
A rapid diagnostic method for E. coli serogroups responsible for gastro-intestinal diseases using loop-mediated isothermal amplification
Matthew L.
Carnevale
*a,
Philip J. R.
Roche
bcd,
Mohamed
Najih
de,
Miltiadis
Paliouras
bcd,
Lenore K.
Beitel
bcd and
Mark A.
Trifiro
bcd
aConcordia University, 7141 Sherbrooke West, Montreal, QC H4B 1R6, Canada. E-mail: matthew_carnevale@hotmail.com; Fax: +1-514-340-7502; Tel: +1-514-340-8222 ext. 5268
bLady Davis Institute, 3755 Côte Ste-Catherine Road, Montreal, QC H3T 1E2, Canada
cJewish General Hospital, 3755 Côte Ste-Catherine Road, Montreal, QC H3T 1E2, Canada
dMcGill University, 845 Sherbrooke West, Montreal, QC H3A 0G4, Canada
eThe Photonic Systems Group, McGill University, 3480 University Street, Montreal, QC H3A 2A7, Canada
First published on 13th November 2014
Abstract
A rapid diagnostic method for Escherichia coli serogroup identification was developed, employing the loop-mediated isothermal amplification reaction (LAMP). Identifying the serogroup responsible for infection is accomplished using primers that are specific to sequences that are exclusive to the particular serogroup. Previous work has primarily focused on detecting one specific sequence or gene as a method of detection of a pathogenic entity. The novel approach involves using primers specific to various genes to determine which are present as a method of identification of the unknown pathogen rather than simple detection. The experiments conducted involved running the LAMP reaction and detecting amplification using gel electrophoresis, fluorescence, and localized surface plasmon resonance. The results obtained demonstrate that the LAMP reaction is efficient, and specific in that it only amplifies target DNA, and that it requires minimal instrumentation in comparison to various other nucleic acid amplification methods. This assay's principle and instrumentation is suited to resource-limited environments and when mobility is required for testing.
1. Introduction
The German E. coli outbreak of 2011 resulted in over 4000 cases of poisoning, including both non-hemolytic-uremic syndrome, and hemolytic-uremic syndrome. These infections resulted in 50 deaths.1 Exposure to the virulent E. coli can occur from consumption of contaminated crops grown with manure that harbored the bacteria. These strains can also be introduced to crops via tainted irrigation systems that were contaminated with cattle feces. One of the steps in lettuce production is rinsing with tap water. This method is ineffective at removing certain strains of E. coli that are capable of penetrating the cells of the plant and are thereby rendered inaccessible by surface cleaners.2 It is quite obvious that exposure to these virulent strains of E. coli is an issue in economically developed nations and not simply something that can be dismissed as a problem that developing countries must deal with, this is reflected by recent changes in government required testing in the US and Canada.3Escherichia coli is a species of Gram-negative bacteria that colonizes regions of the human gastrointestinal tract. Although this is a normal occurrence, there exist some strains of the bacteria that harbor virulent genes. When these pathogenic strains come into contact with humans, in the form of tainted meat or produce, the result is a spectrum of ailments. Certain serogroups that can cause gastrointestinal issues are; the enteropathogenic E. coli (EPEC), enterotoxigenic E. coli (ETEC), enteroaggregative E. coli (EAEC), enteroinvasive E. coli (EIEC), diffusely adherent E. coli (DAEC), and enterohaemorrhagic E. coli (EHEC) that have various attributes that correspond to virulence.4
The EAEC serogroup colonize the colonic mucosa and cause intestinal inflammation and persistent diarrhea.5 They adhere to the mucosa of the intestine by using a fimbrial structure. This structure is approximately 2 nm in diameter and allows the bacteria to aggregate on the epithelial tissue of the intestine.6
ETEC cause illness by producing enterotoxins that can be stable at different temperatures. They produce these toxins without damaging or penetrating the cells of the intestinal mucosa.7 ETEC are the main cause of traveller's diarrhea.
EPEC are also non invasive and cause damage to the apical surface of the enterocyte plasma membrane that results in a rearrangement of the enterocyte cytoskeleton which then leads to a reduction in absorption potential of the intestine.8 This loss in absorption capacity contributes to an electrolyte concentration variance that results in diarrhea.
EIEC are an invasive serogroup. They penetrate the mucosal cells of the colon and then move into adjacent cells. These bacteria can then produce enterotoxins that cause diarrhea.5
DAEC are similar to EAEC however they adhere in a more dispersed pattern to the intestinal mucosal cells than aggregative E. Coli that form colonies of stacked cells.9 This serogroup is believed to cause diarrhea in young children.10
EHEC is a subset of the STEC serotype that produces Shiga toxin or Shiga-like toxins. STEC found in humans are called EHEC. E. coli from each of these serogroups have particular genes for virulence. By detecting the presence of certain genes in a sample from an afflicted patient, it is possible to diagnose a bacterial infection in the patient's gastrointestinal tract. Knowing that certain genes are common only to certain serotypes, the strain of bacteria can be determined, and specific treatments can be applied.
