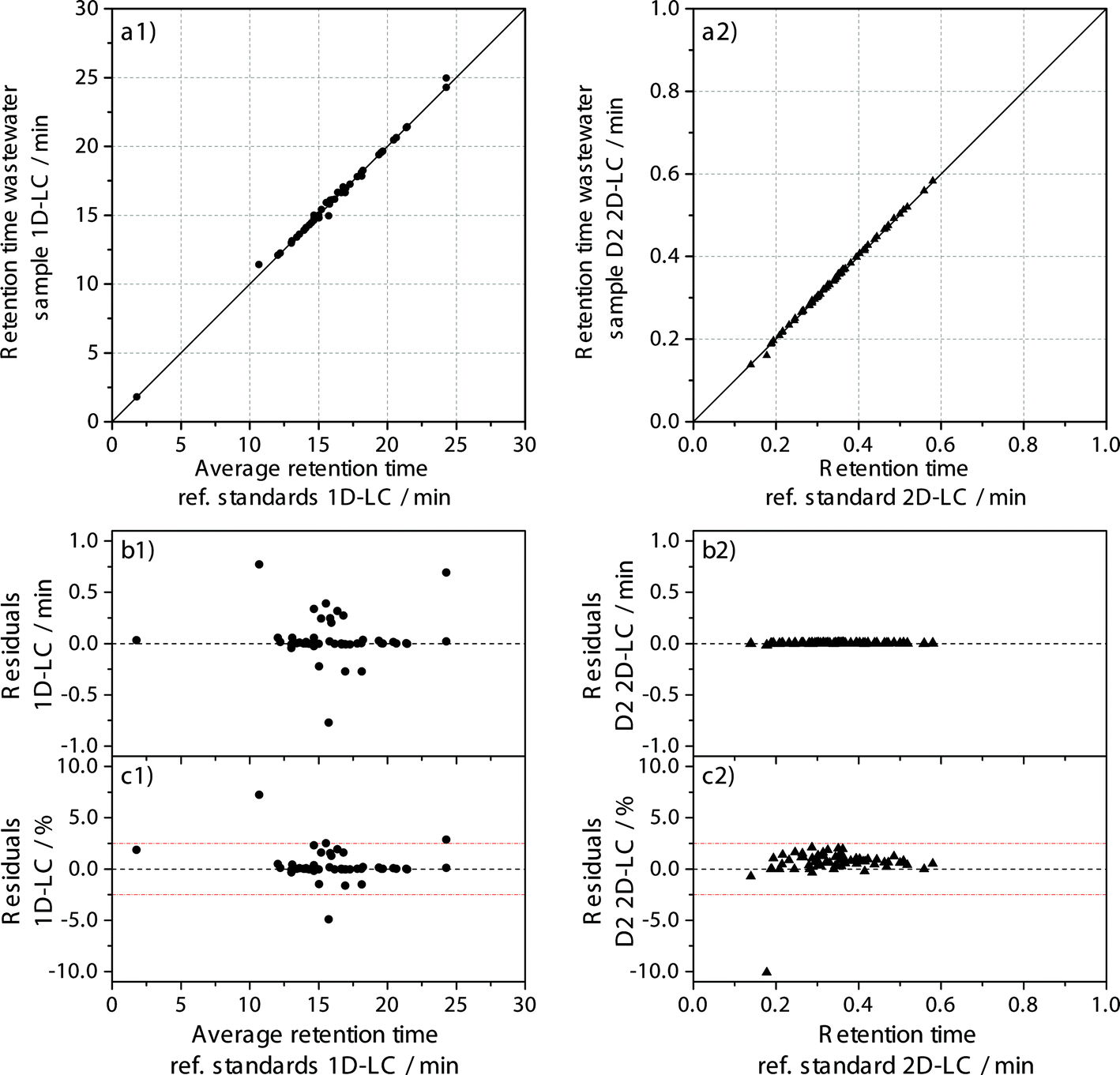 | ||
| Fig. 4 Comparison of retention times measured for the reference standard and the wastewater sample for 1D-LC (a1) and 2D-LC (a2) including the absolute (b1 and b2) and relative (c1 and c2) residuals on the basis of mass accuracy of ±5 ppm and retention time deviation of ±2.5% (left: 1D-HPLC-MS (48 components), right: 2D-nLC × μLC-MS (65 components)). The residuals are differences between measured retention time in wastewater sample and reference standard mix. | ||
For the 1D-LC approach, 44 of 48 possible targets fulfil the additional retention time criterion of ±2.5% with a highest absolute deviation of 0.77 min and the coefficient of determination is R2 = 0.9988. For the 2D-LC approach, 64 of 65 possible targets fulfil the same criterion with a maximum absolute deviation of 0.02 min on the second dimension column and an R2 of 0.9997. Although the R2-values show a very high correlation for both approaches, they contain no information about the scattering. Therefore, the residuals are also included in Fig. 4. Data evaluation shows that the absolute deviation for the 2D-LC approach is much lower, by a factor of 100. However, as is shown by the relative deviation depicted in Fig. 4c, the majority of targets has a deviation smaller than ±0.5% for the 1D-LC approach, while most compounds are spread from 0% to 2.0% for the 2D-LC approach. Although the relative deviation is higher for the 2D-LC approach, it has to be considered that the available solvent gradient window on the D2 column is only 30 seconds long. In contrast, the gradient window for the 1D-LC approach extends to 1440 seconds (24 minutes).
