
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Speciation and quantification of organotin compounds in sediment and drinking water by isotope dilution liquid chromatography-inductively coupled plasma-mass spectrometry
David P.
Bishop
a,
Dominic J.
Hare
 abc,
Adrian
de Grazia
a,
Fred
Fryer
d and
Philip A.
Doble
*a
abc,
Adrian
de Grazia
a,
Fred
Fryer
d and
Philip A.
Doble
*a
aElemental Bio-imaging Facility, University of Technology Sydney, Broadway, New South Wales, Australia. E-mail: philip.doble@uts.edu.au
bThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia
cExposure Biology Laboratory, Frank R. Lautenberg Environmental Health Sciences Laboratory, Department of Preventive Medicine, Icahn School of Medicine at Mount Sinai, New York, USA
dAgilent Technologies, Mulgrave, Victoria, Australia
First published on 11th May 2015
Abstract
A rapid method was developed for the determination of monobutyltin, dibutyltin, tributyltin, monophenyltin, diphenyltin, and triphenyltin by liquid chromatography-isotope dilution-inductively coupled plasma-mass spectrometry in sediments and drinking water. All six species were eluted in less than 6.5 minutes with a binary gradient. Offline solid phase extraction was used to pre-concentrate the organotin compounds for quantification employing two calibration procedures; external standard calibration and isotopic dilution. The external standard calibration approach yielded detection limits in the range of 1.5 to 25.6 ng L−1. The method was linear over four orders of magnitude with regression coefficients greater than 0.99 and a peak area repeatability less than 4.5% RSD (n = 7) for all compounds. The isotopic dilution method was three times more sensitive with detection limits in the range of 0.5–1.2 ng L−1. Recoveries for the external calibration method were from 33–68% with % RSDs of 5.7–12.7%. The isotopic dilution method had recoveries of 70–114% with % RSDs of 1.2–2.9%. The methods were applied to sediments sampled from the Cooks River in Sydney. The isotopic dilution method provided a viable alternative to the more common analysis by gas chromatography-inductively coupled plasma-mass spectrometry for contaminated sediment without the requirement of sample derivatisation.
Introduction
Tri-substituted organotin compounds, such as trialkyl- and triphenyltin (TPhT) have been extensively used as antifungal and antibacterial agents. Other uses of organotin compounds include plastics stabilisers,1 wood preservatives, fire retardants and reducing agents, as well as applications in the pharmaceutical, ceramic and glass industries.2Trialkyltin compounds are extremely toxic biocides that are deliberately and directly released into the environment.3 Trialkyltins, and in particular the tributyl-substituted species (TBT), are the active agents in marine antifouling paints. Their use has been regulated by the International Maritime Organisation (IMO) since 1990![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 due to their extreme toxicity toward aquatic organisms. Organotin compounds are classed as persistent pollutants and remain in coastal waters for many years.5 Organotin compounds are also known to leach from sediments into surrounding water, thereby becoming a steady source of pollution.6 The Australian Drinking Water Guideline is set at a maximum level of 1 μg L−1 for TBT by the National Health and Medical Research Council and the Agriculture and Resource Management Council of Australia and New Zealand, while the Australian Water Quality Guideline for Fresh and Marine Waters of the Australia and New Zealand Environment and Conservation Council set a limit for TBT at 0.004 μg L−1 for a 99% protection level of species in marine waters. The recommended sediment quality guideline for TBT is 5 μg Sn kg−1 normalised to 1% total organic carbon.7 TPhTs are also potent marine toxins and have been shown to be a causal agent in the mutation of Chinese sturgeon.8 TPhT is not readily detected in water and sediment, but through biomagnification can be found at detectable levels in marine organisms.9
4 due to their extreme toxicity toward aquatic organisms. Organotin compounds are classed as persistent pollutants and remain in coastal waters for many years.5 Organotin compounds are also known to leach from sediments into surrounding water, thereby becoming a steady source of pollution.6 The Australian Drinking Water Guideline is set at a maximum level of 1 μg L−1 for TBT by the National Health and Medical Research Council and the Agriculture and Resource Management Council of Australia and New Zealand, while the Australian Water Quality Guideline for Fresh and Marine Waters of the Australia and New Zealand Environment and Conservation Council set a limit for TBT at 0.004 μg L−1 for a 99% protection level of species in marine waters. The recommended sediment quality guideline for TBT is 5 μg Sn kg−1 normalised to 1% total organic carbon.7 TPhTs are also potent marine toxins and have been shown to be a causal agent in the mutation of Chinese sturgeon.8 TPhT is not readily detected in water and sediment, but through biomagnification can be found at detectable levels in marine organisms.9
Traditional approaches for the speciation of organotin compounds involve separation by gas chromatography (GC) with various detection methods including flame ionisation detection (FID), flame photometric detection (FPD) and inductively coupled plasma – mass spectrometry (ICP-MS). Organotin compounds with one to three substituents are polar and non-volatile due to their ionic properties,3 and require derivatisation before analysis by GC. Three common derivatisation methods are normally applied to GC-based analysis of organotin compounds: hydride generation; alkylation by Grignard reagents; and ethylation by sodium tetraethylborate (NaBEt4).10 Hydride generation and NaBEt4 ethylation are particularly useful for aqueous matrices, where direct derivatisation of the sample can be performed. NaBEt4 ethylation is a relatively simple procedure that is principally governed by three factors: acidity of the solution, NaBEt4 concentration and the time of reaction. However, differing degrees of ethylation may be observed depending on the organotin species present. Ethyl- and butyl-substituted tins are both more efficiently derivatised at high pH, whereas methyltin ethylation is preferential at low pH. Methyl- and ethyltin compounds have improved yield with larger concentrations of reagent, yet ethylation of butyltin compounds occurs independent of NaBEt4 concentration.11 Additionally, NaBEt4 decomposes rapidly in air and light and is extremely flammable.12 Derivatisation of organotins in sediments by NaBEt4 requires prior extraction13 unless a large amount of reagent is used to compensate for the side reactions with metals and other matrix components.10
Liquid chromatography (LC-) ICP-MS is an alternative approach for the speciation of organotin compounds, and is an attractive option as derivatisation is not required and the sample preparation procedures are comparatively minimal. Various LC-ICP-MS methods have been used for speciation analysis including micellar,14 ion-pairing2,15–17 and reverse phase6,18–21 separation mechanisms.