Polymerase chain reaction (PCR) is the classical method of DNA amplification. This technique has been used as a diagnostic tool to amplify genes relevant to virulent E. coli.11 Presence of these genes would indicate infection by that particular strain of bacteria. PCR involves cycling between a high temperature (94–98 °C) for DNA denaturation, a lower temperature (50–65 °C) for primer annealing, and a slightly higher temperature (72 °C) to allow a DNA polymerase enzyme to elongate the DNA. This need for thermal cycling requires accurate, expensive instruments thereby limiting the range of use of this diagnostic tool. Loop-mediated isothermal amplification (LAMP) is a novel method of DNA amplification involving the use of four primers plus two loop primers that anneal to the loop structure in LAMP amplicons to accelerate and enhance the sensitivity of the LAMP reaction.12 The fact that the reaction is carried out under isothermal conditions makes it a useful diagnostic tool.
Primers were designed specifically for the LAMP reaction using the Primer Explorer V4 software (http://primerexplorer.jp/elamp4.0.0/index.html). These include outer, inner and loop primers (Fig. 1A). Outer primers were designated as F3 and B3, inner primers were designated as FIP and BIP, and loop primers were LF and LB. These primers were designed to target known genes and plasmids responsible for virulence among the various serogroups of E. coli. Table 1 summarizes these sequences.
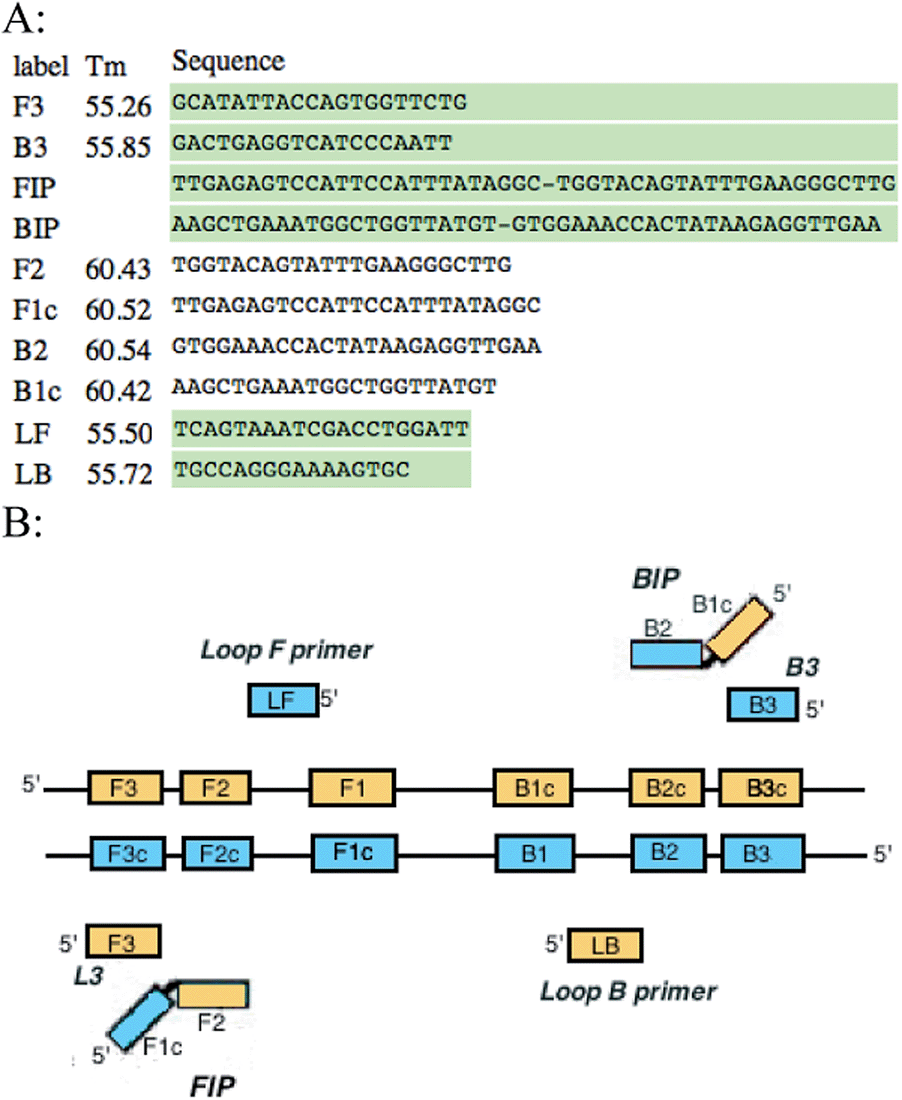 | ||
| Fig. 1 (A) LAMP primers for F41 gene designed with Primer Explorer V4 (B) schematic of LAMP primers aligned with the regions they are complementary to. | ||
| Gene | Function | Serogroup |
|---|---|---|
| STX | Shiga toxin | EHEC |
| STb | Heat labile enterotoxin | ETEC |
| SLT-IIv | Shiga-like toxin | EHEC |
| LT | Heat labile enterotoxin | ETEC |
| K88 | Pilus antigen | ETEC |
| invE | Invasion protein | EIEC |
| F41 | Fimbrial adhesion protein | EIEC |
| etpD | Type II secretion protein | EHEC |
| espP | Serine protease | EHEC |
| aggR | Transcriptional activator of aggregative adherence protein | EAEC |
| eaeA | Adhesion protein | EPEC |
| 987P | Fimbrial adhesion protein | ETEC |
| bfpA | Adhesion protein | EPEC |
| pCVD 432 | EAEC probe | EAEC |
| STp | Porcine heat stable enterotoxin | ETEC |
| STh | Human heat stable enterotoxin | ETEC |
Portions of the genes that were flanked by the primers were verified using NCBI's Basic Local Alignment Search Tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi). This was done to confirm that only the target genes would be amplified and thus false positive were avoided unless through preventable contamination.
Amplification of these genes could be detected by gel electrophoresis, or by visualization of turbidity caused by the magnesium pyrophosphate formed as a byproduct of nucleic acid amplification.16 Amplified DNA can also be detected using fluorescence. A fluorophore such as PicoGreen can selectively bind to dsDNA and by doing so enhances its fluorescence. Since unbound PicoGreen is not fluorescent, only successful amplifications will emit fluorescence. The reaction was carried out on a microtiter plate to allow simultaneous comparison of multiple samples for their presence of the various genes mentioned above.
Presence or absence of these genes allows determination of the pathogen's genotype that translates to its phenotype and ultimately leads to its identification. Knowledge of the identity of the infectious agent is invaluable in diagnosis and treatment of patients.
