
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-elemental analysis of sulfuric acid by oaTOF-ICP-MS after matrix modification with barium bromide†
Lenka
Husáková
*,
Iva
Urbanová
,
Tereza
Šídová
and
Tomáš
Mikysek
Department of Analytical Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentska 573 HB/D, Pardubice, CZ-532 10, Czech Republic. E-mail: Lenka.Husakova@upce.cz; Fax: +420 466 037 068; Tel: +420 466 037 029
First published on 8th May 2015
Abstract
In this work a novel method for the simultaneous multi-elemental analysis of sulfuric acid by inductively coupled plasma orthogonal acceleration time-of-flight mass spectrometry (oaTOF-ICP-MS) after matrix modification with barium bromide was introduced. For this purpose sample aliquots consisting of 10% (w/w) sulfuric acid were mixed with barium bromide and after the precipitation of sulfates, the resulting liquid phase was aspirated with a concentric glass nebulizer for TOF-ICP-MS determination. The separation of sulfur as a precipitated non-soluble barium sulfate enables the elimination of spectral interferences originating from different sulfur containing species like SN+, SNH+, SO+, SOH+, SOH2+, SCO+, SNOH+, SO2+, S2+, SO2H+, or S2H+ on the determination of e.g. Ti, Zn, Ge, and Cu. After that, direct, reliable, and simultaneous determination of 15 elements (Be, Ti, V, Cr, Mn, Ni, Co, Cu, Zn, Ga, Ge, As, Sn, Sb, and Te) at trace and ultra-trace levels in sulfuric acid were performed under optimum instrumental conditions and by using Rh as an internal standard. Accuracy and precision were assessed by analysing sulfuric acid and by comparison of the results of the proposed method with those obtained by the high resolution ICP-MS method. The results of both methods were in good agreement for Ti, V, Cr, Mn, Ni, Co, Cu, Zn, Ga, Ge, and As. The accuracy for all of the 15 elements was also checked by an analytical recovery study. The reported method has a precision greater than 12.5%.
1. Introduction
Large quantities of sulfuric acid are used in many applications in a wide range of industries including the manufacture of fertilizers, lubricants, drugs-pharmaceuticals, rayon, metal processing, batteries, food, dyes and pigments, or chemical manufacturing.1 Due to its aggressive dissolution properties sulfuric acid is prone to acquire metals from other sources and may represent a significant source of contamination in the above applications.2 As the quality and purity of sulfuric acid determines its use for different industrial processes, the targeted trace metal content must be frequently monitored.2–6In order to determine trace element purity levels in sulfuric acid, inductively coupled plasma mass spectrometry is an indispensable analytical tool. However, the direct analysis of even diluted sulfuric acid is not easy as it can produce rapid corrosion of nickel cones which are commonly used in ICP-MS.6 In addition matrix-based interferences make the determination difficult.3,4 Some non-spectroscopic effects can be compensated by dilution of the sample 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (w/w) in high purity water,3–6 evaporation of the sample,3 use of hot-plasma conditions,2 analysis by standard additions or by the use of an internal standard for elements.4 However, spectral interferences7,8 originating from the S-based species produced from matrix components in the plasma can be difficult to overcome,2–6,9 especially with conventional low-resolution quadrupole mass spectrometers.2,3,5 While cool-plasma ICP-MS can be used successfully to remove Ar+, ArH+, ArO+ interfering species on Ca, K, and Fe respectively,3,4 S-based interferences are still important with the elements such as Ti, Cr, Co, Ni, Cu, Zn, As, when direct analysis is performed.2–5 For conventional low-resolution quadrupole ICP-MS the collision/reaction cell ICP-MS is a powerful correction technique to minimize interferences on analytes of interest.2,3 While for older Q-ICP-MS systems the reduction of the multi-element capabilities can be observed, for modern quadrupole ICP-MS systems this problem has been overcome. Sector-field ICP-MS with its high mass resolution can deal with polyatomic interferences originating from major elements even for samples with a complex matrix.4,6 While losing sensitivity in the high resolution mode, the multi-element capability is maintained. These methods may be, however, less accessible to routine analytical laboratories owing to the high cost of instrumentation.
:10 (w/w) in high purity water,3–6 evaporation of the sample,3 use of hot-plasma conditions,2 analysis by standard additions or by the use of an internal standard for elements.4 However, spectral interferences7,8 originating from the S-based species produced from matrix components in the plasma can be difficult to overcome,2–6,9 especially with conventional low-resolution quadrupole mass spectrometers.2,3,5 While cool-plasma ICP-MS can be used successfully to remove Ar+, ArH+, ArO+ interfering species on Ca, K, and Fe respectively,3,4 S-based interferences are still important with the elements such as Ti, Cr, Co, Ni, Cu, Zn, As, when direct analysis is performed.2–5 For conventional low-resolution quadrupole ICP-MS the collision/reaction cell ICP-MS is a powerful correction technique to minimize interferences on analytes of interest.2,3 While for older Q-ICP-MS systems the reduction of the multi-element capabilities can be observed, for modern quadrupole ICP-MS systems this problem has been overcome. Sector-field ICP-MS with its high mass resolution can deal with polyatomic interferences originating from major elements even for samples with a complex matrix.4,6 While losing sensitivity in the high resolution mode, the multi-element capability is maintained. These methods may be, however, less accessible to routine analytical laboratories owing to the high cost of instrumentation.