The perceived disadvantages of LC-ICP-MS are poorer sensitivities and inferior peak capacities compared to GC separation. A typical run time for LC-ICP-MS speciation of organotins in a complex matrix is 20 minutes for six species.6 Further, a higher number of organotin species can be resolved by GC than standard-bore LC.18 Leaching of inorganic and organic tin from the LC column has been reported.3 and metal free LC systems have previously been recommended.20 Regardless, both recent improvements in LC technology and the ease of sample preparation has allowed LC-ICP-MS to remain a viable option for organotin speciation, particularly using isotope dilution as a definitive quantitative methods.22
Isotope dilution (ID) is regarded as the optimal method for trace element analysis. The International Bureau of Weights and Measures classifies isotope dilution mass spectrometry as a primary ratio method of the highest metrological quality.23 ID has been shown to be the method best suited to the certification and characterisation of reference materials.24 The precision of butyltin quantification is increased by one order of magnitude when ID is used, as opposed to standard addition and external calibration.25,26 Spiking of the isotope dilution standard with simultaneous sample equilibration and extraction is now routine.12,27,28 Monperrus et al.29 demonstrated that a seven-minute simultaneous spike equilibration and sample extraction was as effective as a twelve-hour spike equilibration time before extraction, thus greatly enhancing the capacity for ID to be integrated in high-throughput workflows.
The aim of this work was to develop a rapid separation and quantification of the six main organotin species of environmental concern by LC-ICP-MS: monobutyltin (MBT), dibutyltin (DBT), tributyltin (TBT), monophenyltin (MPhT), diphenyltin (DPhT), and triphenyltin (TPhT). Off line solid phase extraction was employed to meet the environmental and drinking water detection limits. External standard calibration and isotopic dilution methods of quantification were compared. The method described here has the potential to dramatically increase sample throughput in high volume laboratories using a relatively simple and robust quantitative LC-ICP-MS approach.
Experimental
Reagents, standards and samples
MBT, DBT, TBT, MPhT, DPhT, and TPhT standards were obtained as chloride salts from Sigma Aldrich (Castle Hill, NSW, Australia). Stock solutions of 100 mg L−1 were prepared in HPLC grade acetonitrile (ACN) (Crown Scientific, Minto, NSW, Australia). A 119Sn-enriched butyltin mix (84.2% 119Sn enriched) was sourced from ISC Science (Oviedo, Spain). All stock solutions were stored in the dark at 4 °C. Tropolone and triethylamine (TEA) were obtained from Sigma Aldrich (Castle Hill, NSW, Australia) and glacial acetic acid (CH3COOH) from Chem Supply (Gillman, South Australia, Australia). Bond Elut C18-500 mg solid phase extraction (SPE) cartridges were obtained from Varian (Melbourne, Victoria, Australia). PACS-2 (Marine Sediment Reference Materials for trace metals and other constituents; MBT, DBT and TBT) was obtained from the NRCC (Ottowa, Canada). Certified values for both the Butyltin QuantID Kit and PACS-2 are shown in Table 1. Sediment samples were collected from the Cooks River, Sydney Australia (39°55′41′′S, 151°9′24′′E).| 119Sn-enriched butyltin mix (ISC Science) | PACS-2 (NRCC) | |
|---|---|---|
| MBT | 0.110 ± 0.005 | — |
| DBT | 0.691 ± 0.009 | 1047 ± 64 |
| TBT | 1.046 ± 0.020 | 890 ± 105 |
Liquid chromatography-inductively coupled plasma-mass spectrometry
The separation conditions and ICP-MS parameters are summarised in Table 2. Separations were performed on either an Agilent Technologies 1200 HPLC or a 1290 UHPLC hyphenated to an Agilent 7500cx ICP-MS (Mulgrave, Victoria, Australia). A binary gradient was employed: mobile phase A consisted of 0.0625% tropolone:0.1% triethylamine:6% glacial acetic acid (v/v) in LC-grade H2O; mobile phase B was 100% acetonitrile. Separation was performed using a Zorbax XDB Eclipse rapid resolution high throughput (RRHT) column. The column eluent was directly inserted into a perfluoroalkoxy (PFA) Burgener nebuliser (Agilent Technologies Australia) by a 60 cm length of 0.13 mm i.d. polyether ether ketone (PEEK) tubing. A standard Scott-type double-pass quartz spray chamber was used. Oxygen was added via a T-piece between the spray chamber and the torch as an 80:20 argon:oxygen blend (BOC Gases Sydney, NSW, Australia), and the spray chamber was Peltier-cooled to −5 °C to facilitate the desolvation process. A 1 mm i.d. tapered torch was used to minimise carbon loading of the plasma. Both m/z 119 and 120 were monitored for ID experiments, and 120 alone for external calibration.