The results clearly point out that the retention time criterion should also be considered for a suspected target screening. Higher deviations in the retention time for the same m/z value could be a hint for false positive hits.
At this point we would also like to discuss the approach of retention time predictions on the basis of octanol–water partition coefficients (logP). This strategy has been proposed for non-target analysis in order to confirm a sum formula on the basis of the accurate mass and a defined mass accuracy. It can be seen from the plot in Fig. S-1,† that there is no correlation when the retention factor is plotted against the logP and thus, the proposed criterion is neither applicable for a suspected-target nor non-target screening approach.
3.6. Implication of peak width and cycle time on MS/MS spectra
A further exclusion of false positive hits could be achieved by MS/MS spectra of the detected targets. This information can be obtained by data dependent MS/MS experiments.The identification of compounds in the wastewater sample on the basis of the three selected criteria (mass accuracy of 5 ppm, retention time deviation lower than 2.5% and confirmation by MS/MS spectra) shows that only 16 targets will be identified using the 1D-LC approach and 31 targets will be identified using the 2D-LC approach. The higher reliability by applying additional criteria for analyte identification always results in a lower number of identified peaks.
It can be deduced from Fig. 5 that the total number of identified peaks is always higher for the 2D-LC approach when the wastewater sample is analysed. Furthermore, it can be observed that the number of excluded targets in Fig. 5 increased by a factor of 2.7 for the 1D-LC approach and a factor of 2.1 for the 2D-LC approach when the additional MS/MS criterion was applied. However, this does not allow the conclusion that the excluded targets are false positive hits. It rather points to a limitation in the mass spectrometer and the chosen mass spectrometric parameters, especially the number of MS/MS experiments which can be made within one cycle. On closer inspection of the data it can be seen that not every precursor ion of a detected target on the basis of the ±5 ppm criterion and 2.5% retention time deviation was selected for a further MS/MS experiment. The reason for the exclusion is the intensity of these target ions, which was lower than the most abundant ions resulting from co-eluting matrix compounds.