ICP time-of-flight mass spectrometers are more accessible to routine analytical laboratories, and can overcome the major limitations associated with the scanning nature of other ICP-MS instrumentation with the advantages of high speed and simultaneous mass determination without any compromise on the mass range, sensitivity or precision.10 Enhanced resolution for the oaTOF mass analyzer is another advantage; however, this is not sufficient to completely resolve interference signals from different polyatomic species during the analysis of complex samples. In order to control the spectral effects during the TOF-ICP-MS analysis the mathematical correction equations11,12 without11 or with the implementation of microwave digestion12 or some other approaches employing matrix modification13 were used for the analysis of urine11 and food samples previously.12,13
Despite the development of methods for the analysis of sulfuric acid by quadrupole ICP-MS which employ collision/reaction cells for interference removal2,3,5 and high resolution ICP-MS instruments,4,6 currently no method is available for this purpose when using TOF-ICP-MS. In this work the modification of the sample preparation procedure by the addition of barium bromide to sulfuric acid samples in order to precipitate and separate sulfates prior to TOF-ICP-MS analysis is introduced. The main advantage of this approach is that it serves as a cheap, rapid and simple solution for overcoming the spectral effects from different S-based polyatomic species on the determination of some elements (e.g. on Ti, Zn, and Cu) whose determination is difficult when using instruments with a low resolving power. Finally, the developed method was found to be accurate and precise for a multi-element determination of 15 trace elements in sulfuric acid.
2. Material and methods
2.1. Instrumentation
The Optimass8000 oaTOF-ICP-MS instrument (GBC Scientific Equipment Pty. Ltd., Australia) of which a detailed description is given in ref.10 was used throughout this study. The details of the instrument and the operating conditions are summarized in Table 1. Using typical operating conditions, a sensitivity of 15000 counts per s per μg per L (mass integrated peak) and resolution (full width at half maximum intensity) of 1800 was achieved for 139La+. For 238U+, the same parameters were 17000 counts per s per μg per L and 2100, respectively.
| Instrument | GBC Optimass8000 |
|---|---|
| Sample uptake rate | 1.0 mL min−1 |
| Sample introduction | Concentric nebulizer with cyclonic spray chamber (70 mL, 10 °C) |
| ICP source | |
| Rf power (27.12 MHz) | 1250 W |
| Plasma gas flow rate | 11 L min−1 |
| Auxiliary gas flow rate | 0.600 L min−1 |
| Nebuliser gas flow rate | 0.980 L min−1 |
| Mass spectrometer | |
| Ion optics | |
| Sampler cone | Nickel |
| Skimmer cone | Nickel-plated copper |
| Extraction | −1300 V |
| Z1 | −190 V |
| Y mean | −495 V |
| Y deflection | −1 V |
| Z lens mean | −1050 V |
| Z lens deflection | −35 V |
| Lens body | −140 V |
| Pulse shaping | |
| Fill | −39 V |
| Fill bias | −0.1 V |
| Fill grid | −2.0 V |
| Pushout plate | 577 V |
| Pushout grid | −473 V |
| Blanker | 200 V |
| Spectral frequency | 33 kHz |
| Reflectron | 600 V |
| Detection | |
| Multiplier gain | 2630 V |
| Ion threshold | 8.4 mV |
| Integration window | Auto |
| Measurement mode | Pulse counting/analog |
| SMARTGATE ion blanking system | |
| Range m/z | Interfering ions |
| 11.5–44.5 | 12C+, 14N+, 16O+, 17OH+, 18H2O+, 28N2+, 30NO+, 32O2+, 32S+, 40Ar+, 41ArH+, 80Ar2+ |
| 55.5–57.0 | 56ArO+, 56Fe+ |
| 66.5–70.0 | 132−138Ba++ |
| 77.5–82.5 | 40Ar2+, 79Br+, 81Br+, 1H81Br+ |
| 94.7–97.5 | 79,81Br16O+ |
| 129.0–140 | 130–138Ba+, 139BaH+ |
| 149.8–155.3 | 154BaO+, 155BaOH+, 150BaO2+, 152BaN+ |
Comparative measurements were performed using a high performance, double focusing magnetic sector field Thermo Scientific™ ELEMENT 2™ ICP-MS (Thermo Scientific, Bremen) attached to a CETAC Aridus II™ Desolvating Nebulizer system (CETAC Technologies Inc., USA). The Aridus II™ couples an Aspire™ low-flow PFA nebulizer and a heated PFA spray chamber with a PTFE membrane desolvator unit. Instrument settings and measurement parameters for the ELEMENT2 are shown in Table 2.
| Instrument | Element 2 attached to the CETAC Aridus II™ desolvating nebulizer system |
|---|---|
| Rf power (27.12 MHz) | 1200 W |
| Plasma gas flow rate | 16 L min−1 |
| Auxiliary gas flow rate | 0.9 L min−1 |
| Nebulizer gas flow rate | 0.9 L min−1 |
| Sweep gas flow rate | 3.6 L min−1 |
| Sample uptake rate | 100 μL min−1 |
| Spray chamber temperature | 110 °C |
| Membrane temperature | 160 °C |
| Sampler cone | Ni 1.1 mm |
| Skimmer cone | Ni 0.8 mm |
| Lens settings | Optimized for 238U |
| Resolution mode | Medium resolution, R = 4000 (Ti, V, Cr, Mn, Co, Ni, Cu, Zn, Ga) |
| High resolution, R = 10000 (Ge, As) |
|
| Total acquisition time per replicate | 0.6 min−1 |
| Number of replicates | 10 |
| Number of measurements per sample | 5 |
The determination of Ba was carried out with an Avanta P double beam atomic absorption spectrometer (GBC Scientific Equipment Pty. Ltd., Australia). A hollow cathode lamp (Photron Pty. Ltd., Australia) was used as the line source (lamp current 15 mA, wavelength 553.6 nm, and spectral bandpass 0.5 nm). The sample solutions were introduced to the spectrometer through a standard GBC pneumatic nebulizer. The gas flame mixture was formed with 10.0 L min−1 nitrous oxide (99.5%, from Linde Gas, Inc., Czech Republic) and 4.5 L min−1 acetylene (99.5%, from Linde Gas, Inc.).
The determination of S was done on an inductively coupled plasma optical emission spectrometer (ICP-OES), Integra XL 2 (GBC Scientific Equipment Pty. Ltd., Australia), by an independent laboratory. The parameters of the instrument setup as well as the wavelength used in these measurements are given elsewhere.14
Centrifugation of the samples was performed using a NF 400 bench top centrifuge (NÜVE INC., Turkey).