| LC: Agilent Technologies 1200SL; 1290 Infinity | ICP-MS: Agilent Technologies 7500cx | ||
|---|---|---|---|
| Column | Zorbax XDB Eclipse C18, 50 × 2.1 mm, 1.8 μm particle size | RF power | 1600 W |
| Flow rate | 0.5 mL min−1 | Carrier gas flow rate | 0.7 L min−1 |
| Injection volume | 5 μL | Auxiliary gas flow rate | 0.2 L min−1 |
| Mobile phase A | 94% H2O, 6% acetic acid, 0.1% triethylamine, 0.0625% tropolone | Option gas flow rate | 0.2 L min−1 (w/0.04 L min−1 O2) |
| Mobile phase B | 100% acetonitrile | Sample and skimmer cones | Pt |
| Gradient | 0–5 s, 45% A-55% B | Monitored masses | 119, 120 |
| Temperature | Ambient |
Passivation
Passivation was performed according to the procedure reported by Gjerde et al.30 to remove potential contaminating species prior to analysis. Briefly, the column was removed from the system and was rinsed with 200 mL deionised water (18.2 MΩ cm−1), followed by 8 M nitric acid for 15 minutes at 1 mL min−1. The system was flushed with deionised water until the pH of the eluent was greater than 5. The LC was then washed with 50 mM EDTA solution for 30 minutes at 1 mL min−1. The final wash was deionised water for 2 hours at 1 mL min−1.Sample preparation and pre-concentration
Aqueous standards were prepared daily from stock solutions. 250 mL of standard or samples solutions were loaded onto the Bond Elut SPE cartridges and were eluted with 4 mL of 80:20 ACN:CH3COOH, 0.1% TEA and 0.0625% tropolone before evaporating to dryness. The samples were then reconstituted in 250 μL of 70:20:10 ACN:CH3COOH:H2O, 0.1% TEA and 0.0625% tropolone. The accuracy was reported as % recovery.
Sediment extraction was based on modification of the procedure by Ruiz Encinar et al.12 The ID standard was diluted 1:10, and 100 μL of the 119Sn butyltin enriched spike was added to the sediment sample (ca. 0.1 g) which was immediately extracted with 2 mL of 75:25 CH3COOH:MeOH. The mixture was extracted in an ultrasonic bath for 30 minutes at approximately 40 °C. The sample was filtered before analysis.
Isotope dilution-inductively coupled plasma-mass spectrometry
The isotopic dilution procedure used the following equation for determination of the concentrations of the Sn species: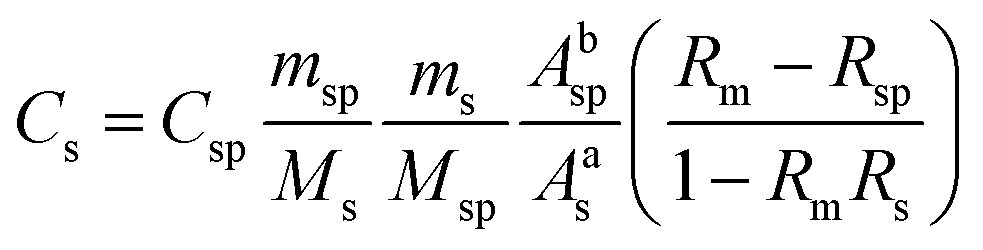 | (1) |
Results and discussion
Mobile phase optimisation
The chromatographic conditions were modified from the method reported by Chiron et al.,6 who reported that plasma stability was superior when methanol was employed as the organic modifier, at the expense of separation efficiency. This was in contrast to Wahlen et al.,19 who employed acetonitrile at lower flow rates with no effect upon the plasma. Tropolone was added to mobile phase A to selectively increase the retention of the di-substituted compounds whilst not affecting the mono- or the tri-substituted compounds.6Triethylamine was also added to mobile phase A to manipulate the selectivity of TBT.20 Acetic acid was added to maintain low pH and manipulate selectivity by complexation. Acetic acid also reduced the adsorption of organotins on the column17 by minimising interactions with the stationary phase,31 significantly reducing peak tailing of di-substituted organotins.20 Increasing acetic acid concentration also reduces the retention time of all organotins.32 In contrast to previous reports,33,34 plasma instability and baseline drift were not observed with this mobile phase composition. The system was very sensitive to small changes in organic modifier concentrations. Preliminary isocratic experiments showed that MBT and MPhT were resolved with a mobile phase of up to 45% ACN. An isocratic separation with 45% acetonitrile was greater than 10 minutes for all compounds. At 55% ACN, DBT, TBT, DPhT and TPhT were resolved within 6 minutes, though MBT and MPhT co-eluted. Therefore, elution with a 45% B-55% B step gradient over 5 seconds from the point of injection was employed.
Fig. 1 shows the separation of the six target organotin species in a 500 μg L−1 standard solution. Complete separation was achieved in under 7 minutes, less than half the time required for methods reported by Chiron et al.6 and Inagaki et al.5 Total separation times were similar to that obtained by GC-ICP-MS.18 However, the re-equilibration time of 2 minutes before sequential runs is less than the time typically required by GC-ICP-MS between injections and temperature ramps.
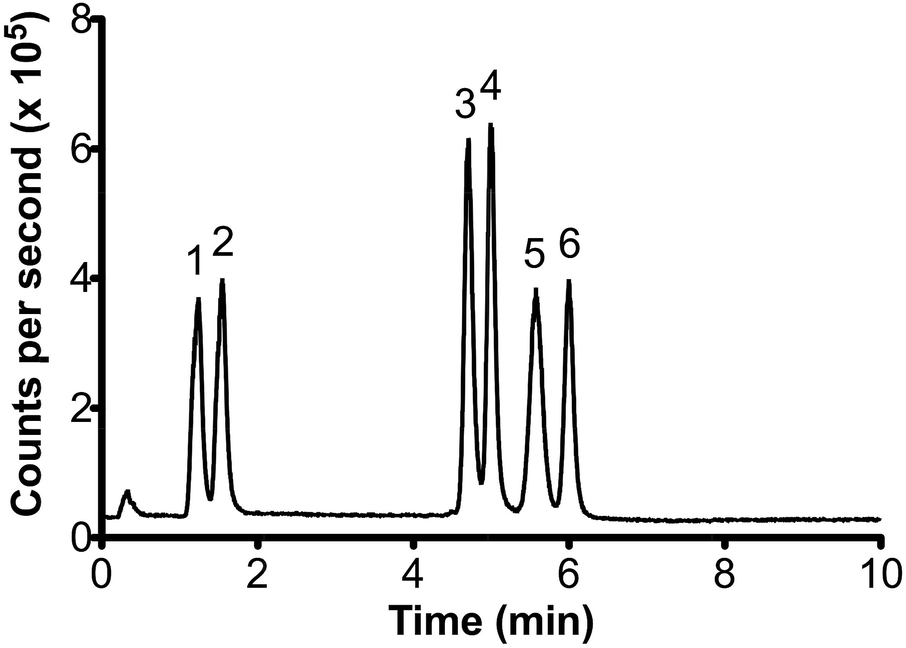 | ||
| Fig. 1 Chromatogram of a 500 μg L−1 mixed standard (5 μL injection volume). Peak order: 1- MPhT, 2- MBT, 3- TBT, 4- DBT, 5- TPhT, 6- DPhT. | ||
External standard calibration and pre-concentration
Analytical performance of external calibration without pre-concentration is outlined in Table 3. Peak area relative standard deviations (RSDs) ranged from 1.5% to 4.4%. Retention time RSDs were less than 0.5% for all peaks. The correlation coefficients (r2) for 6 measured organotin species over a 1 μg L−1 to 1000 μg L−1 concentration range were greater than 0.999. The detection limits for direct injection of the standards ranged from 1.6 to 5 μg L−1.:1 signal to noise ratio. Reported as the concentration of total organotin compound
| LOD (μg L−1) | Peak area RSD (%) n = 7 | Linearity (1 μg L−1 to 1000 μg L−1) | Retention time RSD (%), n = 7 | |
|---|---|---|---|---|
| MBT | 5 | 4.4 | 0.9994 | 0.47 |
| DBT | 3.5 | 2.2 | 0.9992 | 0.40 |
| TBT | 5 | 2.5 | 0.9996 | 0.33 |
| MPhT | 5 | 2.4 | 0.9995 | 0.38 |
| DPhT | 1.6 | 1.5 | 0.9996 | 0.43 |
| TPhT | 3.7 | 2.6 | 0.9999 | 0.47 |
Preliminary off-line preconcentration experiments indicated that 100 mL of a standard 1 μg L−1 organotin solution could be loaded onto the SPE cartridges without breakthrough. A number of elution solvents were trialled to minimise the volume of the SPE eluent for complete recovery of the organotins. Elution of the organotins from the SPE cartridge with solvents that contained greater than 70% ACN produced a negative system peak that interfered with MBT. This interference peak was due to suppression of the background signal from the introduction of the SPE elution solvent to the plasma.35 The optimal elution solvent was 20:10:70 CH3COOH:H2O:ACN and 0.1% TEA and 0.0625% tropolone. All target compounds were eluted from the SPE cartridge with 4 mL of this solution.
Fig. 2 shows a chromatogram of a 1 μg L−1 standard with a 1:25 pre-concentration factor. The peak at 2.5 minutes was a system peak generated from enhancement of the Sn background due to the rapid gradient changing the organic load in the plasma.36 The effect of the ACN gradient on the background Sn intensity is dependent on the lens conditions. Brown et al.37 observed a similar peculiarity in the development of a LC-ICP-MS speciation method for Pb. They experienced baseline suppression at the start of a gradient before a rise and fall similar to that observed here. The ICP-MS lenses were optimised for Sn with a solution containing 50% ACN. The gradient was from 45–55% ACN explaining a rise in the baseline from 45–50% and a fall after 50–55%. This peak was present in the blanks and did not represent an unknown organic Sn species.
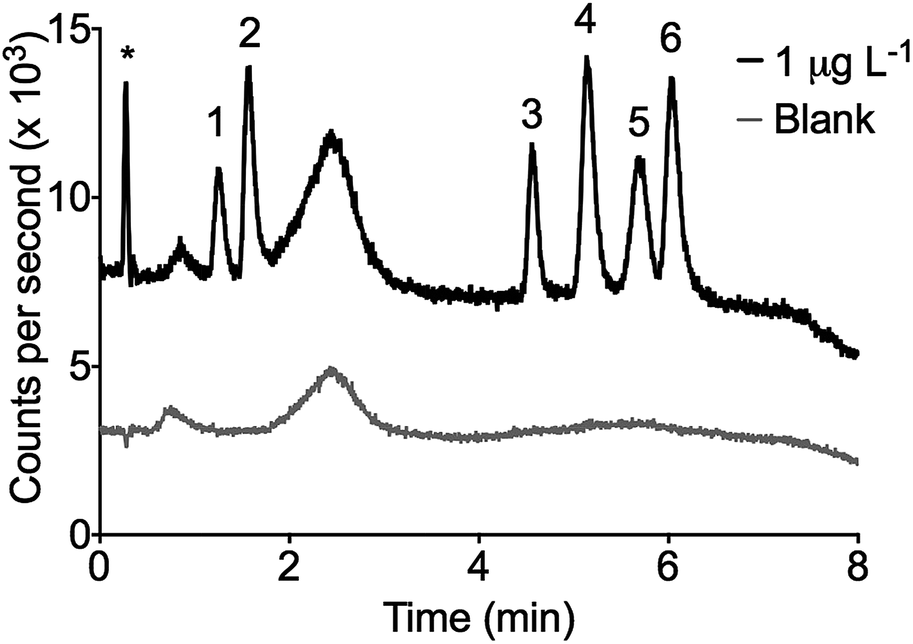 | ||
| Fig. 2 A 1 μg L−1 standard pre-concentrated (concentration factor = 25) and corresponding blank injection (5 μL injection volume). Peak order: (1) MPhT, (2) MBT, (3) TBT, (4) DBT, (5) TPhT, (6) DPhT, * ionic Sn. | ||
Evaporation of the eluent was trialled to further improve detection limits. 250 mL of a 25 ng L−1 standard mix of the target compounds were loaded onto the SPE cartridges. The target compounds were eluted from the SPE cartridges with 80:20 ACN:CH3COOH and 0.1% TEA and 0.0625% tropolone before evaporation to dryness. The increase in ACN concentration resulted in elution of all compounds within a volume of 2 mL. The samples were then reconstituted in 250 μL of 70:20:10 ACN:CH3COOH:H2O water and 0.1% TEA and 0.0625% tropolone before injection.
The analytical performance of the pre-concentration method is detailed in Table 4. The pre-concentration factors for the SPE cartridges ranged from 24 to 32, corresponding to 96–130% recoveries. The pre-concentration factors for the combination of SPE and evaporation ranged from 325 to 677, corresponding to 33–67% recoveries. The % RSDs for these recoveries were 5.7–8.8%, indicating that the method was reproducible. The detection limits for all compounds, calculated as three times the signal-to-noise ratio of a 25 ng L−1 standard, ranged from 1.5 to 25.6 ng L−1. These detection limits are comparable to other methods that have been reported by LC-ICP-MS. Yang et al.17 reported detection limits of 28 ng L−1 and 33 ng L−1 for TPhT and TBT, respectively. Fairman et al.16 reported approximately 2 ng L−1 for both TPhT and TBT. Ugarte et al.38 used SPME-HPLC-ICP-MS for the speciation of tri-substituted organotin compounds reporting detection limits of 11 ng L−1 and 185 ng L−1 for TPhT and TBT, respectively. The 45 minute SPME extraction procedure is significantly longer than aqueous ethylation for GC-ICP-MS detection.
| Solid phase extraction | Solid phase extraction and evaporation | |||||||
|---|---|---|---|---|---|---|---|---|
| CF | % Recovery | DLa (ng L−1) | % RSD | CF | % Recovery | DLa (ng L−1) | % RSDb | |
|
a Determined as 3:1 signal to noise ratio. Reported as the concentration of total organotin compound.
b
n = 5, reported on evaporated pre-concentration factor.
|
||||||||
| MBT | 32 | 128 | 100 | 6.9 | 433 | 43 | 1.5 | 5.7 |
| DBT | 17 | 68 | 100 | 6.7 | 509 | 51 | 3.2 | 9.4 |
| TBT | 27 | 108 | 150 | 9.1 | 477 | 48 | 3.7 | 6.0 |
| MPhT | 28 | 112 | 330 | 5.1 | 476 | 48 | 1.8 | 12.7 |
| DPhT | 29 | 116 | 110 | 8.1 | 677 | 68 | 2.0 | 11.5 |
| TPhT | 24 | 96 | 180 | 12.5 | 325 | 33 | 25.6 | 5.7 |
Isotope dilution LC-ICP-MS
SPE-LC-ID-ICP-MS of a 25 ng L−1 standard mix of MBT, DBT and TBT provided good recoveries of 18–29 ng L−1 with recoveries of 70–114% with % RSDs of 1.2–2.9% (see Table 5). As equilibration of the isotopically enriched species has been achieved, the recovery does not affect the quantitative results. Detection limits were calculated as 0.5–1.2 ng L−1. Detection limits were improved by a factor of three for all compounds when compared to the external calibration method. Degradation of phenyltin species has been shown to be an issue during extraction.39 These species degrade by 2–55% when using mild extraction conditions.40 Phenyltin isotope dilution standards are not readily available.:1 signal to noise ratio. Reported as the concentration of total organotin compound
| CF | % Recovery | DL (ng L−1) | % RSD | |
|---|---|---|---|---|
| MBT | 18 | 72 | 0.5 | 2.9 |
| DBT | 27 | 108 | 1.1 | 1.2 |
| TBT | 29 | 114 | 1.2 | 2.4 |
The LC-ID-ICP-MS method was applied to PACS-2 certified reference material (see Fig. 3). DBT and TBT were in good agreement with the certified values (see Table 6). MBT is not certified in PACS-2 but is known to be present.
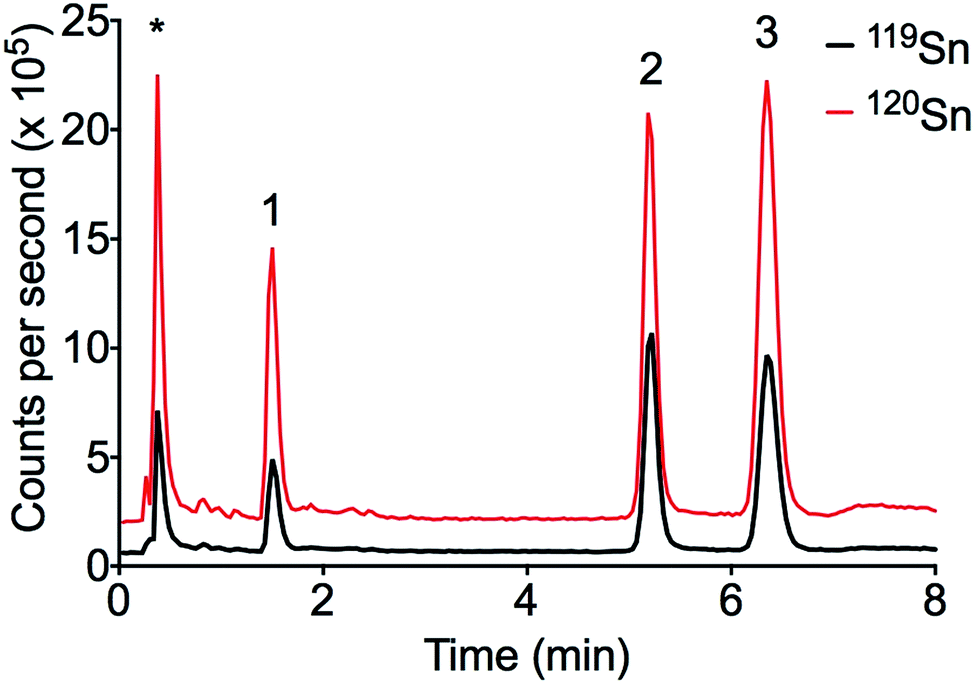 | ||
| Fig. 3 Chromatogram of PACS-2 certified reference sediment. Peak order: (1) MBT, (2) TBT, (3) DBT, * ionic Sn. | ||
| MPhT | MBT | TBT | DBT | TPhT | DPhT | |
|---|---|---|---|---|---|---|
| a Not detected. b Not available. | ||||||
| PACS-2 (ID) | nda | 405 ± 13 | 952 ± 15 | 1044 ± 25 | nd | nd |
| PACS-2 (external calibration) | nd | 259 ± 20 | 889 ± 48 | 790 ± 38 | nd | nd |
| PACS-2 certified | nab | na | 890 ± 105 | 1047 ± 64 | na | na |
| Cooks River (ID) | nd | 771 ± 110 | 2113 ± 205 | 1356 ± 288 | nd | nd |
| Cooks River (external calibration) | nd | 727 ± 172 | 1369 ± 71 | 1410 ± 235 | nd | nd |
Sample analysis
The LC-ID-ICP-MS and the external calibration methods were applied to the analysis of sediment samples collected from Cooks River near the entrance to Botany Bay, Sydney Australia. Three butyltin (MBT, DBT and TBT) species were detected at significant concentrations without the need for SPE (see Fig. 4). Port Botany is the largest container terminal in Sydney and has been in operation since 1930. The regular exposure to a large number of ships and boats over a long period of time may explain the high levels of butyltin compounds.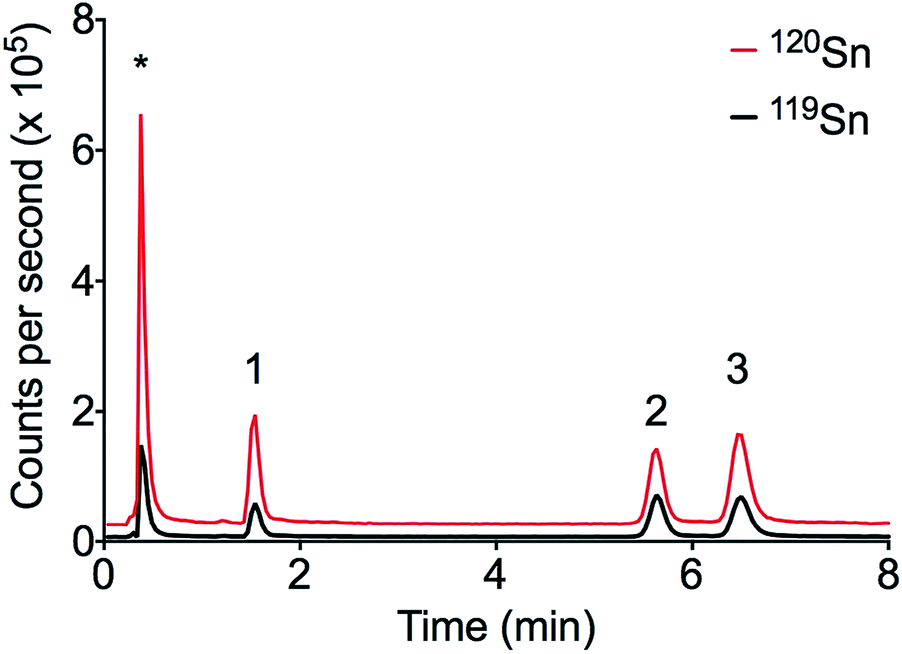 | ||
| Fig. 4 Chromatogram of sediment sample from Cooks River in Sydney, Australia. Peak order: (1) MBT, (2) TBT, (3) DBT, * ionic Sn. | ||
The results shown in Table 5 demonstrate the difficulty of analysing organotins. Extraction of organotins is complex as there are strong interactions with sediment matrices requiring mild conditions to preserve the chemical integrity of the analytes.41 Numerous approaches to the extraction of organotins have been reviewed.42 Many different sediment extraction procedures have been evaluated and were appropriate for the sediment analysed. Abalos et al.43 identified a toluene:acetic acid mixture to yield the highest extraction efficiency while minimising degradation during extraction. They achieved accuracy of 82% and 92% of DBT and TBT in PACS-1 CRM and 70% and 90% of DBT and TBT in CRM-462. Concentrated HBr and tropolone was critical for the extraction of the more polar organotin compounds in sediments collected off the Huelva coast in the southwest of Spain.44 Sediment extraction was based on modification of the procedure by Ruiz Encinar et al.,12 who obtained an extraction yield for DBT and TBT within the certified values for PACS-2. MBT was strongly bound to the matrix and required harsher extraction techniques to recover it quantitatively. Ultrasonic, mechanical, and microwave extractions were compared and all resulted in high extraction efficiencies for MBT, short extraction times and no degradation products.
Extraction optimisation for new samples is less arduous with ID analysis. ID has many advantages over classical calibration procedures such as external calibration and standard addition. These include results not being affected by instrumental instability or matrix effects, and once equilibration has been achieved loss of sample will have no influence on the final result. The same extraction procedure was applied to PACS-2 and the sediment from Cooks River. With external calibration PACS-2 shows good agreement for TBT with the certified value while DBT is underestimated and MBT is lower than the value obtained by isotope dilution. The Cooks River sample shows good agreement for MBT and DBT with the values obtained by ID while TBT is underestimated. Every sediment sample will have a different composition leading to different interactions between the analytes and the sample matrix. The assumption that if an extraction procedure is effective for the CRM it should be able to be applied successfully to the sample is not applicable for organotin speciation. Isotope dilution eliminated the uncertainties due to the extraction procedure and matrix interactions on-column. External calibration relies on complete extraction of the analytes. Isotopic dilution relies on equilibration of the spike after extraction of the natural sample from the matrix is complete. This compensates for incomplete extraction and differences in matrices.
The method described here has several advantages over GC-ICP-MS. The need for derivatisation is removed, eliminating the requirement for the use of hazardous chemicals in ethylation. NaBEt4 is a pyrophoric, unstable compound that is aggressive to the front end of the GC column, leading to faster degradation and reduced column stability over time. The SPE-LC-ICP-MS procedure is shorter than GC-based methods, even when taking into account improvements in GC technology that have improved sample throughput. Sample derivatisation and ethylation can be performed in a similar timeframe, though a standard GC-ICP-MS run is 15 minutes (versus our described 8 minute LC runtime), and an additional 1–3 minutes for inter-sample cooldown make our LC method capable of approximately twice the throughput. Though detection limits using LC-ICP-MS are still an order of magnitude higher than GC,45 this method meets mandated environmental detection limits and is a suitable, rapid screening method that can process large sample volumes in a short timeframe.
Conclusion
LC-ICP-MS was shown to be a viable alternative to GC-ICP-MS for the analysis of organotin pollutants in drinking water and environmental samples without the requirement of pre-column sample derivatisation. The ID method was superior to the external calibration method in terms of accuracy, precision and detection limits. The ID method also met the environmental detection limits of 4 ng L−1 in marine waters. However, the ID method was limited to butyl-substituted species. Significant concentrations of butyltins were found in a sediment sample from the Cooks River.Acknowledgements
Guiseppe Centineo of ISC Science provided PACS-2 certified reference material. Agilent Technologies Australia Pty Ltd provided access to a 1200 series HPLC, a 1290 series UHPLC, and 7500cx ICP-MS.Notes and references
- L. Ebdon, S. J. Hill and C. Rivas, TrAC, Trends Anal. Chem., 1998, 17, 277–288 CrossRef CAS.
- U. T. Kumar, J. G. Dorsey, J. A. Caruso and E. H. Evans, J. Chromatogr. A, 1993, 654, 261–268 CrossRef CAS.
- R. Ritsema and O. F. X. Donard, Tech. Instrum. Anal. Chem., 2000, 21, 1003–1073 CAS.
- International Maritime Organization, International Convention on the Control of Harmful Anti-fouling Systems on Ships 2001.
- K. Inagaki, A. Takatsu, T. Watanabe, Y. Aoyagi, T. Yarita, K. Okamoto and K. Chiba, Anal. Bioanal. Chem., 2007, 387, 2325–2334 CrossRef CAS PubMed.
- S. Chiron, S. Roy, R. Cottier and R. Jeannot, J. Chromatogr. A, 2000, 879, 137–145 CrossRef CAS.
- Australian and New Zealand Environment Conservation Council, Australian and New Zealand Guidelines for Fresh and Marine Water Quality, 2000.
- J. Hu, Z. Zhang, Q. Wei, H. Zhen, Y. Zhao, H. Peng, Y. Wan, J. P. Giesy, L. Li and B. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9339–9344 CrossRef CAS PubMed.
- J. Hu, H. Zhen, Y. Wan, J. Gao, W. An, L. An, A. Fen Jin and X. Jin, Environ. Sci. Technol., 2006, 40, 3142–3147 CrossRef CAS.
- R. Morabito, P. Massanisso and P. Quevauviller, TrAC, Trends Anal. Chem., 2000, 19, 113–119 CrossRef CAS.
- F. M. Martin and O. F. X. Donard, Fresenius' J. Anal. Chem., 1995, 351, 230–236 CrossRef CAS.
