DOI:
10.1039/C4AY02461C
(Paper)
Anal. Methods, 2015,
7, 161-167
A new colorimetric protocol for selective detection of phosphate based on the inhibition of peroxidase-like activity of magnetite nanoparticles†
Received
15th October 2014
, Accepted 5th November 2014
First published on 11th November 2014
Abstract
A simple colorimetric assay for phosphate ion (Pi) has been established based on analyte-induced inhibition of the magnetite nanoparticles (MNPs)-catalyzed oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) in the presence of H2O2. The Fe3O4 MNPs can catalyze the H2O2-mediated oxidation of TMB and yields a blue oxidized product, which exhibits a maximum absorption at 652 nm. Pi could be absorbed on the surface of the Fe3O4 MNPs through coordinating with Fe3+, inducing a reduced colorimetric signal. The colorimetric signal change (ΔA652) in this process was proportional to the concentration of Pi, ranging from 0.2 μM to 200 μM. The limit of detection (S/N = 3) was as low as 0.11 μM. The as-proposed Fe3O4 MNPs–TMB–H2O2 probe exhibited a high selectivity toward Pi over other relevant ions that commonly exist in water and has been applied to Pi detection in drinking water, ground water and lake water samples with satisfactory results.
1. Introduction
Anions are ubiquitous species and play numerous indispensable roles in chemical and biological processes, as well as in environmental pollution,1 thus considerable efforts have been devoted for their simple, rapid and sensitive sensing and detection.2–6 Phosphate (Pi), as an essential component of the nutritional chain of aquatic microorganisms, is a convenient indicator or tracer of pollution in bodies of water.7–10 Phosphate is a well-known contaminant of ground water and stream water. A maximum permissible concentration of phosphate is 0.32 μM for river water and 14.3–143 μM for wastewater.11 The presence of phosphate in drinking water is a health hazard. Furthermore, phosphate concentration in body fluids is an important indicator in the diagnosis of hyperparathyroidism, vitamin D deficiency, and Fanconi syndrome.12 Consequently, Pi detection is of great significance for controlling eutrophication, monitoring drinking water quality and clinical diagnosis. Up to now, several strategies have been established for the detection of phosphate ion, including electrochemical analysis,11,13 chromatography,14–16 spectrophotometry,17 fluorometry,18 and enzymatic biosensors.19,20 However, these methods are usually challenged with sophisticated instruments, tedious experimental procedures, skilled personnel, or high expense. Thus, it is quite necessary to further develop simple, cost-effective and reliable methods for rapid and sensitive determination of Pi in both environmental and biological systems.
Spectrographic technique is simple and can be read out by the naked eye without the aid of sophisticated instruments, thus realizing the visual on-site analysis.21 Moreover, colorimetry by nanomaterials has been exploited for simple and cost-effective sensing of anions owing to their unique chemical, electrical, optical and catalytic properties. Colorimetric methods utilizing nanomaterials for Pi detection have attracted significant attention. For example, Y. Kubo et al. attached isothiouronium groups onto AuNP surface and demonstrated the sensing of oxanions (AcO−, HPO42−, and malonate) in aqueous MeOH solution.22 W. Q. Liu et al. have employed MA-AuNPs as a colorimetric sensing platform for phosphate based on the competition reaction between Pi and carboxylate group-modified AuNPs for Eu3+ ions.23 Nevertheless, those strategies are still not well developed. The use of water-insoluble isothiouronium and the laborious premodification of the AuNPs with sulfhydryl compounds limited their application. To design a novel probe for Pi detection, new types of nanomaterials have been exploited24,25 and the properties, except optical properties, of nanomaterials, such as catalytic activity, may have potential applications in anion detection.
Recently, a landmark work by Yan et al. reported that Fe3O4 magnetic nanoparticles, usually thought to be biologically and chemically inert, possess an intrinsic enzyme mimic activity similar to that found in natural peroxidases.26 In comparison with horseradish peroxidase (HRP),27 Fe3O4 MNPs are low-cost, easy to obtain, more robust, and easy to be separated from the product for recycling.28 These advantages indicate that Fe3O4 MNPs can be powerful tools for potential applications in medicine, biotechnology and environmental chemistry. By employing MNPs as peroxidase mimetics, several novel strategies have been developed for the detection of various chemical and biological entities, such as glucose,29,30 H2O2,29 choline,31 catecholamines,32 melamine,33 nucleic acid,34,35 cells36 and proteins.37,38 However, to date, there is no report on any MNPs used for designing the colorimetric sensing platform for inorganic anions.
Herein, we have developed a novel and convenient label-free colorimetric method for Pi detection, which employed the peroxidase-mimicking activity of MNPs. Fe3O4 MNPs can catalyze the H2O2-medicated oxidation of colorless 3,3′,5,5′-tetramethylbenzidine (TMB) into a blue product (oxTMB), which exhibits a maximum absorption at 652 nm (A652). However, the catalytic activity of Fe3O4 MNPs can be inhibited by Pi due to the coordination effect between Pi and Fe3+ on the surface of Fe3O4 MNPs. The colorimetric signal change in this process can be used for detection of the concentration of Pi. Based on this principle, we have achieved the detection of Pi in different samples with high selectivity and sensitivity. Thus, it is believable that the proposed sensing strategy for Pi will show great prospect in environmental monitoring, clinical diagnostics and scientific research.
2. Experimental
2.1. Reagents and apparatus
Ferric chloride was purchased from Sangon Biotech Co., Ltd. (Shanghai, China). TMB was purchased from Sigma-Aldrich (USA). Analytical grade ferrous sulfate, H2O2, sodium salts of anion (PO43−, H2PO2−, S2−, SO42−, SO32−, F−, Cl−, Br−, BrO3−, SCN−, NO3−, C2O42−), chloride salts of ion (Na+, K+, Al3+, Ba2+, Cu2+, Co2+, Cr3+, Ni2+, Zn2+), Ca(NO3)2, MgSO4, and PbC2O4 were obtained from Beijing Chemical Reagent Co. (China). All reagents were used as received. Acetate buffer solutions with different pH values were prepared by varying the ratio of 0.2 M acetate acid to 0.2 M sodium acetate and diluted to the concentration demanded. The water used throughout the experiment was purified by a Milli-Q water system (resistivity 18.25 MΩ cm, Millipore, ultrapure water).
UV-vis absorption spectra were recorded with a Cary 50 UV-vis spectrophotometer (Varian). High-resolution transmission electronic microscopy (HRTEM) images were obtained using a Tecnai G2 F20 (FEI) transmission electron microscope operated at 200 kV. The sample for TEM characterization was prepared by evaporating a droplet of dilute solution onto double networking carbon-coated copper grids. Images were taken with a commercial digital camera. The X-ray photoelectron spectroscopy (XPS) samples on highly cleaned silicon wafers were analyzed by an ESCALAB MK II spectrometer (VG Scientific) with Al Kα radiation as the X-ray source. Peak positions were internally referenced to the C1s peak at 284.6 eV. X-ray powder diffraction (XRD) study was carried out using Bruker D8 Advance (Bruker AXS, Germany) with a graphite monochromator using Cu Kα radiation.
2.2. Synthesis of Fe3O4 MNPs
Fe3O4 MNPs were synthesized according to Massart's method.39 Under vigorous stirring, 5 mL freshly prepared aqueous mixture of ferric chloride (4 mL, 1 M in 2 M HCl) and ferrous sulfate (1 mL, 2 M in 2 M HCl) was quickly added to ammonia solution (50 mL, 0.7 M). The mixture was stirred continuously for another 30 min at room temperature, and then, the formed black precipitate was collected on the vessel wall by an external permanent magnet, rinsed with ultrapure water four times. The obtained product was then re-dispersed in ultrapure water and subjected to ultrasound for 30 min to make the final concentration ∼4.4 mg mL−1 (referred to as Fe3O4 MNPs stock solution).
2.3. Detection of phosphate anions using Fe3O4 MNPs
5 μL of 1.1 mg mL−1 Fe3O4 MNPs colloidal solution and 20 μL of phosphate solutions with different concentrations were added into 147 μL of 0.2 M acetate buffer (pH 4.0). The mixture was vortexed thoroughly and incubated at ambient temperature for 5 min. Subsequently, 20 μL of 5 mM TMB and 8 μL of 20 mM H2O2 were added into the solution. The mixture was vortex-mixed and incubated in a water bath at 50 °C for 20 min. The final solution was used for absorbance measurement.
2.4. Analysis of real samples
The drinking water and ground water samples were used without further treatment, and the lake water sample was filtered through a 0.22 μm membrane. All the samples were detected according to the following procedure with three replicates: 5 μL of 1.1 mg mL−1 Fe3O4 MNPs colloidal solution and 20 μL real samples were added into 147 μL of 0.2 M acetate buffer (pH 4.0), and then, the mixture was vortexed thoroughly and incubated at ambient temperature for 5 min. Subsequently, 20 μL of 5 mM TMB and 8 μL of 20 mM H2O2 were added into the solution. The mixture was vortex-mixed and incubated at 50 °C in a water bath for 20 min. The final solution was used for absorbance measurement.
3. Results and discussion
3.1. Mechanism of the Pi detection
The formation of Fe3O4 MNPs was confirmed by HRTEM, XPS and XRD characterization. HRTEM images of Fe3O4 MNPs (Fig. S1A, ESI†) show that the diameter of the prepared Fe3O4 MNPs distributed in the range of 6–10 nm and are roughly spherical in shape. The binding energies of Fe 2p3/2 and Fe 2p1/2 for the Fe3O4 MNPs sample were 710.6 and 724.1 eV; simultaneously, 2p3/2 for Fe3O4 did not have a satellite peak (Fig. S1C, ESI†). The XRD pattern shows signals at 2θ = 30°, 35°, 37°, 42°, 56.8, 62.5° (220, 311, 222, 400, 511, 440) (Fig. S1D, ESI†). These diffraction peaks could be characterized as the face-centered cubic phase of Fe3O4, which is consistent with the value reported in a previously studied report (JCPDS file no. 19-629). The HRTEM, XPS and XRD spectra revealed the formation of Fe3O4 MNPs. The aqueous solution of the MNPs was black in color and had no remarkable absorption peak in the wavelength range of 400–800 nm. Although no appreciable change in size or dispersion state was observed using HRTEM (Fig. S1A and B, ESI†), Pi had some enhancement effect on the absorption curve of Fe3O4 MNPs (Fig. S2, ESI†), which preliminarily demonstrated that a binding event occurred between Pi and Fe3+.
Pi can coordinate effectively with Fe3+ on the surface of MNPs based on the fact that Pi has high binding constants with Fe3+.40,41 The coordination of Pi with Fe3+ may affect the surface properties and catalytic ability of Fe3O4 MNPs. Scheme 1 illustrates the basic principle of Pi detection based on the Fe3O4 MNPs–TMB–H2O2 system. In the absence of Pi, Fe3O4 MNPs in the solution maintain their normal peroxidase-mimicking activity, which can catalyze the oxidation of TMB by H2O2, giving a blue oxidation product (oxTMB). However, when Pi is added, Pi will adsorb onto the surface of Fe3O4 MNPs through coordinating with Fe3+. The complexes formed effectively keep the substrate away from the MNPs, which is required for the peroxidase-like reaction. In this manner, Pi inhibits the catalytic activity of Fe3O4 MNPs, thus leading to significantly reduced colorimetric signal. The decrease of the catalytic activity of Fe3O4 MNPs is directly dependent on the concentration of Pi. In this case, the absorption intensity of oxTMB at 652 nm decreases with increase in the concentration of Pi. Therefore, the Fe3O4 MNPs–TMB–H2O2 system can serve as a colorimetric probe for the quantitative detection of Pi.
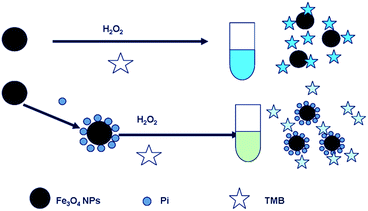 |
| | Scheme 1 Pi detection based on the Fe3O4 MNPs–TMB–H2O2 system. | |
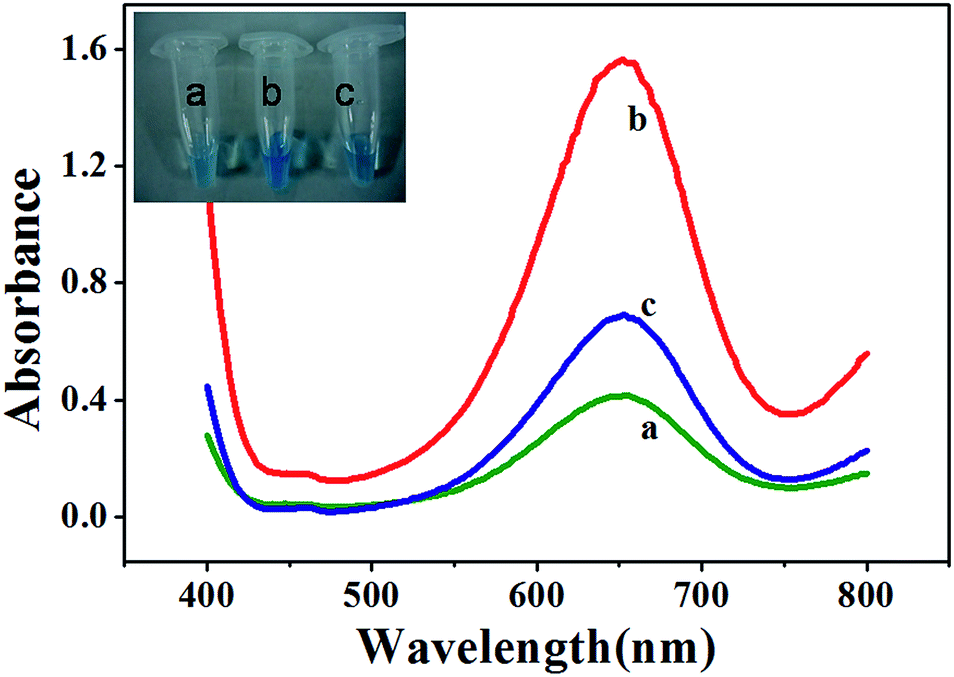
To demonstrate these hypotheses, we monitored Fe3O4 MNPs-catalyzed oxidation of TMB in the absence and presence of Pi. Curve a in Fig. 1 shows that the mixture of 2.5 mM TMB and 4.0 mM H2O2 in 0.2 M NaAc–HAc buffer (pH 4.0) produced a weak absorption at 652 nm (A652). The addition of 27.5 μg mL−1 MNPs to the abovementioned mixture produced a considerably stronger A652 (curve b in Fig. 1), indicating that Fe3O4 MNPs unambiguously catalyzed the oxidation reaction of the substrate TMB with H2O2. However, when 100 μM Pi was present in a solution containing MNPs, the addition of the same concentration of TMB and H2O2 resulted in weaker A652 (curve c in Fig. 1). This could be attributed to the ability of Pi to coordinate on the surface of Fe3O4 MNPs, thereby inhibiting the catalytic activity of Fe3O4 MNPs. From all the abovementioned results, it can be concluded that Fe3O4 MNPs–TMB–H2O2 system is suitable for the sensitive determination of Pi.
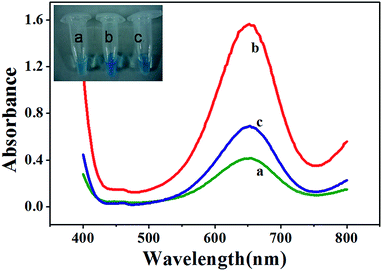 |
| | Fig. 1 UV-vis absorption spectra for reaction solutions of TMB–H2O2 (a), Fe3O4 MNPs–TMB–H2O2 (b) and Fe3O4 MNPs–TMB–H2O2–Pi (c). MNPs: 27.5 μg mL−1, TMB: 2.5 mM, H2O2: 4.0 mM, Pi: 100 μM, and HAc–NaAc buffer solution (0.2 M, pH 4.0). The insets are the images showing respective colorimetric responses. | |
3.2. Optimization of experimental conditions
To find the optimum experimental conditions, the effects of concentration of Fe3O4 MNPs, the pH of the reaction buffer, the incubation temperature and the reaction time were studied in detail.
3.2.1. Effect of concentration of Fe3O4 MNPs.
The concentration of Fe3O4 MNPs is a key factor for this sensing system. In order to choose a suitable concentration of MNPs, the relationship between A652 and the concentration of MNPs was examined. Fig. 2A shows the absorption spectra of the TMB–H2O2 system containing various concentrations of MNPs. As shown in Fig. 2B, the A652 value was directly proportional to the concentration of MNPs ranging from 2.75 to 27.5 μg mL−1. The experimental results revealed that the higher the concentration of Fe3O4 MNPs, the higher the catalytic efficiency of the reaction, and thus, the Fe3O4 MNPs–TMB–H2O2 probe was more sensitive to Pi. While when the concentration of MNPs was higher than 27.5 μg mL−1, A652 almost remained unchanged. Considering these effects, 27.5 μg mL−1 Fe3O4 MNPs solution was selected for the subsequent study in this sensing system.