2.2. Reagents and standards
All solutions were prepared using deionised water purified by means of the UltraClear (SG, Germany) pure water system to 0.05 μS cm−1.Sub-boiled nitric acid was prepared from nitric acid (65%, w/w) of Selectipur quality (Lach-Ner, Neratovice, Czech Republic). Barium nitrate and barium acetate used for interference studies were of TraceSELECT® quality and obtained from Fluka (Fluka, Buchs, Switzerland). Barium bromide 99.999% was purchased from Hi-Chem Ltd. (Czech Republic). Rhodium internal standard solution was prepared from 1 g L−1 Rh solution obtained from SCP Science (Canada). A multi-element stock solution containing 10 mg L−1 of Li, B, Be, Ti, V, Cr, Mn, Ni, Co, Cu, Zn, Ga, Ge, As, Se, Rb, Sr, Zr, Mo, Ru, Pd, Ag, Cd, Sn, Sb, Te, Cs, Hf, Ta, W, Re, Pt, Hg, Tl, Pb, Bi, Th, and U, being routinely used in our laboratory, was prepared from single element standards of 1 g L−1 obtained from Analytika Ltd. (Prague, Czech Republic) or SCP Science. The stock solution of Ba (1 g L−1) was obtained from Analytika Ltd. (Prague, the Czech Republic).
2.3. Samples
The following sulfuric acid samples were used throughout the study: 95–98% (w/w) H2SO4 of TraceSELECT® Ultra grade for ultratrace analysis (Sigma-Aldrich Co. LLC), 96% (w/w) G.R. H2SO4 of pre-analysis grade and 96% (w/w) H2SO4 of pure quality (both from Lach-Ner Ltd., Czech Republic). The accurate sulfuric acid concentrations were determined by titration with sodium hydroxide and with potentiometric indication.15 For H2SO4 of TraceSELECT, pre-analysis grade and of pure quality they were as follows: 96.8 ± 0.1, 96.2 ± 0.4 and 98.6 ± 0.3%. The results are expressed as the mean ± 2SD of two independent determinations.2.4. Sample and calibration standard preparation
The sample preparation for the oaTOF-ICP-MS analysis consisted of the addition of 0.85 mL of 500 g L−1 BaBr2 to 1.25 mL of 10% (w/w) H2SO4. The solution was gently mixed and centrifuged for 5 min at 2500 rpm followed by filtration through a 0.45 μm nylon syringe filter (Whatman Autovial). The sample blanks were prepared by the same method using TraceSELECT® Ultra grade purity level acid.The matrix matched calibration solutions were prepared as follows. To 5 mL of 10% H2SO4 (TraceSELECT® purity level), 3.4 mL of 500 g L−1 BaBr2 were added. The solution was mixed, centrifuged and filtered as described above. The filtrate was divided into seven aliquots. The individual aliquots were spiked with the standard so that they contained 0, 5, 10, 20, 30, 40, and 50 μg L−1 of Li, B, Be, Ti, V, Cr, Mn, Ni, Co, Cu, Ga, Ge, As, Se, Rb, Sr, Zr, Mo, Ru, Pd, Ag, Cd, Sn, Sb, Te, Cs, Hf, Ta, W, Re, Pt, Hg, Tl, Pb, Bi, Th, and U, and 0, 50, 100, 200, 300, 400, and 500 μg L−1 of Zn, and Rh at a final concentration of 1 μg L−1.
The sample preparation for analysis by HR-ICP-MS consisted of dilution of the concentrated acids to obtain 10% solutions of the individual samples. The prepared samples were given to an independent laboratory for reference measurements.
2.5. Sample analysis
2.6. Statistical data treatment
The data were processed using the Statistica 12 computer program (StatSoft, Inc., USA) and QC Expert™ 2.5, TriloByte Statistical Software, (Pardubice, Czech Republic).3. Results and discussion
3.1. Evaluation of spectral interference
In the direct analysis of sulfuric acid, polyatomic species such as SN+, SNH+, SO+, SOH+, SOH2+, SCO+, SNOH+, SO2+, S2+, SO2H+ or S2H+ interfere with important elements such as Ti, Cr, Co, Ni, Cu, Zn or Ga3. Table S1 from the ESI† summarizes potential interferences, which might be encountered during an oaTOF-ICP-MS analysis. For the problematic elements which are listed above, a resolving power higher than 4000 (m/Δm) is needed for successful interference removal as can be seen from the data presented in Table 2 and from the data available in the literature.6,8 Quantitative data of apparent elemental concentration in the presence of sulfuric acid as measured on