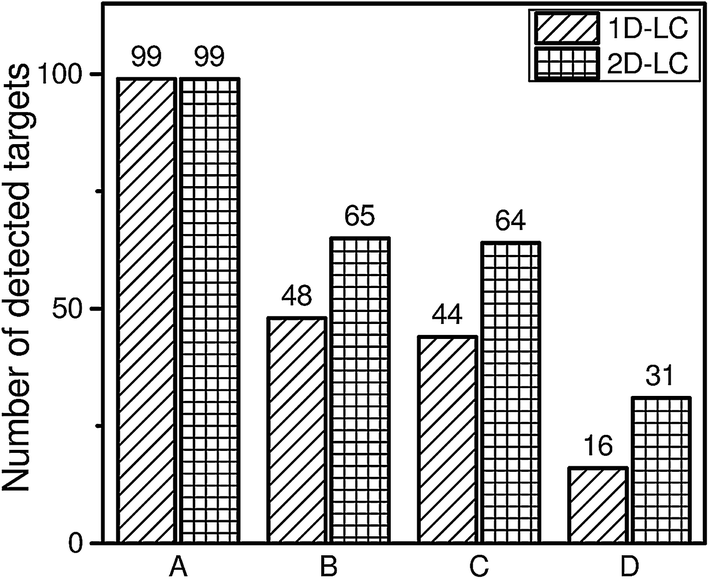 | ||
| Fig. 5 Overview of the identified analytes by 1D-HPLC-MS and 2D-nLC × μLC-MS. Detailed list of detected targets is given in Table S-4.† (A) Detected targets in ref. standard by <5 ppm; (B) detected targets in wastewater sample by <5 ppm; (C) detected targets in wastewater sample by <5 ppm and retention time <2.5%; (D) detected targets in wastewater sample by <5 ppm, retention time <2.5% and MS/MS hit. | ||
In this context it is important to discuss the limitations of hyphenating a chromatographic separation with hybrid high resolution mass spectrometry. For quantitative analysis, usually 15 to 20 data points are required. For qualitative analysis, a lower number of data points over a chromatographic peak is also acceptable. To generate a sufficient number of data points, the cycle time needs to be very short and depends on the m/z range and the number of MS/MS experiments. One MS/MS experiment contains the selection of a specific precursor ion and its subsequent fragmentation. For example, 4 MS/MS experiments are equal to the fragmentation of the 4 most intensive ions that have been detected over a defined m/z range. A larger m/z detection range has only a negligible influence on the cycle time, whereas a higher number of MS/MS experiments with constant dwell time significantly increases the cycle time and reduces the number of data points. Furthermore, the decrease of the dwell time will usually decrease the signal-to-noise ratio, which is not recommended. The 1D-LC approach provides only a short period for the MS/MS experiments during the retention time window of a chromatographic peak. Because of the modulation in 2D-LC, the analyte signal will be cut into several fractions, which increases the possibility for an MS/MS experiment that contain the precursor ion of the target compound.
Generally, the number of MS/MS experiments should be increased with an increasing number of co-eluting signals in order to prevent the loss of MS/MS information. However, this strongly depends on the peak widths which are obtained in 1D-LC and 2D-LC. Table 1 summarizes the calculation of the number of data points over a chromatographic peak in dependence on the peak width and the cycle time.
| Peak width / s | Cycle time / ms | Data points | ||||
|---|---|---|---|---|---|---|
| 1D-LC | 2D-LC | 1D-LC | 2D-LC | 1D-LC | 2D-LCa | |
| a Number of calculated data points for only one fraction. | ||||||
| Scan + 2 MS/MS experiments | 10 | 1 | 500 | 110 | 20 | 9 |
| Scan + 4 MS/MS experiments | 10 | 1 | 700 | 150 | 14 | ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) |
| Scan + 8 MS/MS experiments | 10 | 1 | 1110 | 710 | ![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) |
1 |
| Scan + 12 MS/MS experiments | 10 | 1 | 1500 | 1110 | 7 | 1 |
For the 1D-LC approach, the peak width (at 5% height) was about 10 seconds. Although it would have been possible to reduce the peak width to a few seconds with the available instrumentation by increasing the steepness of the solvent gradient, a smaller peak width inevitably reduces the time for MS/MS experiments. Furthermore, the number of co-eluting peaks would also be higher because of a smaller elution period. This also means that for a suspected target screening, a highly efficient chromatographic separation will lead to a reduced time for obtaining additional MS/MS information that is useful for analyte identification. This limitation is frequently overlooked in the respective literature dealing with ultra-high performance separations for suspected target screening. According to Table 1 the chosen number of MS/MS experiments for both approaches (8 for 1D-LC and 4 for 2D-LC) is an acceptable compromise between the number of data points over a chromatographic peak and the extracted MS/MS information.
Although the number of possible MS/MS experiments per data point for 1D-LC is higher, the total number of MS/MS spectra is not necessarily lower for the 2D-LC approach, which is illustrated on the basis of Fig. S-2.† For the 1D-LC signal 10 data points could be observed, each containing 8 MS/MS spectra. In total it was therefore possible to generate 80 MS/MS spectra over the 1D-LC signal. For the 2D-LC signal in the second fraction, 6 data points could be observed, each containing 4 MS/MS spectra. In total it was possible to acquire 24 MS/MS spectra over the peak in a single fraction. However, the total number of MS/MS spectra consists of the sum of all spectra obtained in each fraction of the 2D-LC signal. This means that the total number of MS/MS spectra for 3 fractions is 72 and thus nearly equal to the 1D-LC approach.