2. Materials and methods
Magnesium chloride (50 mM), Bst 2.0 polymerase (8000 U ml−1), and 10× isothermal amplification buffer were sourced from New England BioLabs. The reaction also required dNTPs (10 mM) from Bio Basic, and RNase-free water from Qiagen. LAMP primers were sourced from Integrated DNA Technologies.2.1 LAMP
Primers for the LAMP reaction were designed with similar melting temperature ranges to allow synchronous testing for multiple sequences. The forward, backward, and loop primers had an average Tm of 55 °C, whereas the inner primers had an average Tm of 61 °C. This 6 °C variance in temperature allowed the inner primers to anneal to the template more frequently and facilitate amplification at the reaction temperature of 66 °C. These six sets of primers recognize eight areas of the target DNA as illustrated in Fig. 1B. The two regions of the inner primers (FIP and BIP) are complementary to two different regions of the target DNA (F1 and F2c for the forward primers, and B1 and B2c for the backward primers). It is these dual complementary primers that allow the loops to form in the reaction. The strand displacement activity of Bst 2.0 polymerase allows the strands of DNA to be separated and for the replication process to be continuous and cyclical without the need for a thermocycler. Primers were resuspended from their lyophilized state and diluted to a final concentration of 25 μmol L−1. The LAMP reaction mixture contained dNTPs, isothermal amplification buffer, magnesium chloride, the six LAMP primers (including two loop primers), RNase-free water, and Bst 2.0 polymerase. The final volume of the reaction mixture was 26 μL including a sample volume of 5 μL. The samples were thermally lysed at a temperature of 96 °C for five minutes then chilled on ice briefly prior to addition of the reaction mixture to prevent DNA polymerase enzyme denaturation. The reaction was then carried out isothermally at 66 °C for a period of 80 minutes, followed by eight minutes at 80 °C to terminate the reaction. Both positive and negative controls were included in the assays. Positive control samples contained wild-type E. coli and negative control samples contained only RNase-free water (no template DNA).2.2 Detection
Approximately 12 μL from each reaction tube was run on a 0.9% agarose gel containing ethidium bromide and visualized under UV light. A DNA ladder was added to the outer lanes of the gel as a reference. Alternatively, a 5 μL aliquot from each tube was added to a microtiter plate along with 1 μL of PicoGreen. This method allowed rapid detection by the naked eye since positive samples were fluorescent green while negative samples remained a dull orange. Exposure to UV light made this contrast more apparent. This fluorescence method was preferred since it allowed comparison between samples in tubes with primers directed against various genes simultaneously. This made it possible to determine which of the virulent genes were present in the sample synchronously.It was also possible to detect amplification using LSPR, which works by measuring the shift in light wavelengths as a result of the changing environment of a plasmonic nanoparticle.17 The addition of 650 nm resonant gold nanoparticles to a reaction mixture made it possible to recognize amplification in the reaction tubes by change in dielectric constant of the solution surrounding the nanorods. This change confirms DNA amplification.