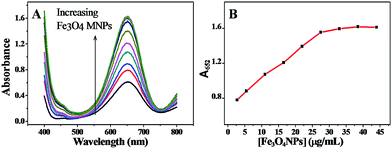 |
| | Fig. 2 (A) UV-vis absorption spectra of the TMB–H2O2 system containing various concentrations of Fe3O4 MNPs. (B) The linear dependence of A652 on the Fe3O4 MNPs concentration. TMB: 2.5 mM, H2O2: 4.0 mM, and HAc–NaAc buffer solution (0.2 M, pH 4.0). | |
3.2.2. Effect of pH.
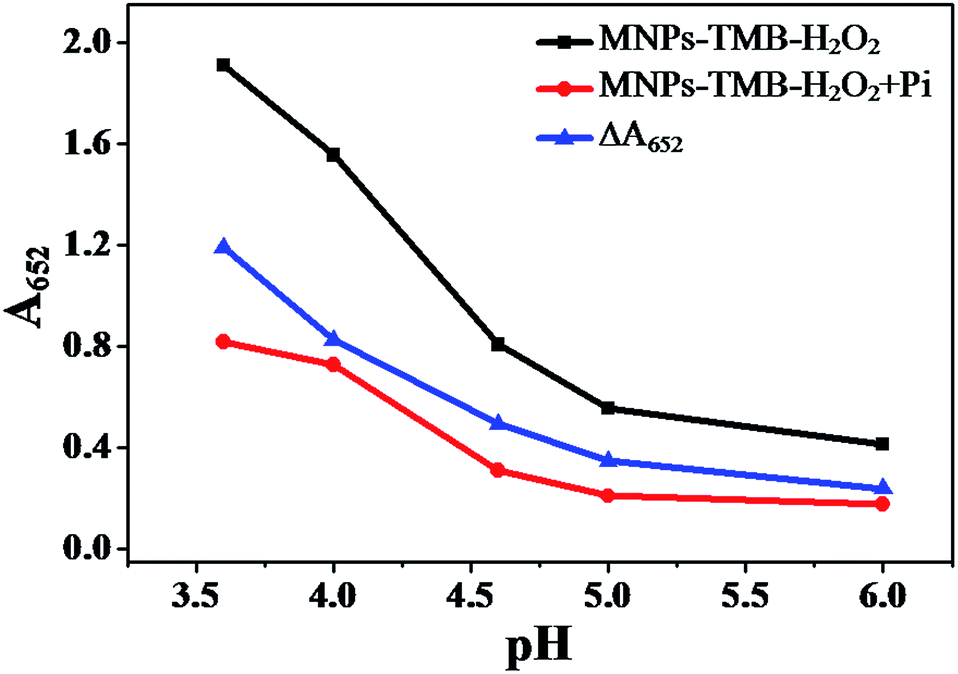
Another driving factor for the as-proposed sensing platform is the buffer pH value as it has effects on the ΔA652 (ΔA652 = A0652 − A652) and the selectivity of the probe toward Pi. A0652 and A652 correspond to the absorbance intensity at 652 nm of the MNPs–TMB–H2O2 probe in the absence and presence of Pi, respectively. The isoelectric point of Fe3O4 MNPs is about 6.8 (Fig. S3, ESI†), which is in accordance with the value reported previously (6.5–6.8).42,43 MNPs are positively charged below pH 6.8 and negatively charged as pH value exceeds 6.8. The high positive value of the zeta potential might mean higher catalytic ability44 and easy adsorption of Pi on the surface of nanoparticles. Therefore, we examined the influence of pH values below 6.8. The relationship between the catalytic ability of the Fe3O4 MNPs and the buffer pH values is shown in Fig. 3. The absorption intensity at 652 nm of Fe3O4 MNPs–TMB–H2O2 and Fe3O4 MNPs–TMB–H2O2–Pi systems both decreased as the pH value increased. This demonstrated that Fe3O4 MNPs exhibited better catalytic ability at relatively lower buffer pH values. In addition, ΔA652 decreased gradually with the increasing value of pH, as shown in Fig. 3. Thus, a lower pH value was preferable to get a higher ΔA652. Nevertheless, S2− would have a significant enhancement effect on this system when buffer pH value is too low (Fig. S4, ESI†), which might be attributed to the formation of the FeS/H2S system.45 Accordingly, to avoid this interference, pH 4.0 was chosen for the following measurement in order to achieve a better sensing signal and selectivity.
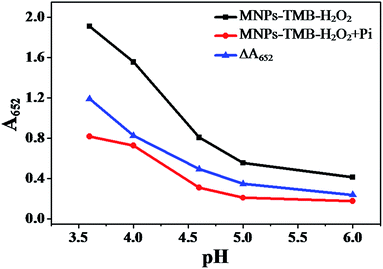 |
| | Fig. 3 Plot of A652 of Fe3O4 MNPs–TMB–H2O2, Fe3O4 MNPs–TMB–H2O2 + Pi system and ΔA652versus pH (0.2 M NaAc–HAc buffer with different pH). MNPs: 27.5 μg mL−1, TMB: 2.5 mM, H2O2: 4.0 mM, and Pi: 100 μM. | |
3.2.3. Effect of the temperature and reaction time.
The effect of the temperature on ΔA652 was investigated and the results are shown in Fig. 4. From Fig. 4, we can see that although A652 values of Fe3O4 MNPs–TMB–H2O2 and Fe3O4 MNPs–TMB–H2O2–Pi systems gradually increased with the increase of temperature in the range of 20–40 °C, ΔA value slightly changed. When the temperature increased from 40 to 60 °C, ΔA652 first increased and then decreased, giving a maximum value at 50 °C. Thus, 50 °C was taken as the optimal reaction temperature.