On the basis of the chosen number of MS/MS experiments a ranking list for all target analytes fulfilling the 5 ppm and the 2.5% retention time criteria was created (see Table S-5†). As can be seen, the priority for triggering an MS/MS experiment depends on the absolute intensity of the precursor ion. A closer look at the results of Table S-5† shows that only 23 of the possible 44 targeted precursor ions of the 1D-LC approach had a sufficient intensity for triggering an MS/MS experiment. An increase of the number of possible MS/MS experiments would not necessarily result in a significant increase of MS/MS information of the selected target compounds. In this case, a doubling of the MS/MS experiments to 16 would result in only one additional MS/MS-spectrum of targeted compounds. For the 2D-LC approach 38 of the possible 64 target compounds had a sufficient intensity for an MS/MS experiment. Further increasing the number of MS/MS experiments to eight in the 2D-LC approach would reduce the number of data points across a chromatographic peak to one and is therefore not acceptable. However, not all MS/MS spectra of targeted compounds could confirm the analyte because of different product ion spectra in comparison to the reference standard. Therefore, the total number of identified targets shown in Fig. 5 is smaller for both approaches in comparison to the number of available MS/MS spectra of the targeted compounds.
The main reason for the very small number of additional MS/MS-spectra of targeted compounds by doubling of the possible MS/MS experiments can be explained on the basis of the 1D-LC and 2D-LC TIC chromatogram of the wastewater sample presented in Fig. 1.
In Fig. 1c the retention time of targets selected for MS/MS experiments is highlighted by green dashed lines for 1D-LC (panel b) and black dots for 2D-LC (panel d). Targets excluded from MS/MS experiments are highlighted by red solid lines for 1D-LC and red stars for 2D-LC. For the 1D-LC chromatogram it can be clearly noticed that at the retention time window of 14 to 17 min and at the maximum of the TIC intensity, a large number of precursor ions of targeted compounds were excluded from MS/MS experiments. At the retention time window of 12 to 14 min and 17 to 22 min where the TIC intensity is rising or falling, many of the precursor ions of targeted analytes were selected for MS/MS experiments. The same observation could be made for the 2D-LC plot. Most selected precursor ions from targets for MS/MS experiments are located around the maximum of the TIC cone, located between the 15th and 40th fraction in D1 and between 0.25 and 0.3 min in D2. Most of the excluded targets from MS/MS experiments could be found at the cone maximum.
The main reason for such an effect is the interfering matrix of the wastewater sample. Such a complex matrix can contain thousands of compounds. Their amount and concentration and/or intensity is often much higher in comparison to the targets of interest, especially at the TIC maxima. Therefore, the precursor ions of targeted analytes in this range often were not selected for an MS/MS experiment and thus excluded. Examples are given in Fig. S-3 and S-4 of the ESI,† where the 1D TIC chromatogram, the MS spectrum at the retention time of the targeted compounds (bisoprolol and iopromide) and their MS/MS spectra for 1D-LC and 2D-LC approaches are shown. The MS spectra contain the selected precursor ions (8 for 1D-LC and 4 for 2D-LC) that were selected for MS/MS experiments on the basis of the absolute intensity. In general, it can be noticed that the MS spectra in both approaches contain a very high number of possible precursors which could be selected for MS/MS experiments.
Bisoprolol elutes at the local maximum inside the TIC chromatogram of both approaches (see Fig. S-3†). As discussed before, the matrix impact in this region is very high and the precursor ion for bisoprolol could not be selected for an MS/MS experiment due to the relatively low signal intensity for the 1D-LC approach. With the 2D-LC approach bisoprolol could be separated chromatographically from the interfering matrix. Therefore, bisoprolol could be selected as a precursor with the highest intensity for an MS/MS experiment.
At this point the 2D-LC system has an advantage compared to the 1D-LC system. In consequence of the much higher chromatographic resolution across the two dimensions, the co-eluting matrix compounds could be much better separated, which can increase the probability of selecting precursor ions from targeted analytes for an MS/MS experiment. Furthermore, by sampling the compounds in more than one fraction, the modulation provides a higher possibility that a relevant precursor ion will be selected for MS/MS experiments during several successive D2 chromatograms. This leads to a higher number of targets which can be additionally identified by their MS/MS spectra for the 2D-LC approach as summerized in Fig. 5.