2.3 Real time lamp instrument
Instrumentation was constructed to achieve a real time LAMP measurement. The instrument is based upon recording the fluorescent signal created as DNA is synthesized and intercalated by fluorophores such as picogreen or sybrgreen. Temperature control comprises a kapton resistive heater to maintain temperature within 1 °C from the optimum, with a thermocouple, that delivers signal to the arduino microprocessor through analog input that runs a digital PID system; thus maintaining a constant temperature by alternating the current supply to the heater using a switch. The power supply required for heating is 24 volts and 0.5 amp. The heater has a machined copper block attached by thermal epoxy for both efficient heat transfer and support of 0.2 ml Eppendorf PCR tubes. Tubes are inserted vertically and images are collected of the internal volume through the reaction using a 5 megapixel camera with UV filter attached. The optics of the Pi camera are adjusted to give a fixed focal length of 8 cm, positioned directly above the heating block and fluorescent excitation is achieved using four 1 mW UV LEDs wired in parallel, running off a 9 Volt battery. LEDs are set at a 45 degree angle to the plane of the heater to illuminate the samples. The data acquisition system runs on a Raspberry Pi B model, with a Pi camera attached. The fluorescence is recorded using a python coded time delay function and stored on a SD drive for later analysis in Image J. All components, optical and electronic, were mounted within a Perspex light excluding box. At the time of writing, this is the first fluorescence real time device to be implemented using a Raspberry Pi system.3. Results
Prior to implementing the cool down step after the initial 96 °C denaturation, there was no amplification. This prompted a reconsideration of protocol and the addition of a cooling step to prevent denaturation of the Bst 2.0 polymerase enzyme. Two different polymerases were tested; Bst polymerase and Bst 2.0 polymerase. Fig. 2A shows an unsuccessful LAMP assay demonstrating no amplification. Fig. 2B shows a successful LAMP assay with amplification. | ||
| Fig. 2 (A) Unsuccessful LAMP assay with no cooling step after initial denaturation at 96 °C. (B) Successful LAMP assay with cooling step after initial denaturation at 96 °C. Lanes Mar: DNA ladder (0.1–10.0 kb), lane 1: empty, lane 2: negative control with Bst polymerase, lane 3: positive control (WT E. coli) with Bst polymerase, lane 4: sample with Bst polymerase, lane 5: empty, lane 6: negative control with Bst 2.0 polymerase, lane 7: positive control (WT E. coli) with Bst 2.0 polymerase, lane 8: sample with Bst 2.0 polymerase. (C) LAMP assay with primers for Wild-type E. coli on the left and primers for 987P gene on the right. (D) LAMP assay with primers for LT gene on the left and primers for STx gene on the right. (E) LAMP assay with primers for etpD gene on the left and primers for bfpA gene on the right. (F) LAMP assay with primers for STp/STh on the left and primers for F41 gene on the right. (G) Microtiter plate with LAMP assay results. Genes are in the same order as in C–F. i.e. fifth row from the top corresponds to etpD gene that is absent in E. (H) Processed microtiter plater results demonstrating relative amounts of gene present in each sample. | ||
The initial E. coli genotyping was done with primers for the 987P, LT, STx, etpD, bfpA, STp/STh, and F41 genes along with primers recognizing wild-type E. coli. The samples tested came from swabs taken from around the lab: #1 was the elevator button, #2 was a vending machine keypad, #3 was the doorknob inside a nearby restroom, and #4 was from the lunch room table. Swabs were immersed in sterile water, which was used as template in the LAMP reaction. From the gel photos (Fig. 2C–E) it was possible to determine that microbes harboring all but the etpD gene were present in the environment, and even contaminated the negative control. From this data, it was concluded that the samples used were not EHEC since the etpD gene that codes for a secretion pathway protein in this serogroup was absent. Also, the sample from the bathroom doorknob did not contain EIEC since the F41 gene was absent.