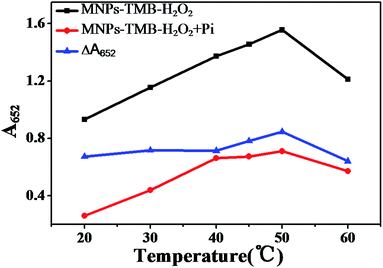 |
| | Fig. 4 Plot of A652 of Fe3O4 MNPs–TMB–H2O2 system and Fe3O4 MNPs–TMB–H2O2 + Pi system versus temperature and ΔA652versus temperature. MNPs: 27.5 μg mL−1, TMB: 2.5 mM, H2O2: 4.0 mM, Pi: 100 μM, and HAc–NaAc buffer solution (0.2 M, pH 4.0). | |
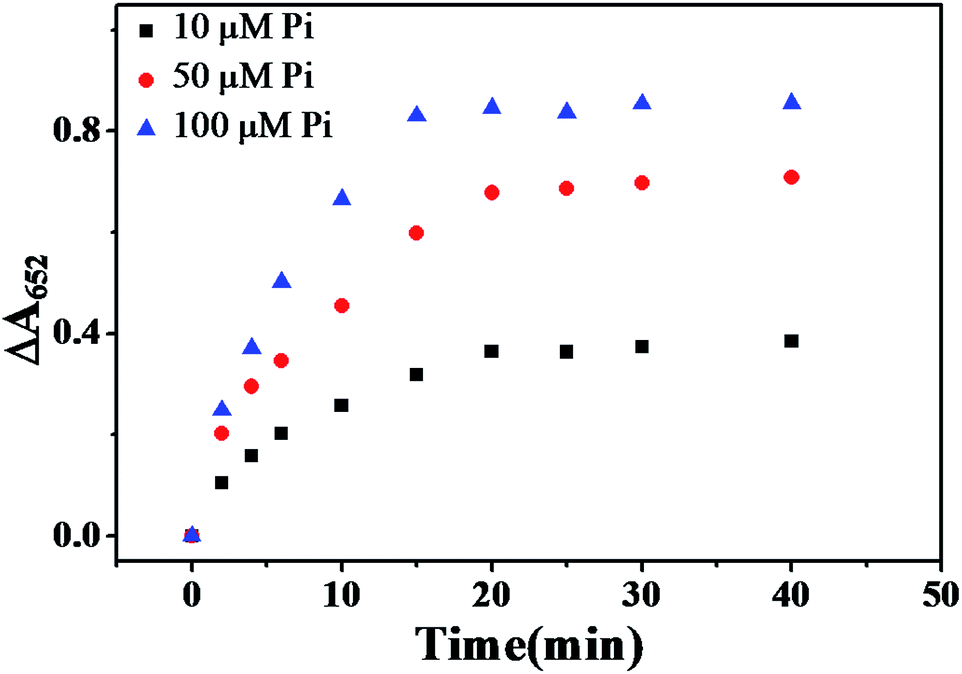
The influence of the reaction time for catalytic oxidation of TMB was also evaluated. Pi and Fe3O4 MNPs were pre-incubated for 5 min to ensure a completed coordination between the Pi and Fe3+ (Fig. S5, ESI†) before the catalytic reaction was carried out. As described in Fig. 5, ΔA652 gradually increased with increased catalytic reaction time, and reached the maximum when reaction time was 20 min, then leveled off. Therefore, 20 min was adopted as the optimal catalytic reaction time.
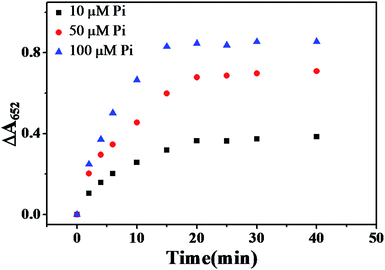 |
| | Fig. 5 Plot of ΔA652versus time of catalytic reaction under various concentrations of Pi at 50 °C. MNPs: 27.5 μg mL−1, TMB: 2.5 mM, H2O2: 4.0 mM, HAc–NaAc buffer solution (0.2 M, pH 4.0). | |
3.2.4. Interference study.
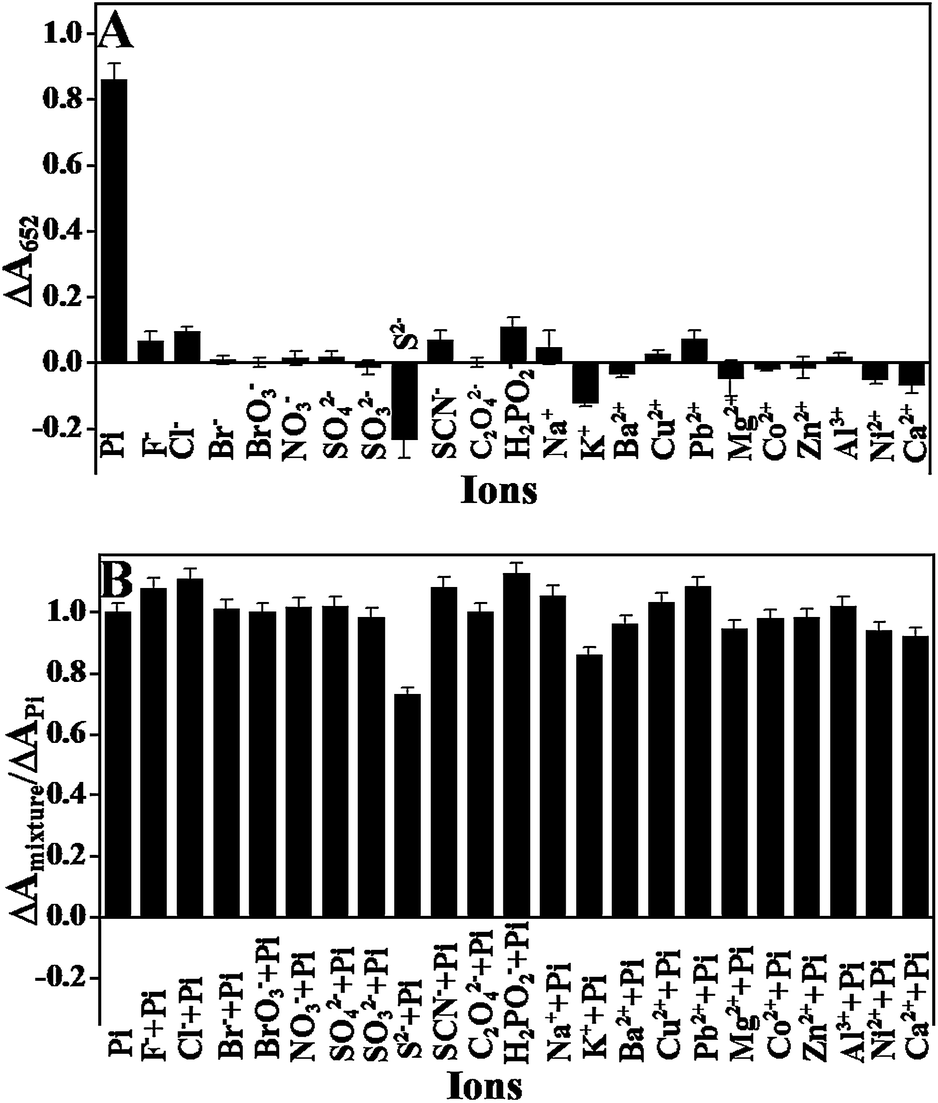
To evaluate the selectivity of this sensing system towards Pi (100 μM), we measured the colorimetric response of this sensing system to some common anions, including Cl−, Br−, BrO3−, NO3−, SO42−, SO32−, SCN−, C2O42−, H2PO2− (all 1.0 mM), F− and S2− (0.1 mM). The results in Fig. 6 show that only Pi could induce a significant decrease in the colorimetric intensity, whereas no evident colorimetric changes were observed in the presence of other ions, even when their concentrations were ten times greater than that of Pi, indicating that the sensing system had a good selectivity towards Pi. This excellent selectivity could be attributed to the coordination reaction between the Pi and Fe3+ as discussed above. In addition, some metallic ions, including Na+, K+, Ca2+, Mg2+, Al3+, Ba2+, Cu2+, Co2+, Ni2+, Zn2+, Pb2+ (all 0.1 mM), were also tested for their colorimetric responses. None of them caused significant colorimetric changes. Because the occurrence of the catalytic reaction is attributed to Fe3+ and Fe2+ ions on the surface of the Fe3O4 nanoparticles, and Fe3+ and Fe2+ interfere greatly with the assay. Nevertheless, the interferences can be ignored when their concentrations were below 10 μM (Fig. S6A†), which was higher than the limit value of iron in surface water (∼5.36 μM) according to the Environmental Quality Standards for Surface Water of the People's Republic of China. Therefore, the interference from iron ions can be neglected for most of the surface water samples. For the detection of Pi in solutions containing high levels of the two ions, pre-treatment of samples to remove them is required. Results in Fig. S6B† illustrated that the interference from ferric ion can be eliminated by a cation-exchange column. Phenol can coordinate with Fe3+ and thus interferes with the assay.32 The threshold for phenol in environmental water is 0.5 mg L−1 (less than 5.3 μM), according to the Chinese National Standards (GB22574-2008). Nevertheless, phenol was generally not detected in tap water, well water, river water and lake water.46–48 For this reason, the interference from phenol can be ignored. In addition to the anions, some metallic ions, including Na+, K+, Ca2+, Mg2+, Al3+, Ba2+, Cu2+, Co2+, Ni2+, Zn2+, and Pb2+ (all 0.1 mM), were also tested for colorimetric responses. None of them caused significant colorimetric changes.
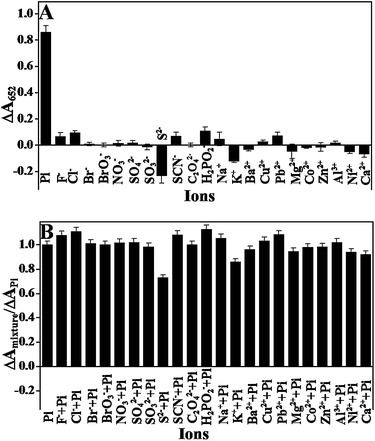 |
| | Fig. 6 (A) The ΔA652 response of the assay system toward phosphate and other anions or metallic ions. (B) Interference study of the sensor in the presence of a mixture of Pi and another anions or metallic ions (0.1 mM of Pi, PPi, F−, S2−, and cations; 1.0 mM of other anions). MNPs: 27.5 μg mL−1, TMB: 2.5 mM, H2O2: 4.0 mM, and HAc–NaAc buffer solution (0.2 M, pH 4.0). | |
3.3. Detection of Pi
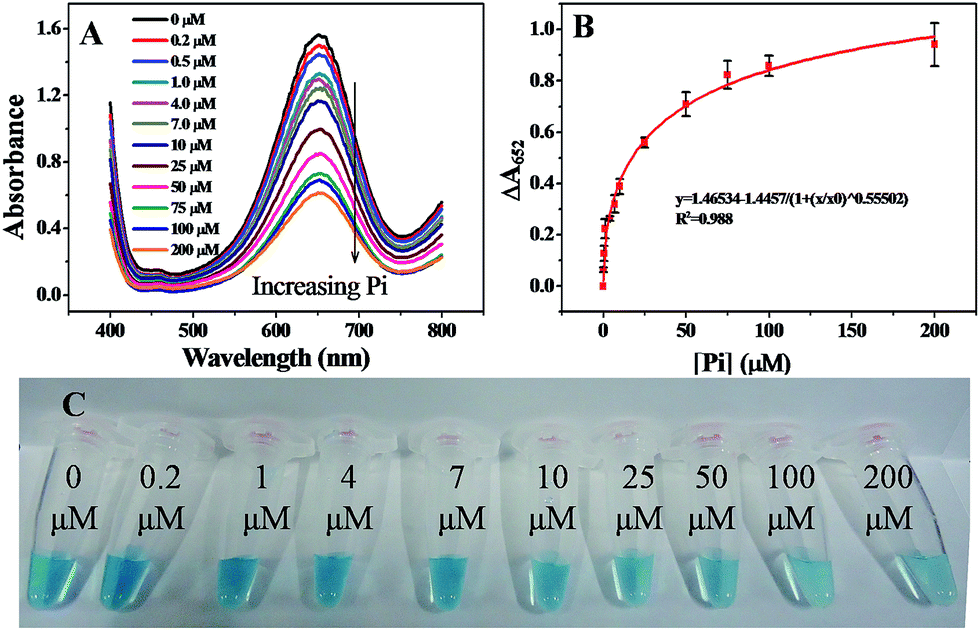
Under the optimized conditions discussed above, the linear response range of the sensing system was measured. As shown in Fig. 7A, the absorption intensity at 652 nm was highly sensitive to Pi and decreased gradually as the concentration of Pi was increased from 0.2 to 200 μM. Accordingly, ΔA652 increased gradually with the concentration of Pi ranging from 0.2 to 200 μM (Fig. 7B) and the calibration curve showed logistic function-shape. Basically, the correlation was ΔA652 = 1.46534 − 1.4457/(1 + ([Pi]/60.807)0.55502), R2 = 0.98797. The theoretical limit of detection, LOD, was calculated to be 0.11 μM, at a signal-to-noise ratio of 3. The value of the LOD was comparable to previously reported values obtained from the electrochemical analysis,11 spectrophotometry,17 fluorometry,18 and enzymatic biosensors.20 The digital image shown in Fig. 7C indicates that a containment level of 4.0 μM can be easily read out by naked eyes.
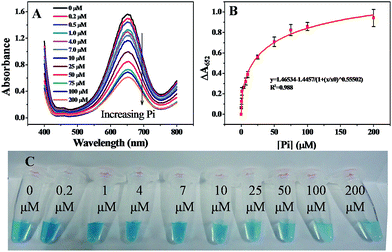 |
| | Fig. 7 (A) UV-vis absorption spectra of assay systems containing different concentrations of Pi. (B) The correlation curve between the change of absorbance at 652 nm (ΔA652) and the concentration of Pi. (C) Images of assay systems containing different concentrations of Pi. MNPs: 27.5 μg mL−1, TMB: 2.5 mM, H2O2: 4.0 mM, and HAc–NaAc buffer solution (0.2 M, pH 4.0). | |
3.4. Detection of Pi in practical samples
To investigate the applicability of the sensing system in real samples, we used the established sensor to determine Pi in drinking water, ground water and lake water. The three water samples were spiked with two concentration levels of Pi anions respectively, and three replicas of each level were analyzed by the proposed method. The experimental results, as shown in Table 1, demonstrated the acceptable quantitative recoveries of all samples. The percentage recoveries were 97.8–113.6% and the RSD was less than 6.5%. In addition, the results obtained by the present method are in accordance with those obtained by the standard molybdenum-blue method,49 indicating the feasibility of the sensing system in practical use.
Table 1 Results of Pi determination and recoveries in water samples
| Samplesa |
The present method (μM) |
Molybdenum-blue method (μM) |
Added (μM) |
Found (μM) |
Recovery (%) |
RSD (n = 3, %) |
|
A is drinking water sample, B is ground water sample, and C is lake water sample.
|
| A |
Undetectable |
Undetectable |
5 |
4.96 |
99.2 |
1.95 |
| 20 |
19.56 |
97.8 |
0.60 |
| B |
3.60 |
3.74 |
5 |
5.68 |
113.6 |
5.57 |
| 20 |
21.8 |
109 |
6.42 |
| C |
Undetectable |
Undetectable |
5 |
5.30 |
106 |
3.90 |
| 20 |
20.66 |
103.3 |
0.71 |
4. Conclusions
In summary, a simple method is proposed herein for the determination of trace levels of Pi based on the shielding of Fe3O4 MNPs' catalytic ability. Assay samples that do not contain Pi display intense colorimetric responses while those containing Pi show significantly reduced colorimetric signals. This difference can be easily detected using UV-vis spectrophotometry and can be used for Pi detection. The change of the colour and the absorption at 652 nm of the Fe3O4 MNPs–TMB–H2O2 sensing system might be attributed to the coordination of Pi and Fe3+. The assay has advantages of rapidity, low cost, high sensitivity and selectivity, thus proving to be a powerful tool in environment science and water quality monitoring.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21435005 and no. 21175124), the Development Project of Science and Technology of Jilin Province (no. 20125090).
Notes and references
- P. D. Beer and E. J. Hayes, Coord. Chem. Rev., 2003, 240, 167–189 CrossRef CAS.
- X. Y. Zhao, Y. Liu and K. S. Schanze, Chem. Commun., 2007, 2914–2916 RSC.
- J. Zhang, C. X. Chen, X. W. Xu, X. L. Wang and X. R. Yang, Chem. Commun., 2013, 49, 2691–2693 RSC.
- Y. L. Liu, K. L. Ai, X. L. Cheng, L. H. Huo and L. H. Lu, Adv. Funct. Mater., 2010, 20, 1–6 Search PubMed.
- F. Qu, N. B. Li and H. Q. Luo, Anal. Chem., 2012, 84, 10373–10379 CrossRef CAS PubMed.
- T. Y. Zhou, M. C. Rong, Z. M. Cai, C. Y. Yang and X. Chen, Nanoscale, 2012, 4, 4103–4106 RSC.
- J. H. Ryther and W. M. Dunstan, Science, 1971, 171, 1008–1013 CAS.
- B. Y. Spivakov, T. A. Maryutina and H. Muntau, Pure Appl. Chem., 1999, 71, 2161–2176 CrossRef CAS.
- C. P. Mainstone and W. Parr, Sci. Total Environ., 2002, 282–283, 25–47 CrossRef CAS.
- P. J. A. Withers and H. P. Jarvie, Sci. Total Environ., 2008, 400, 379–395 CrossRef CAS PubMed.
- W.-L. Cheng, J.-W. Sue, W.-C. Chen, J.-L. Chang and J.-M. Zen, Anal. Chem., 2010, 82, 1157–1161 CrossRef CAS PubMed.
- H. Kawasaki, K. Sato, J. Ogawa, Y. Hasegawa and H. Yuki, Anal. Biochem., 1989, 182, 366–370 CrossRef CAS.
- S. Cosnier, C. Gondran, J.-C. Watelet, W. D. Giovani, R. P. M. Furriel and F. A. Leone, Anal. Chem., 1998, 70, 3952–3956 CrossRef CAS.
- J. B. Quintana, R. Rodil and T. Reemtsma, Anal. Chem., 2006, 78, 1644–1650 CrossRef CAS PubMed.
- M. Colina and P. H. E. Gardiner, J. Chromatogr. A, 1999, 847, 285–290 CrossRef CAS.
- H. M. Qiu, J. J. Geng, C. Han and H. Q. Ren, Chin. J. Anal. Chem., 2013, 12, 1910–1914 Search PubMed.
- C. M. Li, Y. F. Li, J. Wang and C. Z. Huang, Talanta, 2010, 21, 1339–1345 CrossRef PubMed.
- A. K. Dwivedi, G. Saikia and P. K. Iyer, J. Mater. Chem., 2011, 21, 2502–2507 RSC.
- W. C. Mak, C. Y. Chan, J. Barford and R. Renneberg, Biosens. Bioelectron., 2003, 19, 233–237 CrossRef CAS.
- M. A. Rahmana, D.-S. Park, S.-C. Chang, C. J. McNeil and Y.-B. Shim, Biosens. Bioelectron., 2006, 21, 1116–1124 CrossRef PubMed.
- B. Kong, A. W. Zhu, Y. P. Luo, Y. Tian, Y. Y. Yu and G. Y. Shi, Angew. Chem., Int. Ed., 2011, 50, 1837–1840 CrossRef CAS PubMed.
- Y. Kubo, S. Uchida, Y. Kemmochi and T. Okubo, Tetrahedron Lett., 2005, 4, 4369–4372 CrossRef PubMed.
- W. Q. Liu, Z. F. Du, Y. Qian and F. Li, Sens. Actuators, B, 2103, 176, 927–931 CrossRef PubMed.
- H. X. Zhao, L. Q. Liu, Z. D. Liu, Y. Wang, X. J. Zhao and C. Z. Huang, Chem. Commun., 2011, 47, 2604–2606 RSC.
- J.-M. Bai, L. Zhang, R.-P. Liang and J.-D. Qiu, Chem.–Eur. J., 2013, 19, 3822–3826 CrossRef CAS PubMed.
- L. Gao, J. Zhuang, L. Nie, J. B. Zhang, Y. Zhang, N. Gu, T. H. Wang, J. Feng, D. L. Yang, S. Perrett and X. Y. Yan, Nanotechnology, 2007, 2, 577–583 CAS.
- X. J. Chen, Y. Z. Wang, Y. Y. Zhang, Z. H. Chen, Y. Liu, Z. L. Li and J. H. Li, Anal. Chem., 2014, 86, 4278–4286 CrossRef CAS PubMed.
- Y. Z. Wang, K. Qu, L. H. Tang, Z. L. Li, E. Moore, X. Q. Zeng, Y. Liu and J. H. Li, Trends Anal. Chem., 2014, 58, 54–70 CrossRef CAS PubMed.
- H. Wei and E. K. Wang, Anal. Chem., 2008, 80, 2250–2254 CrossRef CAS PubMed.
- C.-J. Yu, C.-Y. Lin, C.-H. Liu, T.-L. Cheng and W.-L. Tseng, Biosens. Bioelectron., 2010, 26, 913–917 CrossRef CAS PubMed.
- C.-H. Liu and W.-L. Tseng, Anal. Chim. Acta, 2011, 703, 87–93 CrossRef CAS PubMed.
- C.-H. Liu, C.-J. Yu and W.-L. Tseng, Anal. Chim. Acta, 2012, 745, 143–148 CrossRef CAS PubMed.
- N. Ding, N. Yan, C. L. Ren and X. G. Chen, Anal. Chem., 2010, 82, 5897–5899 CrossRef CAS PubMed.
- K. S. Park, M. I. Kim, D.-Y. Cho and H. G. Park, Small, 2011, 7, 1521–1525 CrossRef CAS PubMed.
- R. Thiramanas, K. Jangpatarapongsa, P. Tangboriboonrat and D. Polpanich, Anal. Chem., 2013, 85, 5996–6002 CrossRef CAS PubMed.
- C. Kaittanis, S. Santra and J. M. Perez, J. Am. Chem. Soc., 2009, 131, 12780–12791 CrossRef CAS PubMed.
- L. Z. Gao, J. M. Wu, S. Lyle, K. Zehr, L. L. Cao and D. Gao, J. Phys. Chem. C, 2008, 112, 17357–17361 CAS.
- X. N. Li, F. Wen, B. Creran, Y. Jeong, X. R. Zhang and V. M. Rotello, Small, 2012, 8, 3589–3592 CrossRef CAS PubMed.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247–1248 CrossRef.
- C. W. Childs, J. Phys. Chem., 1969, 73, 2956–2960 CrossRef CAS.
- P.-H. Li, J. Y. Lin, C. T. Chen, W. R. Ciou, P.-H. Chan, L. Y. Luo, H.-Y. Hsu, E. W.-G. Diau and Y.-C. Chen, Anal. Chem., 2012, 84, 5484–5488 CrossRef CAS PubMed.
- G. A. Park, Chem. Rev., 1965, 177–198 CrossRef.
- Z. G. Peng, K. Hidajat and M. S. Uddin, J. Colloid Interface Sci., 2004, 271, 277–283 CrossRef CAS PubMed.
- Y. Jv, B. X. Li and R. Cao, Chem. Commun., 2010, 46, 8017–8019 RSC.
- M. Dörr, T. Alpermann and W. Weigand, Origins Life Evol. Biospheres, 2007, 37, 329–333 CrossRef PubMed.
- R. J. Alkasir, M. Ornatska and S. Andreescu, Anal. Chem., 2012, 84, 9729–9737 CrossRef CAS PubMed.
- H. K. Maleh, M. Moazampour, A. A. Ensafi, S. Mallakpour and M. Hatami, Environ. Sci. Pollut. Res., 2014, 21, 5879–5888 CrossRef PubMed.
- R. L. Sun, Y. Wang, Y. N. Ni and S. Kokot, Talanta, 2014, 125, 341–346 CrossRef CAS PubMed.
- C. H. Fiske and Y. Subbarow, J. Biol. Chem., 1925, 66, 375–400 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6. See DOI: 10.1039/c4ay02461c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.