the Optimass 8000 within the range of 0–500 mg L−1 of S were evaluated. Interferences from different sulfur containing species were observed for 48Ti+, 49Ti+, 64Zn+, 66Zn+, 65Cu+ and 82Se+. Fig. 1 shows the influence of S (sulfuric acid) concentration within the range of 0–500 mg L−1 on the relative concentration signal of 5 μg L−1 of 48Ti+, 49Ti+, and 64Zn+ as the most interfered isotopes from the multi-elemental standard. In contrast to the literature5,6 no significant interference was observed on the determination of V, Ge, As or Ga. However, the highest investigated concentration in the solutions was 500 mg L−1, which corresponds to 0.3% (w/w) solution of H2SO4, while the cited interferences were observed during the analysis of 10% (w/w) solutions. Other interferences could be observed during the analysis of more concentrated solutions; however, these investigations were not performed in order to protect the nickel sampler and skimmer cones.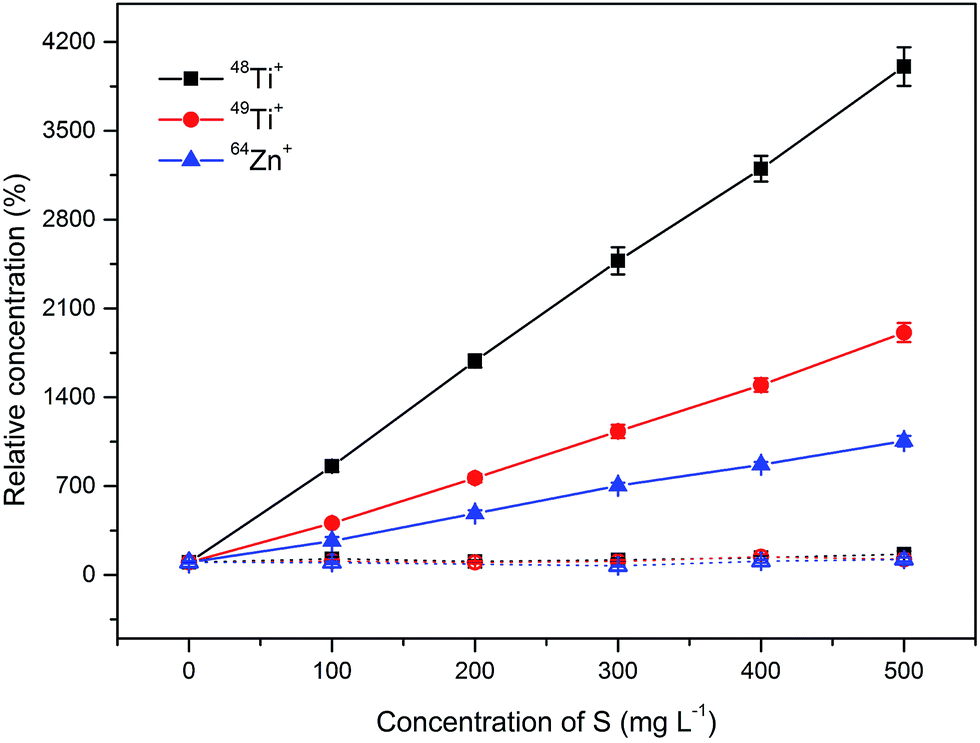 | ||
| Fig. 1 Influence of the amount of S (as H2SO4) on the relative concentrations of 5 μg L−1 of 48Ti+, 49Ti+ and 64Zn+ from the multi-elemental standard in the absence (closed symbols, straight line) and in the presence of barium bromide (open symbols, dotted line). Relative concentration (%) = (concentration evaluated for 5 μg L−1 multi-elemental standard in the presence of barium bromide and sulphur within the investigated range/concentration evaluated for 5 μg L−1 of the standard under conditions without the interfering matrix element) × 100. The bars indicate the range for each determination (n = 10, 2 SD). | ||
3.2. Effect of barium salt on the removal of spectral interference
It has been previously shown that precipitation of the chloride matrix can effectively reduce chloride based interferences16 and the precipitation of Ca2+ as an oxalate may overcome interferences from different calcium containing species on the determination of nickel in serum analysis17 and on the determination of As, Se, Co and Ni in milk13 when using conventional quadrupole17 and TOF-ICP-MS instruments,13 respectively. However, the technique of precipitating interfering species from the matrix is not a commonly used approach in ICP-MS practice for overcoming the spectral effects. There are many sulfates of low solubility,18 such as calcium sulfate, strontium sulfate, or lead(II) sulfate. Barium sulfate is highly insoluble, and is commonly used for the gravimetric analysis of sulfate, and hence was chosen for this study.In this study, barium nitrate was initially employed in order to precipitate and separate sulfate as non-soluble barium sulfate enabling the elimination of the interference. However, the disadvantage of barium nitrate is its lower solubility which limits the concentration of sulfuric acid that can be analyzed. With barium nitrate addition, a sulfuric acid concentration of 2% (w/w) concentration is the maximum that can be used in order to avoid any further dilutions. In addition, significant changes in solubility relating to the temperature are a critical consideration given the cold working environment within the glass spray chamber. Due to this fact Ba(CH3COO)2, and BaBr2 were considered for use. The use of barium acetate led to an excessive input of C into the spectrometer causing interferences at masses 48 and 52 hampering the determination of 48Ti+ and 52Cr+ (data not shown here). Finally, BaBr2 was chosen for the optimization purposes even though interferences from Br+ and BrOH+ were observed at m/z 79, 81, 95–100.
The influence of BaBr2 on the determination of 48Ti+, 49Ti+, 64Zn+ in the presence of 0–500 mg L−1 of S can be seen in Fig. 1, from which it is obvious that the interferences caused by 32S16O+, 33S16O+, 32S2+ and 32S16O2+ were successfully eliminated. Similarly, the suppression of the interference from different S containing species was observed for other interfered isotopes like 82Se+ and 65Cu+ (data not shown here).