To demonstrate the efficiency of the LAMP reaction for sequence amplification, a 5 μL volume of each reaction tube from the assay was aliquotted onto a 60-well microtiter plate along with a 1 μL volume of PicoGreen. The same results from the gel were observed on the microtiter plate (Fig. 2G) proving that only a very small volume of reaction mixture is necessary to visualize results. These images were processed to show relative amount of the target sequences present in a sample. These results have the added convenience that they can be visualized by the naked eye, similar to the case in Fig. 3A and B. Positive wells will be a bright green, whereas wells with no DNA amplification will remain a dull orange. The LAMP assay was also attempted in wells measuring 100 μm wide and 50 μm deep. Visualization on a UV transilluminator showed that wells where DNA amplification had taken place were distinguishable from those where no amplification occurred. This showed that only a very small volume is necessary for successful DNA amplification. The use of a smaller volume format illustrates the compatibility of small volume fluorescent based LAMP for microfluidic chip level diagnostics.
 | ||
| Fig. 3 Naked eye visualization of contrast between negative (−) and positive (+) samples under (A) ambient light, and (B) ultraviolet light. (C) Tubes containing negative control (top), positive control WT E. coli (middle), and swab taken from freezer handle (bottom), along with the reaction mixture (left) as a blank. (D) Data table for fluorescence of controls, sample, and blank. (E) Graph of normalized fluorescence of negative control, positive control, and a swab taken from the freezer handle (mean ± SEM, n = 4). | ||
Results can be obtained colorimetrically with the naked eye in the presence of PicoGreen. Negative results will appear as a dull orange color while samples that underwent amplification will appear a light green (Fig. 3A). Exposure to ultraviolet light intensifies the contrast between samples that have and have not undergone amplification (Fig. 3B). This bright green coloration was due to the addition of PicoGreen that fluoresces in the presence of double stranded DNA, indicating a successful amplification. The LAMP reaction can be used to amplify DNA, and to quantify it. The higher the initial concentration of template DNA in the assay, the higher the fluorescence will be after the reaction is completed. Analysis of relative fluorescence intensity is used to estimate amount of DNA present in the solution. The principle of thresholding can be described as analogous to real time PCR where CT value is dependent on amount of initial template DNA present in a heterogenous mix of bacterial sample. This allows relative estimation of which bacterial serogroup is present, and in what ratio based on the amount of amplified DNA.
The relative fluorescence of resulting LAMP reactions was measured using a UV transilluminator with an exposure time of 40 ms. This instrument recorded the fluorescence of tubes containing negative controls, positive controls (WT E. coli) and the results of a successful LAMP reaction as depicted in Fig. 3. The bottom set of tubes in Fig. 3C clearly shows how a sample for which the LAMP primers are specific for fluoresces much more intensely than the positive control. Fig. 3E illustrates this point graphically with the first bar corresponding to a negative control, the second bar to a positive control, and the final bar to a sample swabbed from the freezer handle that contained DNA for which the LAMP primers used were specific for. The blue line represents the threshold where fluorescence above that point indicates DNA amplification.
Lower cost reagents suitable to resource-limited environments are always required. They reduce overall cost and instrument specifications. Colorimetric analysis of test tubes containing 650 nm resonant gold nanoparticles showed a shift in peak intensity compared to blank and control test tubes. This detection was done using a camera of similar quality to those found in cell phones. This shows how this method of recognition does not require expensive instrumentation.
The significant color shifts from the control and blank tubes meant that the dielectric constant of the medium that the nanorods were immersed in (the reaction mixture) was altered over the course of the reaction. The change in the solution's dielectric constant came from the DNA amplification that took place due to the LAMP reaction. This DNA amplification changed the refractive index of the reaction mixture leading to a color shift in the nanoparticles.
