
Electromembrane extraction using a cylindrical electrode: a new view for the augmentation of extraction efficiency
Yousef Abdossalami
Asl
a,
Yadollah
Yamini
*a,
Maryam
Rezazadeh
a and
Shahram
Seidi
b
aDepartment of Chemistry, Tarbiat Modares University, P.O. Box 14115-175, Tehran, Iran. E-mail: yyamini@modares.ac.ir; Fax: +98-21-82883455; Tel: +98-21-82883417
bDepartment of Analytical Chemistry, Faculty of Chemistry, K.N. Toosi University of Technology, Tehran, Iran
First published on 28th October 2014
Abstract
In the present work, for the first time, a cylindrical electrode that surrounded a hollow fiber membrane was introduced in electromembrane extraction (EME). The setup introduced produces an efficient, inexpensive, stable, and reproducible method for increasing the extraction efficiency of ionizable compounds from different matrices. The method was applied for the extraction of diclofenac and mefenamic acid as model analytes from biological fluids. Effective parameters on EME of the analytes, such as extraction time, applied voltage, and composition of acceptor/donor phases, were investigated and optimized using the experimental design. Under optimized conditions, relative recoveries in the range of 94–105 and preconcentration factors in the range of 50–355 were obtained in various biological matrices. The linear dynamic range of 2.5–500 μg L−1 (with coefficient of determination better than 0.9986) and limit of detection of 0.25 μg L−1 were obtained for both of the analytes in plasma and urine samples. The figures of merit of EME with a cylindrical electrode were compared with the results obtained from conventional EME.
1. Introduction
Electromembrane extraction (EME), as one of the liquid phase microextraction (LPME) methods,1–3 was introduced by Pedersen-Bjergaard et al. in 2006.4 EME has numerous advantages in comparison with hollow fiber-based liquid phase microextraction (HF-LPME) and provides more efficient extractions and sample clean-up.1,2,5–12However, EME suffers from some drawbacks such as low repeatability and extraction recovery, low preconcentration factors, application of high voltages, SLM instability, joule heating, and sparking. Some of the disadvantages are pernicious; thus, several efforts have been made to improve the analytical performance and safety of EME.13 For this purpose, several different EME setups and coupling devices have been introduced. However, to increase repeatability and extraction recovery of EME, Kubáň et al. have exerted a new notion based on the use of stabilized constant direct electrical current.14 Traditional EME setups were used for the separation of only acidic or basic drugs; thus, simultaneous extraction of both acidic and basic analytes is one of the EME challenges. Fakhari et al. introduced a new method named dual hollow fiber electromembrane extraction for simultaneous extraction and preconcentration of acidic and basic drugs in a single step.15 Basheer et al. designed an interesting EME setup using four sheets of porous polypropylene membrane for the simultaneous extraction of acidic and basic drugs.16 Yamini et al. applied a simple setup for the simultaneous extraction of acidic and basic analytes. In this method, two separate pieces of hollow fibers, each of which contained one of the electrodes, were used for the simultaneous extraction of acidic and basic drugs.17 Petersen et al., for the first time, have presented miniaturized EME using flat membranes.18 In another work, nano-electromembrane extraction (nano-EME) was introduced for the extraction of the analytes from 200 μL of sample solution into approximately 8 nL acceptor phase.19
However, in all reported EMEs, by applying the voltage, double-layer thickness around the hollow fiber increases over time, which causes reduction in the extraction efficiency.20 For the first time, Rezazadeh et al. have introduced pulsed electromembrane extraction (PEME) for the reduction of double-layer thickness.21 However, this method did not yield the expected performance.
Moreover, in all EME setups, two wire electrodes are used to create an electrical field. Thus, a planar electrical field is formed, which causes transmission of a number of species in a certain direction around the hollow fiber; therefore, the double-layer thickness increases over time, and Joule heating, SLM instability, and sparking result from the creation of the double layer. However, to the best of our knowledge, so far, there is no report on employing electrodes with different geometric shapes for the enhancement of extraction efficiency.
In this work, electrodes with two shapes (cylindrical versus wire electrode) were used as the outer electrode (the electrode in the donor phase), and the effect of electrode shape on the performance of EME extraction was examined. Mefenamic acid and diclofenac have been used as model analytes. Extraction behavior of these analytes was previously investigated by several EME setups.22–25
2. Experimental
2.1. Chemicals and reagents
Standard materials of mefenamic acid (MEF) and diclofenac (DIC) were obtained from the Department of Medical Sciences of Tehran University (Tehran, Iran). 1-Octanol was obtained from Fluka (Buchs, Switzerland). Other reagents were of analytical grade and purchased from Merck (Darmstadt, Germany). The porous hollow fiber used for the SLM was a PPQ3/2 polypropylene hollow fiber from Membrana (Wuppertal, Germany) with the inner diameter of 0.6 mm, wall thickness of 200 μm and pore size of 0.2 μm. The ultra-pure water was provided by a model Aqua Max-Ultra Youngling ultra-pure water purification system (Dongan-gu, South Korea). HPLC-grade acetonitrile and methanol were purchased from Caledon (Ontario, Canada). Stock standard solutions of each analyte (1000 μg L−1) were separately prepared by dissolving proper amounts of each analyte in methanol and stored at 4 °C. Mixtures of standard working solutions were prepared by dilution of stock solution with ultra-pure water.2.2. Apparatus
Chromatographic separation was performed with a HPLC instrument, including a Varian 9012 HPLC pump (Walnut Creek, CA, USA), a six-port Cheminert HPLC valve from Valco (Houston, TX, USA) with a 20 μL sample loop and equipped with a Varian 9050 UV-Vis detector. Chromatographic data was recorded and analyzed using ChromanaCH software (version 3.6.4). An ODS-3 column (250 mm × 4.6 mm, with 5 μm particle size) from MZ-Analysentechnik (Mainz, Germany) was used to separate DIC and MEF under isocratic elution conditions. A mixture of 50 mM ammonium acetate (pH 5.2) and acetonitrile (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55) was used as the mobile phase and the analytes were detected at 285 nm. Microliter syringes (25–500 μL) were purchased from Hamilton (Bonaduz, Switzerland).
:55) was used as the mobile phase and the analytes were detected at 285 nm. Microliter syringes (25–500 μL) were purchased from Hamilton (Bonaduz, Switzerland).
2.3. Equipment for electromembrane extraction (EME)
The equipment for the EME procedure is shown in Fig. 1. A glass vial with internal diameter of 15 mm and height of 5 cm was used. The electrodes used in this work were platinum wires as anode with the diameter of 0.2 mm, and were obtained from Pars Pelatine (Tehran, Iran). Stainless steel wire with the diameter of 0.5 mm and stainless steel cylindrical electrodes as cathode with the inner diameter of 10.0 mm, outer diameter of 11.0 mm, and height of 3.0 cm were obtained from Iran Alloy Steel Company (Yazd, Iran). The electrodes were coupled to a power supply model 8760T3 with a programmable voltage in the range of 0–600 V and with a current output in the range of 0–500 mA from Paya Pajoohesh Pars (Tehran, Iran). During the extraction, the EME unit was stirred with a stirring speed in the range of 0–1250 rpm by a heater-magnetic stirrer model 3001 from Heidolph (Kelheim, Germany) using a 5 × 2 mm magnetic bar. | ||
| Fig. 1 The equipment used for electromembrane extraction with a cylindrical cathode. | ||
2.4. EME procedure
Fifteen milliliters of the sample solution containing the analytes was transferred into the sample vial. To impregnate the organic solvent in the pores of the hollow fiber wall, a 4 cm piece of the hollow fiber was cut and dipped in the solution for 5 s, and then the excess of organic solvent was gently wiped away by blowing air with a Hamilton syringe. The upper end of the hollow fiber was connected to a medical syringe needle tip as a guide tube, which was inserted through the rubber cap of the vial. Ten microliters of 100 mM NaOH (acceptor solution) was introduced into the lumen of the hollow fiber by a microsyringe, and the lower end of the hollow fiber was mechanically sealed. One of the electrodes, the anode, was introduced into the lumen of the fiber. The fiber containing the anode, SLM and the acceptor solution was later directed into the cylindrical cathode that was located in the sample solution. The electrodes were subsequently coupled to the power supply, and the extraction unit was placed on a stirrer with the stirring speed of 700 rpm. The predetermined voltage was turned on and extraction was performed for a prescribed time. When the extraction was completed, the end of the hollow fiber was adjoined to the Hamilton syringe, and then the acceptor phase was collected and injected into HPLC. The final acceptor volume was 10 ± 2 μL.2.5. Real samples
2.6. Calculation of preconcentration factor, extraction recovery, and relative recovery
The preconcentration factor (PF) was defined as the ratio of the final analyte concentration in the acceptor phase (Cf,a) and the initial concentration of analyte (Ci,s) in the sample solution: | (1) |
2.7. Data analysis and statistical methods
Response surface methodology (RSM) is effective for responses that are influenced by many factors and their interactions. The method was originally described by Box and Wilson.26 Several studies indicated that it is useful for developing, improving, and optimizing processes.27,28 Optimization of effective parameters on the extraction of MEF and DIC by EME was performed by a face-centered central composite design (FCCCD). In all the cases, design generation and statistical analyses were performed by means of the software package Statgraphics Plus version 5.1 for Windows (Rockville, MD, USA).3. Results and discussion
3.1. Theoretical aspect
Since 2006, many works have been carried out using the EME method. In all the works, to obtain high extraction efficiency, high voltage and long extraction time have been used.29–35 However, electrical potential differences above 300 V were found to be inappropriate due to the system suffering from bubble formation at the electrode surfaces, instability problems such as increasing current amplitude, punctuation of the SLM, and sparking.21In conventional EME methods, an electrical field is applied only in one angular direction by a two-wire electrode. Thus, analyte molecules could transfer across the SLM only in one degree around the hollow fiber. Current phenomenon leads to mass transfer resistance through the SLM, sparks, and decreases the extraction efficiency. Using the cylindrical electrode instead of a wire electrode that is placed in the outer solution (donor phase), a cylindrical electrical field may be constructed at all angles around the inner electrode (acceptor phase). Thus, SLM stability increased and extraction efficiency improved. By employing a cylindrical electrode, the potential difference required to reach a high extraction efficiency may be very low and in this method, several problems with EME could be minimized.
3.2. Effect of stirring speed and SLM composition
The effects of stirring speed and SLM composition on the extraction efficiency of the analytes with EME was studied using one variable at a time method. Stirring speed has a basic role in increasing the efficiency of extraction and kinetics of EME by increasing the mass transfer and reducing the thickness of the double layer around the SLM.17 The effect of stirring speed on extraction efficiency was studied up to 1000 rpm while 1-octanol was used as the SLM to migrate the analyte from 10 mM NaOH (donor) to 100 mM NaOH (acceptor). To this end, 30 V was applied for extraction for 20 min. A stirring rate of 700 rpm yielded the best results and it was selected as the optimal stirring rate.According to previous reports, the chemical nature of the SLM is a highly critical parameter in EME extraction efficiency. Organic solvents for SLM in EME should have several specific properties.17 The water-immiscible SLM should have polarity similar to that of the polypropylene fiber; so that it can be easily immobilized within the pores of the fiber. The SLM should have an appropriate electrical resistance to keep the electrical current of the system at its lowest possible level and it should have certain chemical properties to enable electrokinetic migration of the model analytes. Moreover, charged analytes should have suitable solubility in the SLM. It was shown that long-chain alcohols are the alternatives for this purpose.20 1-Octanol, undecanol, dihexyl ether, dodecanol, and octanoic acid were investigated as SLM composition while 30 V was applied as the driving force to migrate the analyte from 10 mM NaOH (donor) to 100 mM NaOH (acceptor) for 20 min. The results of this study exhibit that 1-octanol was the best candidate for the analytes in EME (Fig. 2A).
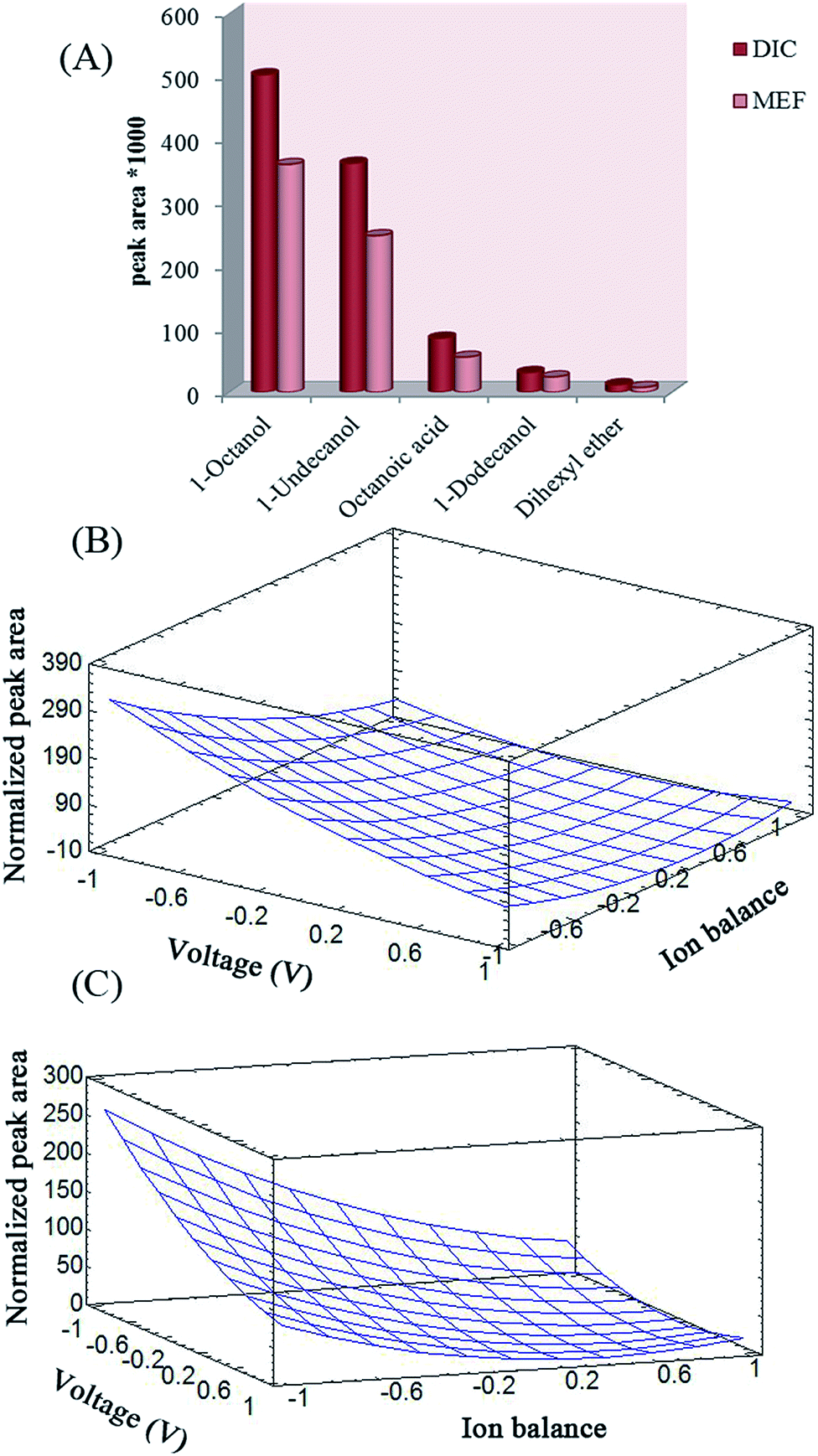 | ||
| Fig. 2 (A) Optimization of membrane composition. Three-dimensional representation of the response surfaces where extraction time remained at its central level and the others varied within the experimental range for (B) cylindrical and (C) wire electrodes. | ||
3.3. Effect of coexisting ions
In EME, the effect of coexisting ions on extraction efficiency is investigated as ion balance.36 According to the literature, the presence of a high content of ionic substances leads to an increase in the value of the ion balance (χ) in the system, which in turn decreases the flux of analytes across the SLM.37,38 This fact may be attributed to the competition of salt ions with analyte ions to pass through the SLM, increasing the thickness of the double layer around the SLM and therefore, decreasing the mass transfer of analytes through the SLM into the acceptor phase.20 Moreover, according to the literature, increasing the ionic strength of the sample solution increases the instability of the SLM due to Joule heating phenomenon.20 Here, the effect of coexisting ions was investigated by addition of 0.1% (w/v) of NaCl and the results were in full agreement with the previous studies. Thus, migration of the analytes would be more efficient in the absence of salt.3.4. Optimization of effective parameters using central composite design (CCD)
The main variables that can affect the extraction efficiency of EME are SLM composition, compositions of the donor and acceptor phases, extraction time, applied voltage, and stirring rate. Based on earlier experiments, 1-octanol was the best organic membrane for extraction of these acidic drugs, and the optimal stirring rate of 700 rpm was obtained. Therefore, central composite design (CCD) was employed to optimize three parameters: applied voltage (V), extraction time (t), and ion balance, in order to maximize the experimental response. Ion balance is determined mainly by the HCl concentration in the sample solution and acceptor phase. In order to optimize this parameter, the HCl concentration of the acceptor phase was kept constant (100 mmol L−1) and its concentration in the donor phase was varied within 0–100 mmol L−1 range.38Different series of experiments were designed to determine how the effective parameters influence the final response using wire and cylindrical electrodes. The low (−1), central (0), and high (+1) levels of these variables are given in Table 1. Normalized peak area for each run was selected as the response objective for the study. To normalize the peak areas, all the experiments were first run and the peak area of each analyte was divided by its smallest peak area afterwards. Normalized peak area was subsequently added for each run and used in calculations of total normalized peak area. Using multiple regression analysis, the experimental responses were correlated with the significant factors. The goodness of fit of the quadratic polynomials is expressed by the coefficient of determination, R2, which should be at least 0.8.28 The coefficients of determination were 0.9613 and 0.9467 for cylindrical and wire electrodes, respectively, which indicates that the equation obtained has good adequacy for correlating the experimental results.
| Factors | Symbol | Levels | ||
|---|---|---|---|---|
| Low (−1) | Center (0) | High (+1) | ||
| Extraction time (min) | t | 5 | 12.5 | 20 |
| Voltage (V) | V | 20 | 45 | 70 |
| Ion balance | χ | 0 | 0.5 | 1 |
In order to investigate model fitness, analysis of variance (ANOVA) was performed, which proved that the model was significant and the “lack of fit” was not significant (P = 0.05). The Pareto charts show the main effects and their influence on the response (Fig. 3A and B). The bar lengths of the charts are proportional to the absolute value of the estimated main effects. The vertical lines correspond to 95% confidence interval. An effect that exceeds this reference line is a significant parameter. The color of the bars indicates whether the response would be improved or not by changing a given factor from the lowest to the highest level. The results show that only applied voltage and ion balance affect the final response. The effect of independent variables on the response was analyzed using RSM. The results given in Fig. 2B and C illustrate the relationship between the explanatory and response variables in a three-dimensional representation of the response surface. To this end, one variable was kept at its central level, and the others varied within the experimental range. Based on the ANOVA and the plots, the normalized peak areas of the drugs for both the cylindrical and wire electrodes are increased by decreasing the applied voltage and ion balance. Increasing the voltage may result in decreasing the recoveries due to mass transfer resistance caused by the build-up of a boundary layer of ions at the interfaces at both sides of the SLM or saturation of the analyte in the acceptor phase. On the other hand, electrolysis reactions cause decreasing of the pH of acceptor phase. Therefore, analytes back-extraction into the donor phase may occur due to their neutralization. Hence, extractability decreased by increasing the strength of electrical field. Based on previous studies on the role of ion balance,36 it was anticipated that the decrease in ion balance caused an increase in the flux. Therefore, high recoveries may be obtained if ion concentration in the acceptor solution is high compared to that in the donor solution. Ionic concentration of the phases is determined mainly by their pH values. Thus, a pH gradient is necessary for an excellent extraction. As can be seen in Fig. 2B and C, the maximum response was yielded by application of 20 V electrical potential and selection of 100 mM NaOH as the acceptor solution.
 | ||
| Fig. 3 Pareto charts of the main effects, A; voltage, B; time, C; ion balance for (A) cylindrical and (B) wire electrodes. | ||
Optimal conditions were attained via using 10 mM and a 100 mM NaOH solution as donor and acceptor phases, respectively, and applying a 20 V electric potential for 15 min. The sample solution was agitated by stirring at a rate of 700 rpm. In addition, the SLM composition was pure 1-octanol for DIC and MEF.
3.5. Method performance
Figures of merit of the proposed EME using cylindrical and wire electrode were investigated in human plasma and urine samples.To reduce matrix effect, a match matrix method was used to obtain calibration curves of DIC and MEF using drug-free urine and plasma samples. Linearity was studied by analysis of eight extracts obtained from the aliquots of each sample in triplicates using a cylindrical electrode. To improve mass transfer of the analytes, human plasma and urine samples were diluted 1:4 and 1:3, respectively, with pure water and the pH value was adjusted to 7.00 by addition of proper amounts of hydrochloric acid and/or sodium hydroxide solutions. As shown in Table 2, an acceptable linear range and linearity (coefficient of determination greater than 0.9943) were obtained. Repeatability of the method was examined by five replicate measurements of the drugs in the samples at a concentration level of 10 μg L−1. The %RSDs were found to be lower than 10.8. Limits of detection (LODs) and limits of quantification (LOQs) obtained for both setups are provided in Table 2. To this end, LOD and LOQ were determined considering 3 S/N and 10 S/N, respectively. In another experiment, EME with a wire electrode was carried out for extraction of the analytes from human plasma and urine samples (pH = 7.00). Optimal conditions for extraction of both drugs were applied and preconcentration factors were calculated. As provided in Table 2, the cylindrical electrode offered higher preconcentration factors in comparison with the wire electrode, which may be caused by the electrical field construction in all angles around the inner electrode.
| Cylindrical electrode | Wire electrode | ||||
|---|---|---|---|---|---|
| Plasma | Urine | Plasma | Urine | ||
| a Preconcentration factor at 10 μg L−1. b Based on seven replicated measurements at concentration of 10 μg L−1. | |||||
| LOD (μg L−1) | DIC | 0.20 | 0.20 | 1.00 | 1.00 |
| MEF | 0.50 | 0.50 | 2.50 | 2.00 | |
| LOQ (μg L−1) | DIC | 1.0 | 1.0 | 5.0 | 2.5 |
| MEF | 2.0 | 2.0 | 10.0 | 5.0 | |
| Linearity (μg L−1) | DIC | 1.0–250 | 1.0–250 | 5.0–250 | 2.5–250 |
| MEF | 2.0–250 | 2.0–250 | 10.0–250 | 5.0–250 | |
| Calibration equation | DIC | y = 423.3x (μg L−1) + 187.2 | y = 475.9x (μg L−1) + 201.5 | y = 269.9x (μg L−1) + 153.2 | y = 286.4x (μg L−1) + 139.5 |
| MEF | y = 421.8x (μg L−1) + 165.3 | y = 468.7x (μg L−1) + 193.4 | y = 257.4x (μg L−1) + 142.9 | y = 261.5x (μg L−1) + 149.8 | |
| R 2 | DIC | 0.9988 | 0.9992 | 0.9943 | 0.9998 |
| MEF | 0.9986 | 0.9995 | 0.9974 | 0.9992 | |
| PFa | DIC | 355 | 50 | 173 | 27 |
| MEF | 116 | 47 | 39 | 18 | |
| RSDb | DIC | 3.5 | 4.6 | 8.1 | 6.2 |
| MEF | 5.7 | 7.3 | 5.9 | 9.3 | |
3.6. Analysis of real sample
In order to compare applicability of the presented EME setup to analysis of real samples, concentrations of the analytes in various human plasma and urine samples were analyzed. All samples were prepared as explained in the experimental section. For this purpose, plasma and urine samples were diluted 1:4 and 1:3, respectively, with pure water and the pH values were adjusted to 7.00 by addition of proper amounts of hydrochloric acid (100 mM) and/or sodium hydroxide (100 mM) solutions. For quantitative analysis, optimal conditions were applied. Chromatograms obtained after extraction from human plasma and urine samples are shown in Fig. 4. Afterward, to determine the method accuracy, each sample was spiked at three concentration levels of the analytes and EME was carried out to calculate the concentration of the analytes. According to the FDA definition, a matrix effect is the direct or indirect alteration or interference in response to presence of inadvertent analytes or other interfering substances in the sample.39Table 3 illustrates that results of four replicate analyses of each sample obtained by the proposed method are in satisfactory agreement with the spiking amounts. No substantial matrix effect was observed for the real samples studied and the method yielded propones with acceptable accuracy.
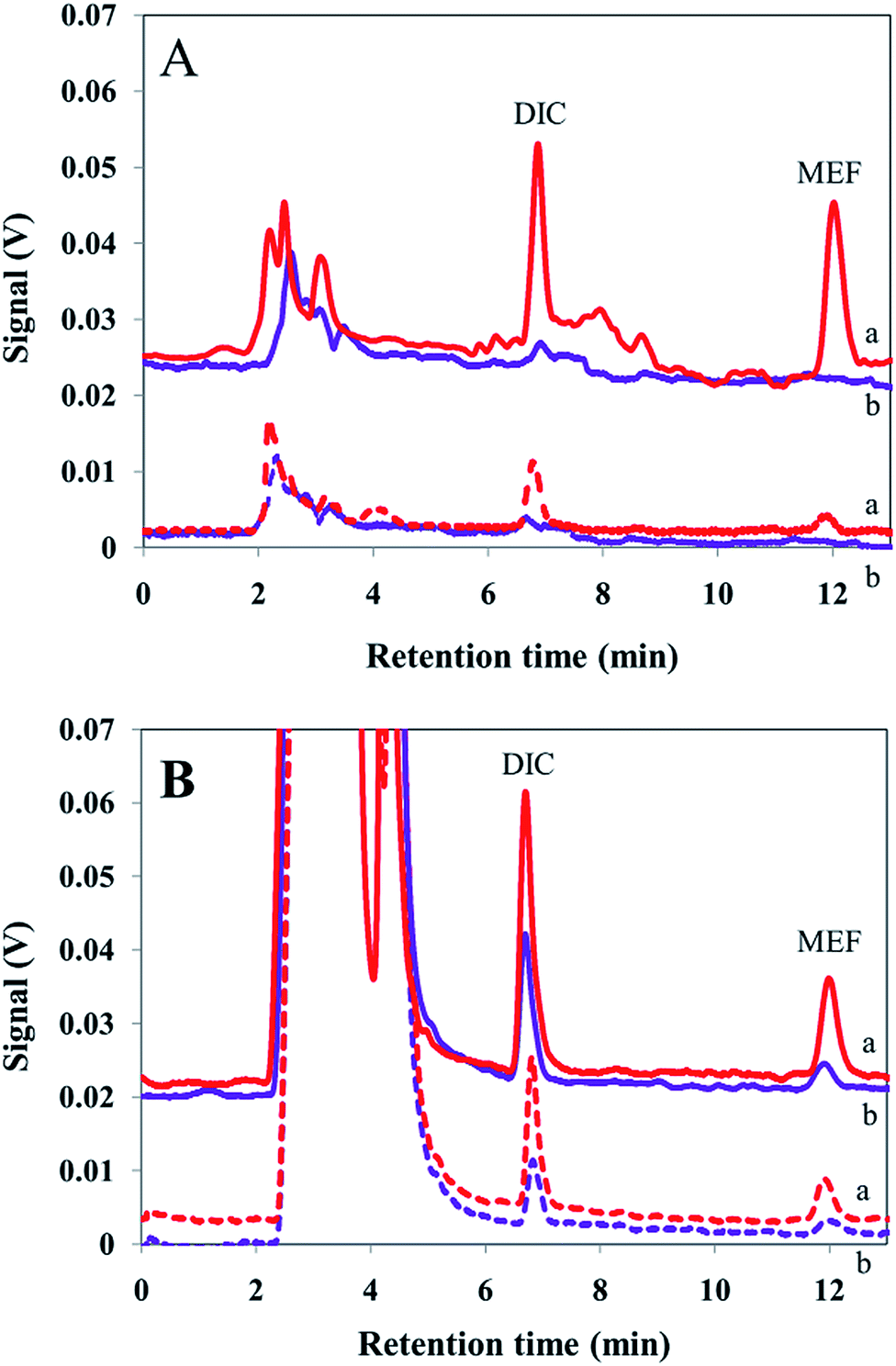 | ||
| Fig. 4 Chromatograms obtained after EME of (A) plasma sample, (B) urine sample (solid and dash line for cylindrical and wire electrodes, respectively, and (a) non-spiked sample, (b) spiked sample at a concentration level of 10 μg L−1). | ||
| Samplea | Type of electrode | Analyte | C real (μg L−1) | C added (μg L−1) | C found (μg L−1) (n = 4) | RSD% | RR% |
|---|---|---|---|---|---|---|---|
| a The results are related to the urine sample collected from a volunteer and a drug-free plasma (blood group A+) sample (see, Section 2.5). b Not detected. | |||||||
| Urine | Cylindrical | DIC | 12.2 | 10 | 22.6 | 6.2 | 104.2 |
| 100 | 115.0 | 6.9 | 102.8 | ||||
| 200 | 224.2 | 7.2 | 106.0 | ||||
| MEF | 5.6 | 10 | 15.7 | 5.2 | 101.0 | ||
| 100 | 112.4 | 6.2 | 106.8 | ||||
| 200 | 214.9 | 6.3 | 104.7 | ||||
| Wire | DIC | 9.1 | 10 | 19.5 | 8.2 | 103.1 | |
| 100 | 112.4 | 6.2 | 106.8 | ||||
| 200 | 218.3 | 7.4 | 104.6 | ||||
| MEF | 4.3 | 10 | 13.6 | 7.0 | 94.6 | ||
| 100 | 101.2 | 7.4 | 96.1 | ||||
| 200 | 198.9 | 8.7 | 97.3 | ||||
| Plasma | Cylindrical | DIC | <LOQ | 10 | 10.4 | 6.6 | 104.3 |
| 100 | 106.0 | 5.9 | 106.0 | ||||
| 200 | 208.2 | 6.2 | 104.1 | ||||
| MEF | ndb | 10 | 10.3 | 7.1 | 103.7 | ||
| 100 | 109.2 | 7.1 | 109.2 | ||||
| 200 | 216.8 | 6.7 | 108.4 | ||||
| Wire | DIC | <LOQ | 10 | 10.1 | 9.2 | 102.1 | |
| 100 | 106.9 | 7.4 | 106.9 | ||||
| 200 | 218.8 | 6.7 | 109.4 | ||||
| MEF | ndb | 10 | 9.6 | 8.1 | 97.5 | ||
| 100 | 95.6 | 8.1 | 95.6 | ||||
| 200 | 194.9 | 7.6 | 97.6 | ||||
4. Conclusions
A new EME method using a cylindrical electrode around hollow fiber for the extraction of analytes from different matrices has been presented for the first time. The new EME technique makes the extraction system stable and repeatable, unlike conventional EME methods. Moreover, the use of a cylindrical electrode has increased the extraction efficiency and preconcentration factor. The proposed method offers more efficient extraction and higher stability, because the boundary layer thickness is very low. The cylindrical electrode has the ability to prevail over the perturbation of interfering ions in the analysis of real samples.References
- M. Ghambarian, Y. Yamini and A. Esrafili, J. Pharm. Biomed. Anal., 2011, 56, 1041–1045 CrossRef CAS PubMed.
- M. Ghambarian, Y. Yamini and A. Esrafili, J. Chromatogr. A, 2012, 1222, 5–12 CrossRef CAS PubMed.
- B. Hu, M. He, B. Chen and L. Xia, Spectrochim. Acta, Part B, 2013, 86, 14–30 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2006, 1109, 183–190 CrossRef CAS PubMed.
- A. Esrafili, Y. Yamini, M. Ghambarian and B. Ebrahimpour, J. Chromatogr. A, 2012, 1262, 27–33 CrossRef CAS PubMed.
- S. S. H. Davarani, A. Morteza-Najarian, S. Nojavan, A. Pourahadi and M. B. Abbassi, J. Sep. Sci., 2013, 36, 736–743 CrossRef CAS PubMed.
- K. S. Hasheminasab, A. R. Fakhari and M. H. Koruni, J. Sep. Sci., 2014, 37, 85–91 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini, A. Saleh and M. Moradi, J. Sep. Sci., 2011, 34, 585–593 CrossRef CAS PubMed.
- K. F. Seip, J. Stigsson, A. Gjelstad, M. Balchen and S. Pedersen-Bjergaard, J. Sep. Sci., 2011, 34, 3410–3417 CrossRef CAS PubMed.
- B. Ebrahimpour, Y. Yamini, S. Seidi and F. Rezaei, Anal. Methods, 2014, 6, 2936–2942 RSC.
- H. Ebrahimzadeh, Y. Yamini, K. M. Ara and F. Kamarei, Anal. Methods, 2011, 3, 2095–2101 RSC.
- M. Moradi, Y. Yamini, A. Vatanara, A. Saleh, M. Hojati and S. Seidi, Anal. Methods, 2010, 2, 387–392 RSC.
- H. Ahmar, A. R. Fakhari, H. Tabani and A. Shahsavani, Electrochim. Acta, 2013, 96, 117–123 CrossRef CAS PubMed.
- A. Šlampová, P. Kubáň and P. Boček, J. Chromatogr. A, 2012, 1234, 32–37 CrossRef PubMed.
- H. Tabani, A. R. Fakhari and A. Shahsavani, Electrophoresis, 2013, 34, 269–276 CrossRef CAS PubMed.
- C. Basheer, J. Lee, S. Pedersen-Bjergaard, K. E. Rasmussen and H. K. Lee, J. Chromatogr. A, 2010, 1217, 6661–6667 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini, M. Rezazadeh and A. Esrafili, J. Chromatogr. A, 2012, 1243, 6–13 CrossRef CAS PubMed.
- N. J. Petersen, H. Jensen, S. H. Hansen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2009, 1216, 1496–1502 CrossRef CAS PubMed.
- M. D. Ramos-Payán, B. Li, N. J. Petersen, H. Jensen, S. H. Hansen and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2013, 785, 60–66 CrossRef PubMed.
- Y. Yamini, S. Seidi and M. Rezazadeh, Anal. Chim. Acta, 2014, 814, 1–22 CrossRef CAS PubMed.
- M. Rezazadeh, Y. Yamini, S. Seidi and A. Esrafili, J. Chromatogr. A, 2012, 1262, 214–218 CrossRef CAS PubMed.
- F. Rezaei, Y. Yamini, M. Moradi and B. Ebrahimpour, Talanta, 2013, 105, 173–178 CrossRef CAS PubMed.
- M.-R. Rouini, A. Asadipour, Y. H. Ardakani and F. Aghdasi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 800, 189–192 CrossRef CAS PubMed.
- I. Niopas and K. Mamzoridi, J. Chromatogr. B: Biomed. Sci. Appl., 1994, 656, 447–450 CrossRef CAS.
- S. Seidi, Y. Yamini, A. Heydari, M. Moradi, A. Esrafili and M. Rezazadeh, Anal. Chim. Acta, 2011, 701, 181–188 CrossRef CAS PubMed.
- V. Kiyanpour, A. R. Fakhari, R. Alizadeh, B. Asghari and M. Jalali-Heravi, Talanta, 2009, 79, 695–699 CrossRef CAS PubMed.
- Y. Yamini, M. Rezaee, A. Khanchi, M. Faraji and A. Saleh, J. Chromatogr. A, 2010, 1217, 2358–2364 CrossRef CAS PubMed.
- C. C. Loi, H. C. Boo, A. S. Mohamed and A. A. Ariffin, J. Am. Oil Chem. Soc., 2010, 87, 607–613 CrossRef CAS.
- S. S. H. Davarani, H. R. Moazami, A. R. Keshtkar, M. H. Banitaba and S. Nojavan, Anal. Chim. Acta, 2013, 783, 74–79 CrossRef CAS PubMed.
- N. C. Domínguez, A. Gjelstad, A. M. Nadal, H. Jensen, N. J. Petersen, S. H. Hansen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2012, 1248, 48–54 CrossRef PubMed.
- L. E. E. Eibak, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Pharm. Biomed. Anal., 2012, 57, 33–38 CrossRef PubMed.
- K. S. Hasheminasab and A. R. Fakhari, Anal. Chim. Acta, 2013, 767, 75–80 CrossRef CAS PubMed.
- S. Nojavan, A. Pourahadi, S. S. Hosseiny Davarani, A. Morteza-Najarian and M. Beigzadeh Abbassi, Anal. Chim. Acta, 2012, 745, 45–52 CrossRef CAS PubMed.
- M. Safari, S. Nojavan, S. S. H. Davarani and A. Morteza-Najarian, Anal. Chim. Acta, 2013, 789, 58–64 CrossRef CAS PubMed.
- J. Wang, G. Wu, W. Shi, X. Liu, C. Ruan, M. Xue and D. Ge, J. Membr. Sci., 2013, 428, 70–77 CrossRef CAS PubMed.
- T. M. Middelthon-Bruer, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Sep. Sci., 2008, 31, 753–759 CrossRef CAS PubMed.
- J. Lee, F. Khalilian, H. Bagheri and H. K. Lee, J. Chromatogr. A, 2009, 1216, 7687–7693 CrossRef CAS PubMed.
- M. Rezazadeh, Y. Yamini, S. Seidi and L. Arjomandi-Behzad, J. Chromatogr. A, 2014, 1324, 21–28 CrossRef CAS PubMed.
- Guidance for Industry; Bioanalytical Method Validation, U.S. Department of Health and Human Services, Food and Drug Administration, Rockville, 2001.
| This journal is © The Royal Society of Chemistry 2015 |