3.3. Application to the analysis of sulfuric acid
The concentration of sulfuric acid used for precipitation was optimized in order to analyze solutions with the acid content as high as possible to avoid further dilutions which would negatively impact the detection limits of the method. The optimization was performed for 40, 30, 20, 10, 5, and 2% solutions of sulfuric acid, which were spiked with 0, 10, and 20 μg L−1 of multi-elemental standard described in Section 2.2. Each concentration was prepared in two sets. The first set was spiked with the standards before the precipitation with BaBr2, while the other set was spiked with the standards after the precipitation of BaSO4 and its removal from the solution. This approach helped to both monitor the influence of the sample matrix on the analyte sensitivity and to reflect the influence of co-precipitation and/or inclusion of the analyte in the precipitated matrix. It was found that the determination of elements in sulfuric acid of a concentration higher than 10% led to irreproducible results, which is in contrast to the analysis of lower concentrations of sulfuric acid (10, 5, and 2%). Good agreement between the slopes of the solutions, which were constructed for solutions spiked before or after the precipitation, was obtained for 10, 5, and 2% (w/w) sulfuric acid with differences less than 5% between the slopes and thus 10% solutions were used for the analysis. Fifteen elements (9Be+, 48Ti+, 51V+, 52Cr+, 55Mn+, 58Ni+, 59Co+, 63Cu+, 64Zn+, 71Ga+, 72Ge+, 75As+, 118Sn+, 123Sb+, 128Te+) could be determined using the 10% solution, however, co-precipitation or inclusion of elements such as Cd, Pb, Ag, Li, Rb, Hf, or Hg occurred even for the lowest tested concentration of sulfuric acid. Co-precipitation of elements such as Ag, Pb, Hg, or Tl could be expected as these elements form sparingly soluble bromides. Besides that, for Sr, Pb or Hg, it is also possible that they co-precipitate during the formation of barium sulfate.18 With the excess of BaSO4, isomorphic substitution where the position replacement of one cation by another of a similar size leading to the incorporation of a variety of cations into solid crystals can take place. This would lead to the formation of rhombic crystals similar to those of BaSO4. As for the other elements, for which we did not obtain reproducible results, the whole problem will be much more complex and was not part of this study.The comparison of slopes of aqueous calibration and the standard additions method, which is a robust strategy for compensating for matrix effects is shown in Fig. 2. In addition, the slope values of matrix matched calibration are also included in this figure. As can be seen from Fig. 2, the slope values of matrix matched calibration and standard additions technique are in a good agreement with the biggest apparent difference being only 9.2% for 59Co. The approach using matrix matched calibration for quantification is less time consuming than the standard additions method.
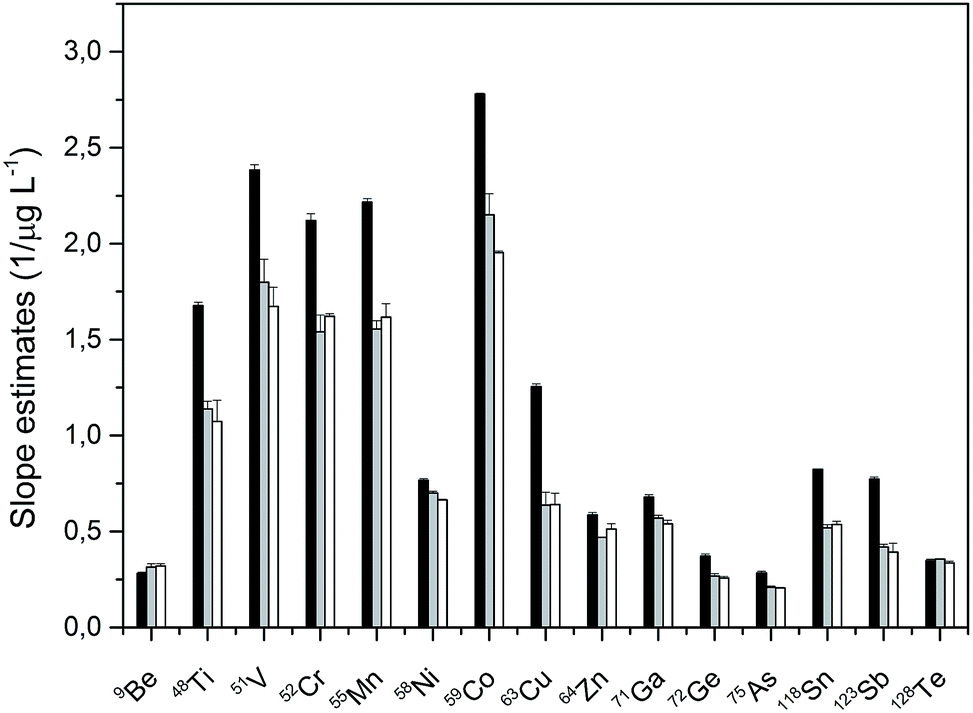 | ||
| Fig. 2 Comparison of the slopes (1/μg L−1) of aqueous calibration (black bar) and that of matrix matched calibration (grey bar) with standard additions method (white bar) evaluated with the use of Rh as the internal standard. The bars indicate the standard deviation error of the estimate of the slope. | ||
3.4. Drawbacks and solutions of the use of barium bromide
There are several drawbacks associated with the employment of barium bromide in ICP-MS analysis to which attention must be paid in order to avoid them. A high concentration of the residual barium salt may both contaminate the spectrometer and be a source (precursor) of other interfering species and/or compounds like Ba++, BaO+, BaH2+, BaOH+, BaO2+, or BaN+, interfering the determination of 66–68Zn+, 133Cs+, 152Sm+, 153Eu+, 155Gd+. However, Zn, Ga, and Ge could be determined using different isotopes, namely 64, 71, and 72, respectively. In this study, the barium salt was calculated to be added in strictly equimolar amounts to that of sulfuric acid. Using these proportions, the majority of barium was precipitated as barium sulfate and separated from the analyzed solution. Solutions of 10% of H2SO4 were precipitated with an increasing amount of BaBr2 starting at 825 μL of 500 g L−1, which was calculated to be equimolar to 1.25 mL of 10% H2SO4, up to an excess of 25 μL. During this experiment, the decrease in the signal at m/z = 48 and increase in the signal at m/z = 217 corresponding to 138Ba79Br+ was monitored. This peak was first observed with an excess of 20 μL compared to the calculated amount while the signal at m/z = 48 remained constant with further addition of BaBr2, which suggests that the interfering sulfur has been removed and the signal corresponds to only 48Ti+. The change in the signal for the monitored masses can be seen in Fig. 3. Mass spectra for the analysis of sulfuric acid without and with the employment of an optimal amount of barium bromide for matrix modification at masses of 48 and 49 for Ti and within the range of 216 and 220 amu reflecting the formation of BaBr+ species are shown in Fig. 4. The concentration of the remaining S determined by ICP-OES in the samples prepared using the optimized amount of BaBr2 was for all samples less than 9.5 mg L−1, which suggests that the removal of the interfering S as BaSO4 was efficient. The remaining concentration of Ba2+ in filtrates determined by the FL-AAS method did not exceed the value of 4 mg L−1. However, this concentration may still influence the analysis. Due to this fact, specific SMARTGATE ion blanking setting (see Table 1) was used in order to avoid these species reaching the detector and thus prolong its life. At this point, any unwanted ions can be rejected from the flight path by deflection plates in the SMARTGATE, while ions of interest are passed on for detection.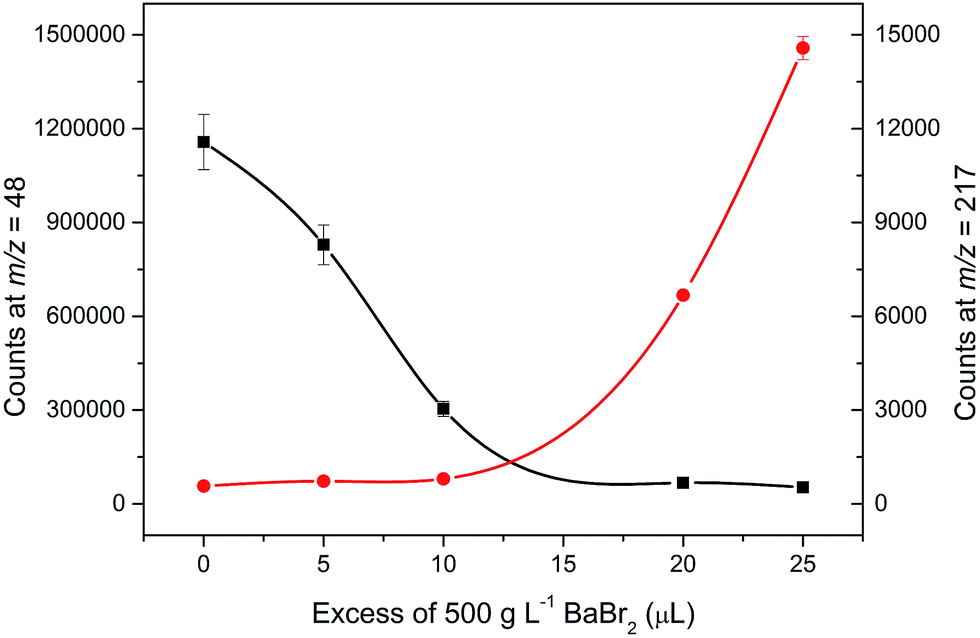 | ||
| Fig. 3 Optimization of barium bromide (500 g L−1) amount, more exactly of the volume being necessary to be added over those to be calculated as equimolar to 10% (w/w) H2SO4via the monitoring of the signal at the mass of 48 and 217, corresponding to the decrease of interference at mass 48 (black line) and excess of barium bromide via the 138Ba79Br+ creation (red line), respectively. The bars indicate the range for each determination (n = 10, 2 SD). | ||
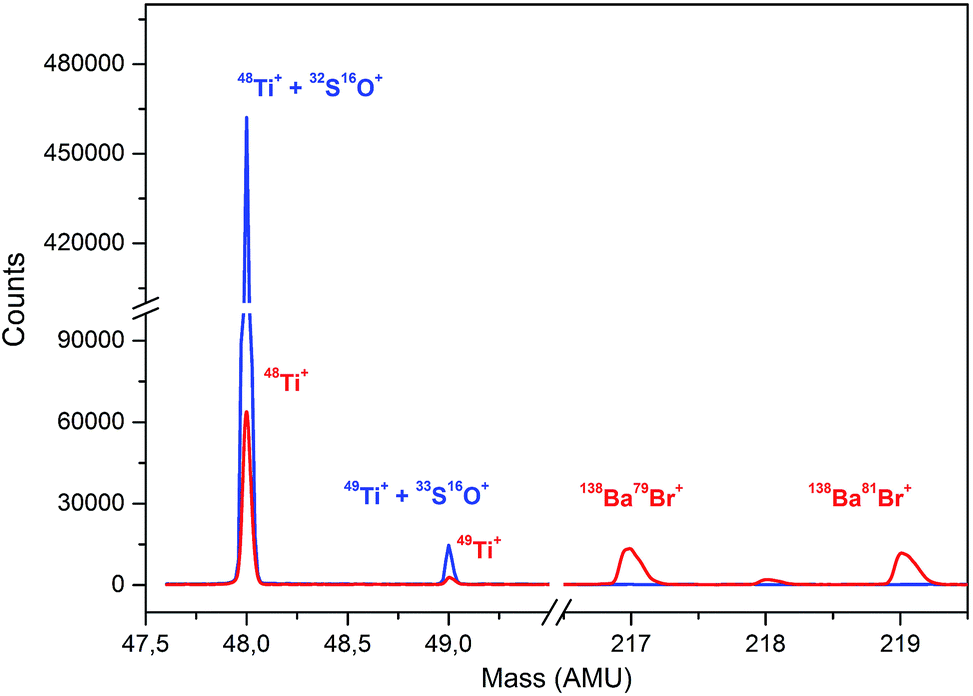 | ||
| Fig. 4 Mass spectra for the analysis of sulfuric acid without (blue line) and with (red line) the employment of the optimal amount of barium bromide for matrix modification at masses of 48 and 49 for Ti and within the range of 216 and 220 amu reflecting the formation of the BaBr+ species. | ||
3.5. Analytical figures of merit
| H2SO4, p.a. | H2SO4, pure | H2SO4, TraceSELECT® LODbc μg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentrationabc μg L−1 | R | RSDc (%) | Concentrationabc μg L−1 | R | RSDc (%) | |||||||
| HR-ICP-MS | TOF-ICP-MS | % | HR-ICP-MS | TOF-ICP-MS | % | HR-ICP-MS | TOF-ICP-MS | |||||
| a Mean ± 2 SD (n = 3). b All values are reported in 10 w/w H2SO4. c Values for HR-ICP-MS and that of TOF-ICP-MS method were evaluated with the use of In and Rh, respectively, as internal standard elements. d Analytical recovery, R (%) evaluated for the TOF-ICP-MS method and the samples spiked with 4 μg L−1 for the listed elements except for Zn, which was spiked with 40 μg L−1. e Relative standard deviation (%) expressing the precision of the method in terms of intra-day precision. For the analytes whose determination was below the LOD the results are shown for the samples spiked similarly as described (seed). f Relative standard deviation (%) expressing the precision of the method in terms of inter-day precision. For the analytes whose determination was below the LOD the results are shown for the samples spiked similarly as described (seed). g Not determined. | ||||||||||||