4. Discussion
The LAMP reaction was successful at amplifying specific regions of the target DNA and thus indicated the presence of a particular strain of microorganism. A contamination of environmental E. coli in the lab setting caused most of the negative controls to show some amplification, however this problem was overcome by realizing that this contamination only caused a very small amount of amplification, as the primers were not specific for the contaminant's DNA. This issue was overcome by implementing a statistical thresholding system where only values exceeding 3191.05 on the relative fluorescence scale for images taken with a 40 ms exposure time were considered to be a result of specific DNA amplification. Fig. 4A and B show how a positive amplification of DNA for which the LAMP primers are specific for fluoresce more intensely than the positive control that contained DNA for which the LAMP primers were not specific.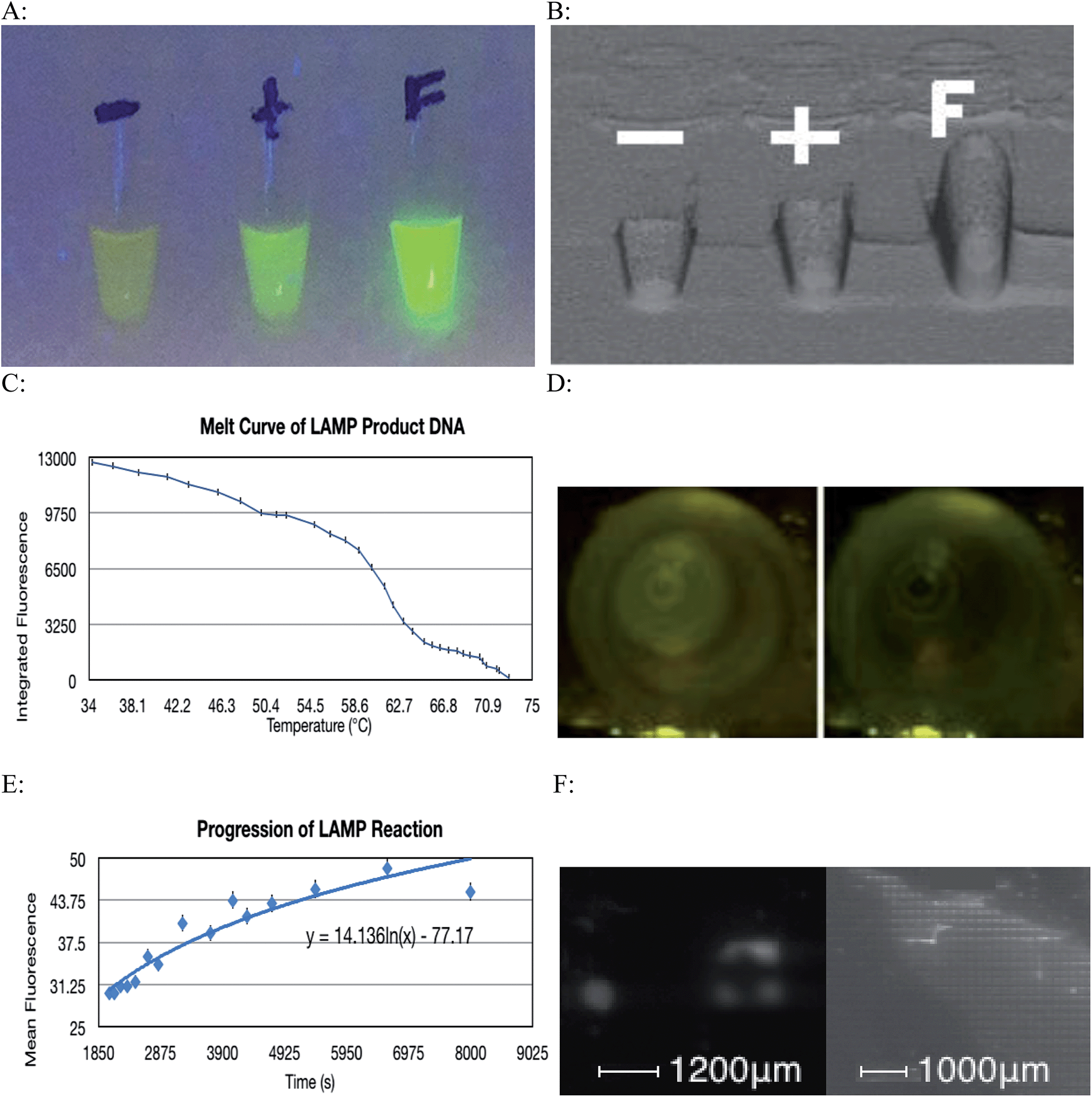 | ||
| Fig. 4 (A) Ultraviolet visualization of results including negative and positive controls. (B) Comparison of fluorescence intensity between control tubes and positive sample. (C) Melt curve of DNA product of LAMP reaction (mean ± SEM, n = 3). (D) Comparison of fluorescence at 34.31 °C (first frame) and 72.86 °C (second frame). (E) Fluorescence as a function of progression of LAMP reaction (mean ± SEM, n = 3). (F) Side by side comparison of successful LAMP assays done in 400 μm and 100 μm wells respectively. Wells that showed amplification are brighter than those that showed no amplification like the ones found in the upper right region of the photo. | ||
Environmental bacteria could have been introduced to the reaction through contaminated micro-tubes, pipette tips or reagents. Also, repeated amplification of the same target sequence for which the LAMP primers are specific for, leads to accumulation of amplification products in the laboratory. Over the course of the experiment, aerosolized amplification products will eventually build up to a level that will contaminate ventilation systems, laboratory reagents, and laboratory equipment.18 The Bst 2.0 polymerase sourced from New England BioLabs is purified from E. coli cells. It is possible that a small amount of bacterial nucleic acids were co-purified with the enzyme, eventually becoming a contaminant. To minimize this type of contamination, all plasticware should be treated with ultraviolet radiation to cross-link any contaminant DNA thus inhibiting it from being replicated. UV light creates pyrimidine dimers and other covalent modifications on the contaminating DNA and thereby inactivates it as a template for future amplification. Oligonucleotide primers should not be exposed to UV radiation as they will experience cross-linking and be rendered ineffective. There is also evidence that polymerase exposure to UV radiation will have deleterious effects on its function.19 Making a new dilution from the stock primer solution also eliminates contamination found in primer solutions that have been used multiple times. Finally, working in a PCR hood, and wiping gloves, and tube rack with a surface decontaminant further reduces the probability of introducing a contamination to the reaction. Using a 10% sodium hypochlorite solution followed by 70% ethanol to wipe work area and equipment has been shown to be effective at removing nucleic acids. The use of sodium hypochlorite causes oxidative damage to nucleic acids thus preventing it from amplification in subsequent reactions.20