- J. R. Encinar, P. R. Gonzalez, J. I. G. Alonso and A. Sanz-Medel, Anal. Chem., 2002, 74, 270–281 CrossRef.
- C. Carlier-Pinasseau, G. Lespes and M. Astruc, Environ. Technol., 1997, 18, 1179–1186 CrossRef CAS PubMed.
- Y. Inoue, K. Kawabata and Y. Suzuki, J. Anal. At. Spectrom., 1995, 10, 363–366 RSC.
- N. P. Vela and J. A. Caruso, J. Anal. At. Spectrom., 1996, 11, 1129–1135 RSC.
- B. Fairman, T. Catterick, B. Wheals and E. Polinina, J. Chromatogr. A, 1997, 758, 85–92 CrossRef CAS.
- H. J. Yang, S. J. Jiang, Y. J. Yang and C. J. Hwang, Anal. Chim. Acta, 1995, 312, 141–148 CrossRef CAS.
- R. Wahlen and C. Wolff-Briche, Anal. Bioanal. Chem., 2003, 377, 140–148 CrossRef CAS PubMed.
- R. Wahlen and T. Catterick, J. Chromatogr. B: Biomed. Sci. Appl., 2003, 783, 221–229 CrossRef CAS.
- S. White, T. Catterick, B. Fairman and K. Webb, J. Chromatogr., A, 1998, 794, 211–218 CrossRef CAS.
- C. Rivas, L. Ebdon and S. J. Hill, J. Anal. At. Spectrom., 1996, 11, 1147–1150 RSC.
- P. Rodriguez-Gonzalez, J. M. Marchante-Gayon, J. I. G. Alonso and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef PubMed.
- T. J. Quinn, Metrologia, 1997, 34, 61–65 CrossRef.
- J. Vogl, J. Anal. At. Spectrom., 2007, 22, 475–492 RSC.
- C. Bancon-Montigny, P. Maxwell, L. Yang, Z. Mester and R. E. Sturgeon, Anal. Chem., 2002, 74, 5606–5613 CrossRef.
- L. Yang, Z. Mester and R. E. Sturgeon, J. Anal. At. Spectrom., 2002, 17, 944–949 RSC.
- M. Monperrus, O. Zuloaga, E. Krupp, D. Amouroux, R. Wahlen, B. Fairman and O. F. X. Donard, J. Anal. At. Spectrom., 2003, 18, 247–253 RSC.
- J. R. Encinar, P. Rodriguez-Gonzalez, J. Rodriguez Fernandez, J. I. Garcia Alonso, S. Diez, J. M. Bayona and A. Sanz-Medel, Anal. Chem., 2002, 74, 5237–5242 CrossRef.
- M. Monperrus, R. C. R. Martin-Doimeadios, J. Scancar, D. Amouroux and O. F. X. Donard, Anal. Chem., 2003, 75, 4095–4102 CrossRef CAS.
- D. T. Gjerde, C. P. Hanna and D. Hornby, DNA Chromatography, Wiley VCH, 2002 Search PubMed.
- C. F. Harrington, G. K. Eigendorf and W. R. Cullen, Appl. Organomet. Chem., 1996, 10, 339–362 CrossRef CAS.
- E. Rosenberg, V. Kmetov and M. Grasserbauer, Fresenius' J. Anal. Chem., 2000, 366, 400–407 CrossRef CAS.
- E. Gonzalez-Toledo, R. Compano, M. Granados and M. Dolors Prat, TrAC, Trends Anal. Chem., 2003, 22, 26–33 CrossRef CAS.
- E. H. Larsen, Spectrochim. Acta, Part B, 1998, 53, 253–265 CrossRef.
- Y. Nygren and E. Björn, J. Chromatogr. A, 2010, 1217, 4980–4986 CrossRef CAS PubMed.
- N. Forsgard, E. Nilsson, M. Andersson and J. Pettersson, J. Anal. At. Spectrom., 2006, 21, 305–310 RSC.
- A. A. Brown, L. Ebdon and S. J. Hill, Anal. Chim. Acta, 1994, 286, 391–399 CrossRef CAS.
- A. Ugarte, N. Unceta, M. C. Sampedro, M. A. Goicolea, A. Gomez-Caballero and R. J. Barrio, J. Anal. At. Spectrom., 2009, 24, 347–351 RSC.
- S. J. Kumar, S. Tesfalidet, J. P. Snell, D. N. Van and W. Frech, J. Anal. At. Spectrom., 2004, 19, 368–372 RSC.
- D. N. Van, T. T. X. Bui and S. Tesfalidet, Anal. Bioanal. Chem., 2008, 392, 737–747 CrossRef PubMed.
- E. Graupera, C. Leal, M. Granados, M. D. Prat and R. Compano, J. Chromatogr. A, 1999, 846, 413–423 CrossRef CAS.
- C. Dietz, J. Sanz, E. Sanz, R. Munoz-Olivas and C. Camara, J. Chromatogr. A, 2007, 1153, 114–129 CrossRef CAS PubMed.
- M. Abalos, J. M. Bayona and P. Quevauviller, Appl. Organomet. Chem., 1998, 12, 541–549 CrossRef CAS.
- J. L. Gomez-Ariza, E. Morales, I. Giraldez and R. Beltran, Int. J. Environ. Anal. Chem., 1997, 66, 1–13 CrossRef CAS PubMed.
- P. Rodríguez-González, J. R. Encinar, J. I. G. Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2002, 17, 824–830 RSC.
| This journal is © The Royal Society of Chemistry 2015 |