3.7. Comparison of detected targets for 1D-LC and 2D-LC approaches
To complete the comparison it is important to know how many of the detected targets could be identified with both approaches, only with 1D-LC and only with 2D-LC. The results are shown in Fig. 6.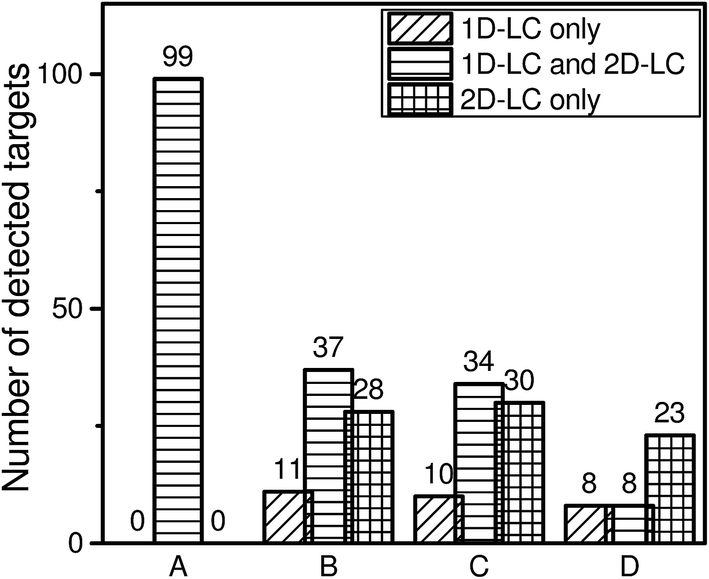 | ||
| Fig. 6 Overview of detected targets with both approaches, only with 1D-LC and only with 2D-LC. (A) Detected targets in ref. standard by <5 ppm; (B) detected targets in wastewater sample by <5 ppm; (C) detected targets in wastewater sample by <5 ppm and retention time <2.5%; (D) detected targets in wastewater sample by <5 ppm, retention time <2.5% and MS/MS hit. | ||
Data evaluation shows that all 99 targets of the reference standard could be identified with both approaches. In the case of the wastewater sample, there are some compounds which will be either detected by the 1D-LC or by the 2D-LC approach. This is especially apparent when all three criteria which have been defined for analyte identification are applied. Here, only 8 targets could be identified with both approaches and the remaining 8 and 23 targets were only identified by either 1D-LC or 2D-LC, respectively. The comparison in Fig. 5 and 6 clearly reveals that the number of compounds which can be detected only with the 1D-LC approach is rather low.
If a too small mass accuracy value such as ±1 ppm is used, the number of detected targets that fulfill all three criteria (mass accuracy of ±1 ppm, retention time deviation of ±2.5% and positive MS/MS spectra) is reduced to 4 for 1D-LC and 21 for 2D-LC. This means that for the selected conditions the higher mass accuracy has a significant negative influence on the absolute number of identified targets for both approaches. This finding underlines that for reliable analyte identification a higher mass accuracy value like ±5 ppm is more suitable in combination with additional criteria such as retention time and MS/MS information.
4. Conclusion and outlook
In this work a comparison of 1D-LC-MS and 2D-LC-MS approaches was performed on the basis of a multi component reference standard and a complex wastewater sample. For the comparison three different criteria for compound identification on the basis of mass accuracy of ±5 ppm, retention time deviation of ±2.5% and MS/MS information were chosen. At this point we would like to point out that the comparison was done under a “worst-case-scenario” for the 2D-LC set-up. The reason is that the inner diameter of the second dimension column is much smaller than that of the 1D-LC set-up. Therefore, only a much smaller sample volume could be injected onto the first dimension column. Consequently, peak concentrations are not directly comparable. However, the fact that the number of identified target peaks is always higher for the microscale LC × LC approach underlines the superior performance of the miniaturized comprehensive 2D-LC separation over a classical one-dimensional separation.Furthermore, it could be shown that the accurate mass criterion of ±5 ppm in combination with further criteria as, e.g., retention time deviation and MS/MS information leads to a higher reliability of identified compounds for both approaches. This does not imply that an identification of a compound should be always based on all selected criteria. Since there is currently no regulative guideline for environmental screening analyses, a point-by-point decision about the appropriate strategy22 is placed in the hands of the operator.
The comparison also reveals the limitations of modern mass spectrometers in terms of compound identification. A reasonable way to increase the number of MS/MS experiments and therefore the possibility to obtain MS/MS information of all detected targets is the continuous increase in data acquisition rate of the mass spectrometer.
Finally, the results of the comparison could provide valuable information for further technical developments as well as data processing strategies.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.References
- M. Krauss, H. Singer and J. Hollender, Anal. Bioanal. Chem., 2010, 397(3), 943–951 CrossRef CAS PubMed.
- M. Zedda and C. Zwiener, Anal. Bioanal. Chem., 2012, 403(9), 2493–2502 CrossRef CAS PubMed.
- U. D. Neue, J. Chromatogr. A, 2008, 1184(1–2), 107–130 CrossRef CAS PubMed.
- S. Di Palma, M. L. Hennrich, A. J. Heck and S. J. Mohammed, Proteomics, 2012, 75(13), 3791–3813 CrossRef CAS PubMed.
- L. Mondello, C. A. Lewis and D. K. Bartle, Multidimensional Chromatography, John Wiley & Sons, Ltd, Chichester, UK, 2001 Search PubMed.
- S. R. Groskreutz, M. M. Swenson, L. B. Secor and D. R. Stoll, J. Chromatogr. A, 2012, 1228, 31–40 CrossRef CAS PubMed.
- S. Wang, L. Qiao, X. Shi, C. Hu, H. Kong and G. Xu, Anal. Bioanal. Chem., 2015, 407(1), 331–341 CrossRef CAS PubMed.
- D. Li, C. Jakob and O. Schmitz, Anal. Bioanal. Chem., 2015, 407(1), 153–167 CrossRef CAS PubMed.
- D. Li and O. J. Schmitz, Anal. Bioanal. Chem., 2015, 407(1), 231–240 CrossRef CAS PubMed.
- S. Schiesel, M. Lämmerhofer and W. Lindner, J. Chromatogr. A, 2012, 1259, 100–110 CrossRef CAS PubMed.
- P. Dugo, F. Cacciola, P. Donato and L. Mondello, Comprehensive chromatography in combination with mass spectrometry, Wiley, Hoboken, NJ, 2011, pp. 331–390 Search PubMed.
- P. Dugo, F. Cacciola, P. Donato and L. Mondello, Comprehensive chromatography in combination with mass spectrometry, Wiley, Hoboken, NJ, 2011, pp. 391–427 Search PubMed.
- D. R. Stoll, X. Wang and P. W. Carr, Anal. Chem., 2007, 80(1), 268–278 CrossRef PubMed.
- I. Francois, K. Sandra and P. Sandra, Comprehensive chromatography in combination with mass spectrometry, Wiley, Hoboken, NJ, 2011, pp. 281–330 Search PubMed.
- J. Haun, J. Leonhardt, C. Portner, T. Hetzel, J. Tuerk, T. Teutenberg and T. C. Schmidt, Anal. Chem., 2013, 85(21), 10083–10090 CrossRef CAS PubMed.
- J. Leonhardt, T. Hetzel, T. Teutenberg and T. C. Schmidt, Chromatographia, 2015, 78(1–2), 31–38 CAS.
- T. N. Decaestecker, K. M. Clauwaert, J. F. van Bocxlaer, W. E. Lambert, E. G. van den Eeckhout, C. H. van Peteghem and A. P. de Leenheer, Rapid Commun. Mass Spectrom., 2000, 14(19), 1787–1792 CrossRef CAS.
- T. N. Decaestecker, S. R. Vande Casteele, P. E. Wallemacq, C. H. van Peteghem, D. L. Defore and J. F. van Bocxlaer, Anal. Chem., 2004, 76(21), 6365–6373 CrossRef CAS PubMed.
- B. O. Keller, J. Sui, A. B. Young and R. M. Whittal, Anal. Chim. Acta, 2008, 627(1), 71–81 CrossRef CAS PubMed.
- C. Moschet, A. Piazzoli, H. Singer and J. Hollender, Anal. Chem., 2013, 85(21), 10312–10320 CrossRef CAS PubMed.
- T. Kind and O. Fiehn, BMC Bioinf., 2006, 7, 234 CrossRef PubMed.
- E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer and J. Hollender, Environ. Sci. Technol., 2014, 48(4), 2097–2098 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay01143d |
| This journal is © The Royal Society of Chemistry 2015 |