Genotyping of infectious bacteria from samples taken from patients could lead to suitable and informative identification of the bacteria. This identification could be used to specifically target that strain of bacteria and thus improve treatment efficacy by using narrow spectrum rather than broad-spectrum antibiotics. This is a preferred treatment since the medication will not target as much of the body's natural flora and prevent the risk of superinfection. Also, the narrow spectrum antibiotic will limit selection for antibiotic tolerant bacteria, as would be the case with broad-spectrum antibiotics. The most obvious benefit the LAMP reaction has over other diagnostic tools such as PCR is the fact that it is carried out under isothermal conditions. This is highly beneficial in areas that lack the means to purchase expensive thermocyclers such as developing nations. The reaction can be carried out in a water bath as long as the reaction temperature of 66 °C is maintained. The use of PicoGreen also means that visualization of results can be done without instrumentation such as a transilluminator or gel electrophoresis machine. Using gold nanoparticles to detect DNA amplification was possible however, it had a much poorer signal to noise ratio than the fluorescent method as illustrated in Table 2.
| Method | Mean signal | Noise | SNR |
|---|---|---|---|
| Plasmonics | Red: 166.02 | 152.04 | 1.09 |
| Green: 177.52 | 171.06 | 1.03 | |
| Blue: 168.13 | 160.1 | 1.05 | |
| Fluorescence | 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 431.5 431.5 |
4713.75 | 19.82 |
The LAMP reaction is extremely versatile. Although the main focus of this experiment was pathogenic E. coli, the primers used in the reaction can be designed to bind to the DNA of any organism or virus. Areas of high mutation should be avoided to ensure primer complementarity, and prevent false negatives, though with careful design point mutations could be identified also. It is even possible to detect subspecies of particular microorganisms.
The fact that only a very small volume is required for the LAMP reaction to occur means that the whole process can be miniaturized. Since a 100 μm well can successfully accommodate amplification (Fig. 4F), many genes can be tested on a very small area, as each well of the plate will pertain to a specific gene. This can be combined with a means of maintaining constant temperature, along with a camera and a microprocessor that can distinguish successful amplification from failed amplification to create a diagnostic instrument. This tool would be capable of analyzing multiple wells and determining the identity of a particular pathogenic organism by its genotype.
Images used to generate the melt curve (Fig. 4C) were analyzed using ImageJ software. Although the change in fluorescence in consecutive images is difficult to visualize with the naked eye, comparing the first and final images used shows a considerable change in fluorescence of 12581 relative units (Fig. 4D). Baseline fluorescence was determined to be 9782.86. This figure corresponds to three standard deviations above the fluorescence detected in the Blank tube (9632.67). Integrated fluorescence was calculated by subtracting baseline fluorescence from computed fluorescence.
The progression of the LAMP reaction could be seen when the fluorescence data was graphed (Fig. 4E). As before the subtle visual differences in consecutive images are difficult to discern with the naked eye however with image analysis, the pattern of increased fluorescence over time is demonstrated. The cyclic nature of the fluorescence is due to the slight fluctuations in temperature. These slight deviations from optimal reaction temperature caused some DNA denaturation. The fluorophore used binds to dsDNA and so the denaturation resulted a transient loss of fluorescence.