| 9Be+ | <LOD | 108 | 3.5 | 3.4 | <LOD | 102 | 0.7 | 5.5 | 0.19 | |||
| 48Ti+ | 0.43 ± 0.05 | 0.44 ± 0.05 | 98 | 5.7 | 4.5 | 0.84 ± 0.04 | 0.97 ± 0.15 | 102 | 7.7 | 9.8 | 0.012 | 0.44 |
| 51V+ | 0.72 ± 0.12 | 0.82 ± 0.04 | 98 | 2.5 | 6.2 | 0.18 ± 0.03 | 0.21 ± 0.05 | 91 | 11.2 | 4.7 | 0.009 | 0.09 |
| 52Cr+ | 0.31 ± 0.08 | 0.28 ± 0.02 | 101 | 3.6 | 5.9 | 33.5 ± 1.2 | 28 ± 6 | 91 | 10.7 | 4.4 | 0.009 | 0.21 |
| 55Mn+ | 0.44 ± 0.04 | 0.46 ± 0.05 | 90 | 5.4 | 3.5 | 3.7 ± 0.2 | 3.7 ± 0.2 | 106 | 2.7 | 5.1 | 0.006 | 0.26 |
| 58Ni+ | 0.63 ± 0.13 | <LOD | 94 | 4.5 | 9.8 | 20.6 ± 0.7 | 20.9 ± 1.4 | 90 | 3.3 | 3.2 | 0.012 | 0.56 |
| 59Co+ | <LOD | <LOD | 95 | 1.9 | 7.6 | 0.21 ± 0.02 | 0.23 ± 0.04 | 97 | 8.7 | 5.3 | 0.006 | 0.04 |
| 63Cu+ | 0.1 ± 0.06 | <LOD | 93 | 2.6 | 4.7 | 0.75 ± 0.03 | <LOD | 101 | 3.9 | 4.0 | 0.012 | 0.90 |
| 64Zn+ | 3.4 ± 0.2 | 3.6 ± 0.4 | 98 | 5.6 | 7.7 | 0.15 ± 0.05 | <LOD | 106 | 2.1 | 3.3 | 0.018 | 0.34 |
| 71Ga+ | 0.35 ± 0.06 | 0.38 ± 0.04 | 99 | 5.3 | 12.0 | <LOD | <LOD | 100 | 4.7 | 7.0 | 0.012 | 0.15 |
| 72Ge+ | <LOD | <LOD | 98 | 0.3 | 4.6 | <LOD | <LOD | 101 | 7.6 | 8.7 | 0.012 | 0.71 |
| 75As+ | 271 ± 3 | 270 ± 28 | 102 | 5.2 | 1.2 | 2.7 ± 0.2 | 2.7 ± 0.6 | 93 | 10.3 | 10.3 | 0.027 | 0.54 |
| 118Sn+ | 1.07 ± 0.20 | 97 | 9.3 | 2.2 | 0.82 ± 0.20 | 105 | 12.1 | 3.5 | 0.29 | |||
| 123Sb+ | 0.86 ± 0.04 | 96 | 2.3 | 5.7 | 1.69 ± 0.28 | 91 | 8.3 | 7.8 | 0.56 | |||
| 128Te+ | <LOD | 99 | 3.7 | 9.1 | 0.82 ± 0.12 | 96 | 7.3 | 9.1 | 0.25 | |||
A second approach for the accuracy assessment involved the computation of analytical recoveries determined in samples of p.a. and pure H2SO4 (n = 3) using standard additions at concentrations as indicated in Table 3. The concentration of the standard additions was chosen as a compromise to be at the same concentration level as the analytes and to cover the whole range of the elements present in the analyzed samples. The recoveries obtained were in all cases between 90 and 108% for all elements, indicating that the method is accurate to within 10%.
The precision of the method was evaluated in terms of intra-day and inter-day comparison. Intra-day precision was determined as the relative standard deviation (RSD) of analyses of the sample prepared independently three times during the same day, while inter-day precision (as RSD) was evaluated based on the results of analyses of the samples on three different days over a period of one month. Every solution was analysed in ten replicates yielding a mean value used for the calculations. The RSD values of intra-day and inter-day studies were typically found to be below 12.1% and 10.3%, respectively, which show that the precision of the method was satisfactory (see Table 3).
Also of note is that barium bromide used in this study was not of the highest purity available, which will further significantly impact the LOD values. It is also important to note that direct analysis of 10% (w/w) sulfuric acid is also possible with TOF-ICP-MS ensuring approximately ten times better LOD values for non-interfered elements like Li, Be, Cd, Mo, Sn, Sb, W, or Bi when compared with the values given in Table 3. However we did not use this approach in a higher extent in order to protect the nickel cones as platinum ones were not available. In this case, also determination of elements such as Li, Be, Cd, Mo, Sn, Sb, W, or Bi, whose determination is impacted by the matrix modification method will ensure analytical recoveries within 86–112%. The recoveries were calculated for the samples of 10% H2SO4 spiked with 4 μg L−1 of the analytes.
The detection capability of the proposed method, however, is adequate to determine the impurities of H2SO4 of Grade 1 and 2, as suggested by SEMI. The obtained detection limits are one to two orders of magnitude lower than the maximum permissible content of elements for Grade 2 and Grade 1 purity being in the range of 5 to 300 ppb in concentrated sulfuric acid.
4. Conclusions
The proposed method employing barium bromide for the precipitation of sulfates prior to the analysis of sulfuric acid offers a simple and efficient way for the accurate quantification of 15 elements in this difficult matrix by the TOF-ICP-MS method. One advantage is that using this approach the interference of the matrix is decreased significantly and accurate analysis with the use of the matrix matched calibration method can be performed even for elements like Ti, Cu, or Zn, which are more troublesome if ICP-MS instruments with an insufficient resolving power are used. Another benefit of this procedure is its simplicity, speed and low price of the analysis. Moreover, all the elements are determined simultaneously without the need of specific setting for individual analytes which may be another benefit over the Q-ICP-MS or HR-ICP-MS methods. The proposed method using precipitation with the barium salt is expected to be applied for TOF- as well as for Q-ICP-MS as the purpose of this procedure is to get rid of spectral interferences caused by sulfur-based polyatomic species, which is beneficial for both of these instrumentations. However, the detection limits are limited by the quality of barium bromide and additionally some elements like Cd, Pb, Hg, Sr, Bi, or Ag cannot be determined properly with the proposed methodology due to the problems with co-precipitation and/or inclusion in the precipitated matrix. Despite the above mentioned drawbacks, for TOF-ICP-MS instrumentation this procedure is currently the only solution to attenuate the interferences to a manageable level.Acknowledgements
Financial support from the project no. SGSFChT 2014007 of the University of Pardubice (Czech Republic) is gratefully acknowledged.References
- K. Wetzel, Sulfuric Acid Handling, http://www.ams.usda.gov/AMSv1.0/getfile?dDocName=STELPRDC5101325, cited 10.2.2015.
- K. Kawabata, Y. Kishi and R. Thomas, Spectroscopy, 2003, 18, 16–29 CAS.
- J. Takahashi, K. Youno, Analysis of impurities in semiconductor grade sulfuric acid using the Agilent 7500cs ICP-MS, http://www.chem.agilent.com/Library/applications/5988-9190EN.pdf, cited 2.2.2015.
- J. D. Wills, M. Hamester, T. Lindemann, L. Rottman, Analysis of Sulfuric Acid to Meet SEMI Tier C Requirements Using a Single Set of Operating Conditions with Sector Field ICP-MS, https://static.thermoscientific.com/images/D14427~.pdf, cited 2.2.2015.
- K. Ong, Determination of Impurities in Semiconductor-Grade Sulfuric Acid with the NexION 300S ICP-MS, http://www.perkinelmer.com/cmsresources/images/44-135838app_010282_01nexion300s-impuritiessemiconh2so4.pdf, cited 3.2.2015.
- TJASolutions, Trace Metal Analaysis of Ultrapure Sulphuric Acid Using the VG Axiom High Resolution ICP-MS, https://www.yumpu.com/en/document/view/15353014/trace-metal-analysis-of-ultrapure-sulphuric-acid-using-the-vg-axiom-, cited 2.2.2015.
- E. H. Evans and J. J. Giglio, J. Anal. At. Spectrom., 1993, 8, 1–18 RSC.
- N. M. Reed, R. O. Cairns, R. C. Hutton and Y. Takaku, J. Anal. At. Spectrom., 1994, 9, 881–896 RSC.
- S. H. Nam, H. Chung, J. J. Kim and Y. I. Lee, Bull. Korean Chem. Soc., 2008, 29, 2237–2240 CrossRef CAS.
- R. E. Sturgeon, J. W. H. Lam and A. Saint, J. Anal. At. Spectrom., 2000, 15, 607–616 RSC.
- L. Husakova, I. Urbanova, L. Audrlicka-Vavrusova, J. Sramkova, T. Cernohorsky, M. Bednarikova and L. Pilarova, Microchim. Acta, 2011, 173, 173–181 CrossRef CAS.
- L. Husakova, I. Urbanova, J. Sramkova, T. Cernohorsky, A. Krejcova, M. Bednarikova, E. Frydova, I. Nedelkova and L. Pilarova, Food Chem., 2011, 129, 1287–1296 CrossRef CAS PubMed.
- L. Husakova, I. Urbanova, J. Sramkova, M. Konecna and J. Bohuslavova, Talanta, 2013, 106, 66–72 CrossRef CAS PubMed.
- T. Černohorský, A. Krejčová, M. Pouzar and L. Vavrušová, Food Chem., 2008, 106, 1246–1252 CrossRef PubMed.
- R. de Levie, Aqueous Acid-base Equilibria and Titrations, Oxford University Press, Inc., New York, 1999 Search PubMed.
- T. D. B. Lyon, G. S. Fell, R. C. Hutton and A. N. Eaton, J. Anal. At. Spectrom., 1988, 3, 265–271 RSC.
- S. X. Xu, L. Stuhnesekalec and D. M. Templeton, J. Anal. At. Spectrom., 1993, 8, 445–448 RSC.
- Handbook of Chemistry and Physics, Internet Version 2005, ed. D. R. Lide, CRC Press, Boca Raton, 2005 Search PubMed.
- Teledyne_CETAC_Technologies, Signal Enhancement and Solvent-Based Interference Reduction for Quad-ICP-MS with the CETAC Aridus II™ Desolvating Nebulizer System, http://www.cetac.com/__documents/TN01_AridusII.pdf, cited 17.4.2015.
- H. Wildner, J. Anal. At. Spectrom., 1998, 13, 573–578 RSC.
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53, 1739–1763 CrossRef.
- S. I. Standards, SEMI C44-0708-Specifications and Guidelines for Sulfuric Acid, http://ams.semi.org/ebusiness/standards/SEMIStandardDetail.aspx?ProductID=211%26DownloadID=107, cited 16.2.2015.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay00582e |
| This journal is © The Royal Society of Chemistry 2015 |