The principle of microfluidics can be applied to the LAMP reaction, and could potentially lead to fewer incidences of contamination. Having LAMP primers lyophilized inside a lab-on-a-chip (LOC) device, and using fresh reagents would decrease the probability of environmental contamination. The in vitro design of the assay implies a system to which contamination is decreased due to a physical barrier from the environment, preventing any aerosolized nucleic acids from contacting the reagents. LOC's also require less reagent volumes thus lowering the cost per assay. This principle is also extremely convenient for point-of-care testing, where LOC's containing miniaturized assays for the detection of particular pathogens can be used by healthcare professionals in poorly equipped settings to perform molecular diagnostic tests.21 The low cost associated with LOC's means that healthcare providers can use and dispose of them at the patient's bedside. Including the time required for preparation, results are obtained in under 2 hours. This is obviously very useful in an emergency room setting where cutting down time between infection and treatment is paramount. This is also very valuable in the food industry, where early detection of tainted meat, poultry, or produce would prevent the transmission of infection and limit expensive recalls. All together the LAMP reaction has proven to be a reliable source for DNA amplification. Its added strengths of speed, specificity, and instrument simplicity make the LAMP reaction a dependable and efficient method for molecular diagnostics. Current efforts are ongoing towards an intra-serogroup identification approach for STEC as this is the most threatening class to human health and agribusiness profitability.
References
- D. A. Rasko, D. R. Webster, J. W. Sahl, A. Bashir, N. Boisen, F. Scheutz, E. E. Paxinos, R. Sebra, C. S. Chin, D. Iliopoulos, A. Klammer, P. Peluso, L. Lee, A. O. Kislyuk, J. Bullard, A. Kasarskis, S. Wang, J. Eid, D. Rank, J. C. Redman, S. R. Steyert, J. Frimodt-Møller, C. Struve, A. M. Petersen, K. A. Krogfelt, J. P. Nataro, E. E. Schadt and M. K. Waldor, N. Engl. J. Med., 2011, 365, 709–717 CrossRef CAS PubMed.
- E. B. Solomon, S. Yaron and K. R. Matthews, Appl. Environ. Microbiol., 2002, 68, 397–400 CrossRef CAS.
- M. T. Osterholm, N. Engl. J. Med., 2011, 364, 889–891 CrossRef CAS PubMed.
- J. P. Nataro and J. B. Kaper, Clin. Microbiol. Rev., 1998, 11, 142–201 CAS.
- J. P. Nataro, T. Steiner and R. L. Guerrant, Emerging Infect. Dis., 1998, 4, 251–261 CrossRef CAS PubMed.
- J. P. Nataro, Y. Deng, D. R. Maneval, A. L. German, W. C. Martin and M. M. Levine, Infect. Immun., 1992, 60, 2297–2304 CAS.
- J. M. Wilkes and J. A. DiPalma, Pract. Gastroenterol., 2002, 26, 14–23 Search PubMed.
- S. C. Clarke, R. D. Haigh, P. P. Freestone and P. H. Williams, Clin. Microbiol. Rev., 2003, 16, 365–378 CrossRef CAS.
- I. C. Scaletsky, M. L. Silva and L. R. Trabulsi, Infect. Immun., 1984, 45, 534–536 CAS.
- J. A. Girón, T. Jones, F. Millán-Velasco, E. Castro-Munoz, L. Zarate, J. Fry, G. Frankel, S. L. Moseley, B. Baudry, J. B. Kaper, G. K. Schoolnik and L. W. Riley, J. Infect. Dis., 1991, 163, 507–513 CrossRef PubMed.
- M. J. Brian, M. Frosolono, B. E. Murray, A. Miranda, E. L. Lopez, H. F. Gomez and T. G. Cleary, J. Clin. Microbiol., 1992, 30, 1801–1806 CAS.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, H. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- R. A. Schneider and S. C. To, Infect. Immun., 1982, 36, 417–418 CAS.
- K. Ogata, R. Kato and S. Yamada, Jpn. J. Infect. Dis., 2002, 55, 14–18 CAS.
- D. Orth, K. Grif, M. P. Dierich and R. Würzner, Epidemiol. Infect., 2006, 134, 719–723 CrossRef CAS PubMed.
- Y. Mori, K. Nagamine, N. Tomita and T. Notomi, Biochem. Biophys. Res. Commun., 2001, 289, 150–154 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- D. H. Persing, J. Clin. Microbiol., 1991, 29, 1281–1285 CAS.
- C. C. Pao, J. J. Hor, P. L. Tsai and M. Y. Horng, Mol. Cell. Probes, 1993, 7, 217–219 CrossRef CAS PubMed.
- H. Hayatsu, S. Pan and T. Ukita, Chem. Pharm. Bull., 1971, 19, 2189–2192 CrossRef CAS.
- X. Fang, Y. Liu, J. Kong and X. Jiang, Anal. Chem., 2010, 82, 3002–3006 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |