
Rapid determination of N-methylpyrrolidine in cefepime by combining direct infusion electrospray ionisation-time-of-flight mass spectrometry with field asymmetric waveform ion mobility spectrometry
Robert W.
Smith
a,
Lisa B.
Cox
a,
Aswandi
Yudin
a,
James C.
Reynolds
a,
Mark
Powell
b and
Colin S.
Creaser
*a
aCentre for Analytical Science, Department of Chemistry, Loughborough University, LE11 3TU, Loughborough, UK. E-mail: C.S.Creaser@lboro.ac.uk; Fax: +44 (0) 1509 223925; Tel: +44 (0)1509 222552
bMark Powell Scientific Limited, Neston, Cheshire CH64 9XA, UK
First published on 4th November 2014
Abstract
The determination of N-methyl pyrrolidine, a potential impurity in the cephalosporin antibiotic cefepime, by direct infusion ESI combined with field asymmetric waveform ion mobility spectrometry-mass spectrometry (ESI-FAIMS-MS) is demonstrated. The addition of a chip-based FAIMS separation prior to detection by time-of-flight mass spectrometry enables selective transmission of NMP in the presence of cefepime without interference from NMP formed by CID in the mass spectrometer interface. The limits of detection and quantification of NMP in cefepime were 0.011% (w/w) and 0.036% (w/w) NMP in cefepime respectively, well below the 0.3% (w/w) threshold concentration for NMP in cefepime. The % relative standard deviation was 3.9% with linearity for standard additions in the range 0.005–0.5 μg ml−1 NMP.
1. Introduction
Cefepime (I) is a fourth-generation cephalosporin antibiotic that was developed as a broad range activity antibiotic in the 1990s.1,2 Cefepime was found to be particularly effective against Pseudomonas aeruginosa strains which exhibited resistance against other cephalosporin antibiotics, such as ceftazidime and cefotaxime.3 Cefepime has greater activity against Gram-negative bacteria compared to other cephalosporins,4 and is used to treat a wide degree of infectious diseases.5 However, cefepime is unstable and slowly degrades over time. The degradation rate is greatly increased at higher temperatures (25–37 °C) and in an aqueous solution, the loss affects the antibiotic activity significantly enough that it is recommended cefepime not be used for clinical use in aqueous conditions at >25 °C for more than a few hours.6
The impurity N-methyl pyrrolidine (NMP, II) may be present in cefepime samples as a result of degradation of the drug. NMP has been reported as having unknown toxicity by US Environmental Protection Agency,7 and the potential for toxicity to patients receiving the drug is yet to be determined. An experimental study performed on monkeys, in which NMP (50 mg kg−1) was administered over 28–30 consecutive days resulted in them developing esotropia and ataxia.5 The dosage of NMP used in the study was much greater than the amount expected to be received in a daily dose of cefepime (∼6 g) administered to patients, but the presence of the potentially harmful impurity is still undesirable. It has been reported that an increase in the NMP level is proportional to the decline in the antibiotic potency of cefepime.8,9 Monitoring the level of NMP in cefepime is, therefore, important for ensuring drug quality for clinical use and a threshold concentration of NMP in cefepime has been set at 0.3% (w/w).
The determination of NMP in cefepime presents an analytical challenge because of the rapid degradation of cefepime to NMP in aqueous solution above 4 °C and outside the pH range 4–6, resulting in the presence of higher levels of NMP in solution than are present in the solid cefepime sample. At present, there are four main techniques employed for NMP determination: high performance liquid chromatography (HPLC), ion chromatography (IC), capillary electrophoresis (CE) and gas chromatography (GC).5,6,8–10 There are limitations associated with all these techniques. The HPLC method requires a mobile phase of pH 2 and the IC method uses a column temperature of 40 °C; both of these conditions could lead to degradation of cefepime and reduce measurement accuracy. Poor detection and quantification limits for CE and the use of a solvent with high toxicity, pyridine, to extract NMP for the GC technique makes these methods less desirable for routine analysis of NMP. The preparation of a cefepime sample for analysis therefore requires either the extraction of the NMP from the cefepime matrix to minimise the contribution from NMP produced by degradation in solution prior to analysis, or rapid analysis of the sample solution before significant degradation occurs.11
A further challenge in the determination of NMP in cefepime is that the singly and doubly protonated cefepime generated by electrospray mass spectrometry (ESI-MS) are readily fragmented in the intermediate pressure region of the mass spectrometer interface by collision-induced dissociation (CID), also known as in-source CID, to form protonated NMP. The NMP generated by CID interferes with response from the protonated NMP formed from NMP present in the cefepime sample, since NMP resulting from the sample and via CID cannot be distinguished by mass spectrometry alone.
Field asymmetric waveform ion mobility spectrometry (FAIMS) is a rapid gas phase separation technique in which the transmission of ions is based on differences in mobility in low and high electric fields; a separation dimension that is highly orthogonal to m/z.12–14 A FAIMS device is therefore able to transmit ions of a selected differential mobility, in the similar way that a quadrupole mass analyser may be used to transmit ions of a selected m/z. The combination of a FAIMS device with electrospray ionization and mass spectrometry (ESI-FAIMS-MS) has been shown to improve selectivity and limits of detection/quantification without loss of linear dynamic range.15–18
Miniaturised chip-based FAIMS-MS has been used for a range of pharmaceutical applications, including the separation of pharmaceutical excipients17 and isobaric potentially genotoxic impurities,18 and the reduction of chemical noise in the analysis of biofluids.16,17 In this paper, the potential for combining a miniaturised FAIMS device with direct infusion-ESI-MS to remove interference from NMP fragment ions generated by CID in the mass spectrometer interface is demonstrated, enabling the determination of NMP in cefepime.
2. Materials and methods
2.1 Chemicals
HPLC grade methanol, water and formic acid were obtained from Fisher Scientific (Loughborough, UK). Cefepime was obtained from Orchid Chemicals and Pharmaceuticals Limited (Chennai, India). N-Methyl pyrrolidine was supplied by Sigma-Aldrich Limited (Gillingham, UK).2.2 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). The stock was subsequently diluted to 1 μg ml−1 with methanol–water (50:50) + 0.1% formic acid. A series of dilutions in the range 0.01–1.0 μg ml−1 (2-fold at each step) were used for standard addition experiments.
:50) + 0.1% formic acid (990 μl), or NMP solution (500 μl + 490 μl diluent) for the standard additions calibration, was performed immediately before analysis. NMP and methanol–water (50:50) + 0.1% formic acid solutions were combined before preparation of cefepime samples so that weighing and dissolving cefepime, and transferring an aliquot of cefepime solution (10 μl) to the diluted NMP solution were the only steps performed between acquisitions.
:50). The stock was subsequently diluted to 1 μg ml−1 with methanol–water (50:50) + 0.1% formic acid. A series of dilutions in the range 0.01–1.0 μg ml−1 (2-fold at each step) were used for standard addition experiments.
:50) + 0.1% formic acid (990 μl), or NMP solution (500 μl + 490 μl diluent) for the standard additions calibration, was performed immediately before analysis. NMP and methanol–water (50:50) + 0.1% formic acid solutions were combined before preparation of cefepime samples so that weighing and dissolving cefepime, and transferring an aliquot of cefepime solution (10 μl) to the diluted NMP solution were the only steps performed between acquisitions.
2.3 Instrumentation
Sample solutions and standards were infused (15 μl min−1) using a syringe pump into the JetStream ESI source of an Agilent 6230 TOF MS (Agilent Technologies, UK) operated in positive ion mode. The ESI source was operated with a grounded nebuliser pressure of 35 psig, a nozzle voltage of 2000 V, a sheath gas temperature of 250 °C at a flow of, 8 l min−1, a drying gas temperature of 150 °C at a flow of 7 l min−1 and a spray shield voltage of 2500 V.A prototype chip-based FAIMS device (Owlstone Ltd., Cambridge, UK) was fitted at the inlet to the mass spectrometer (Fig. 1), behind the spray shield and in front of the MS inlet transfer capillary, which was operated at 3000 V with a nebulizer voltage of 2000 V. The chip-based FAIMS consists of multiple parallel electrode gaps (100 μm) with a short path length (700 μm), allowing high field strengths (<300 Td) and short ion residence times (50–250 μs). The fragmentor voltage applied at the TOF inlet was varied between 75 and 375 V (25 V steps), but set to 200 V for quantitative measurements. The MS acquisition rate was set to 10 scans per s for FAIMS scanning experiments and 1 scan per s for quantification using static FAIMS.
 | ||
| Fig. 1 Schematic diagram of the ESI-FAIMS-TOFMS interface. | ||
Scanning FAIMS experiments were carried out in the compensation field (CF) range −2 to 5 Td (scan speed 0.5 Td s−1) at each dispersion field (DF), which was incremented in the range 180–230 Td in 5 Td steps to determine optimum conditions to separate NMP from cefepime. Static FAIMS experiments used a fixed CF (0 Td) and DF (180 Td) to pre-select NMP generated in the ESI ion source. Cefepime solutions were infused into the source and data acquired for 2 min per sample. Data were acquired using Agilent MassHunter Acquisition B.05.00 and processed using Agilent MassHunter Qualitative Analysis B.05.00. Microsoft Excel 2010 was used to produce CF plots.
3. Results and discussion
Cefepime degrades rapidly in solution, so the time between preparing a cefepime solution for analysis and acquiring data for the determination of NMP can potentially influence the accuracy of the quantification of NMP in the original cefepime sample. Sample preparation and analysis time therefore needs to be minimised, making the direct infusion of the sample into the ESI source of a mass spectrometer a potentially rapid alternative to conventional approaches for NMP determination. However, the conditions used for ESI-MS may lead to fragmentation of cefepime ions to protonated NMP in the mass spectrometer interface, which will increase the NMP response. The potential of FAIMS to separate the response for NMP in the original cefepime sample from NMP generated by in-source CID was therefore investigated.3.1 ESI-MS of cefepime: in-source CID
ESI-MS analysis of a cefepime solution spiked with NMP (0.1% w/w), shows the presence of intense doubly (m/z 241) and singly (m/z 481) protonated cefepime peaks in the spectrum, with a relatively weak response for protonated NMP (m/z 86; Fig. 2). The voltage applied to a lens in the intermediate pressure region of the mass spectrometer interface affects transmission of ions into the mass spectrometer and can also cause collision-induced dissociation (CID).15,16 The response for NMP in Fig. 2 may therefore be attributed to contributions from NMP in the cefepime sample and from CID of protonated cefepime in the mass spectrometer interface. Optimising the lens voltage (termed the fragmentor voltage for the mass spectrometer used in this study) is important for the analysis because it has a significant impact on the analyte response. | ||
| Fig. 2 Electrospray mass spectrum of cefepime sample with NMP spiked at 0.1% (w/w). | ||
The effect of the fragmentor voltage on the intensity of the NMP (m/z 86) and cefepime (m/z 481) ions was investigated to determine the extent of cefepime fragmentation by CID in the mass spectrometer interface. NMP and cefepime were infused separately into the ESI source and the fragmentor voltage varied in the range 75–375 V (Fig. 3). The maximum response for transmission of the NMP ion was at a fragmentor voltage of 200 V (Fig. 3a). However, infusion of a cefepime standard also resulted in a response for NMP, which increased at fragmentor voltages above 200 V as a result of CID in the mass spectrometer interface at all fragmentor voltages studied (Fig. 3b). The NMP response in the cefepime sample directly infused into the ESI source therefore arises from a combination of the NMP in the cefepime sample and CID of the cefepime ion generated by ESI, preventing the direct determination of NMP by ESI-MS. It would be possible, in principle, to subtract the percentage contribution to the total NMP response from in-source CID in order to determine the NMP in cefepime, but separation of these contributions allows the direct determination of NMP.
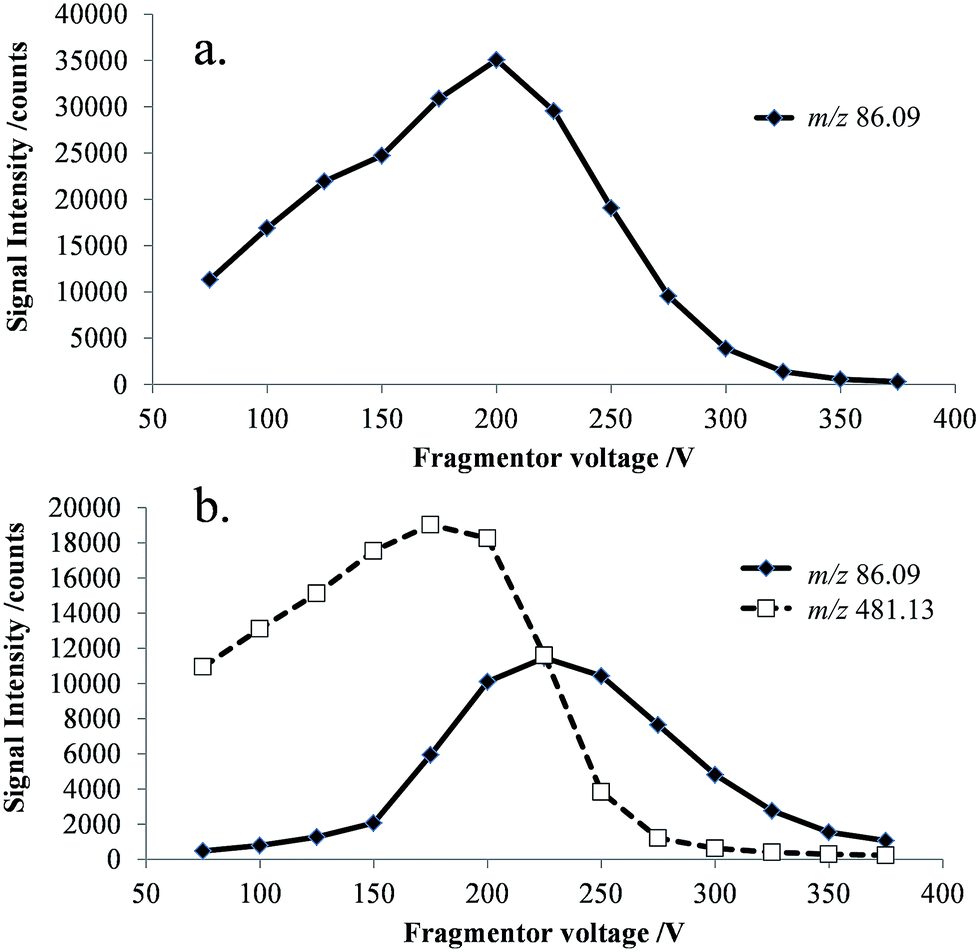 | ||
| Fig. 3 Selected ion responses for [NMP + H]+ (m/z 86) and [cefepime + H]+ (m/z 481) at 25 V increments of the fragmentor voltage (75–375 V), (a) NMP standard; and (b) cefepime standard. | ||
3.2 Direct infusion ESI-FAIMS-MS studies
The FAIMS dispersion field (DF) and compensation field (CF) characteristics of protonated cefepime and NMP were investigated to determine whether these ions could be separated on the basis of differences in their high field and low field mobility. Fig. 4 shows the mass-selected ion responses for cefepime (m/z 241 and 481, solid grey and dashed line) and NMP (m/z 86, solid black line) in the FAIMS compensation field (CF) spectrum (−1.5 to 2 Td) of a solution of cefepime containing 0.1% (w/w) NMP. A DF stepping experiment was used to determine the optimum DF (180 Td) for the separation of NMP and cefepime. The protonated NMP selected ion response shows two peaks at CF 0 Td and at CF 0.7 Td. The first peak (CF 0 Td, Fig. 4 insert) matches the CF spectrum of an NMP standard (data not shown) indicating that this peak arises from NMP in the solution subjected to the ESI. The second Td peak, centred at CF 0.7 Td, peak is associated with NMP formed by CID of the singly and doubly protonated cefepime in the mass spectrometer interface after transmission of the intact cefepime ions through the FAIMS, which therefore appears in the FAIMS spectrum at the same CF as cefepime. There was no evidence of significant fragmentation of the cefepime within the FAIMS device. Separation of NMP (0 Td) and cefepime (0.7 Td) with NMP at 0.1% (w/w) demonstrates the effectiveness of FAIMS to transmit NMP in the presence of a 1000 fold excess of cefepime.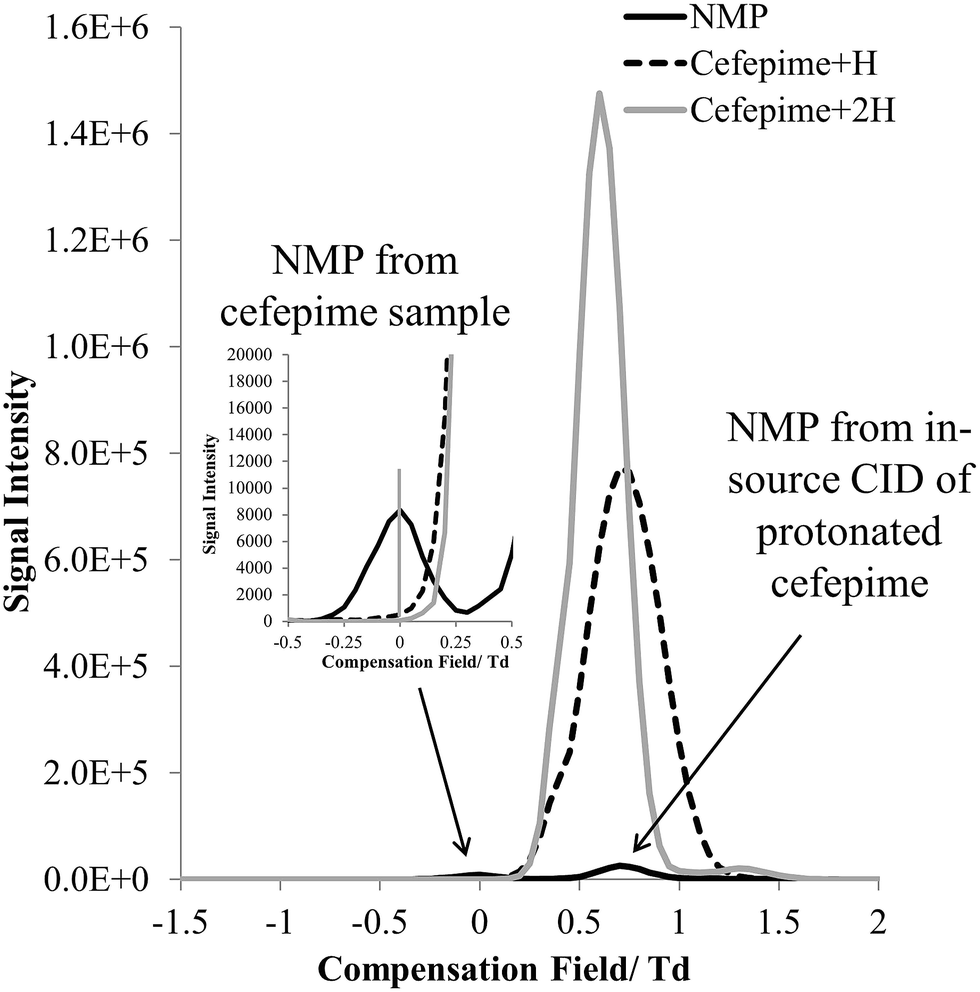 | ||
| Fig. 4 CF spectra showing selected ion response (SIR) of [NMP + H]+ (m/z 86) and [cefepime + H]+ (m/z 481) at a 1:1000 ratio (w/w) at DF 180. | ||
The mass spectrum extracted from the FAIMS CF scan at a CF of 0 Td shows the protonated NMP to be the base peak in the mass spectrum (Fig. 5). There is a weak response from the protonated cefepime at this CF, because of the 1000 fold excess of the drug, but this is small compared to the relative responses for NMP and cefepime in the absence of a FAIMS separation. Fig. 5 shows no response for the doubly protonated cefepime. The FAIMS device is therefore able to transmit the NMP generated in the ESI source from the original cefepime solution selectively, with no contribution from the NMP formed by in-source CID, making it possible to determine NMP in cefepime by direct ESI-FAIMS-MS without prior chromatographic separation.
 | ||
| Fig. 5 Mass spectrum of an infused sample of cefepime spiked with NMP at 0.1% (w/w) obtained by ESI-FAIMS-MS (DF 180 Td; CF 0 Td). | ||
The FAIMS was operated in static mode (CF 0.1 Td, DF 180 Td) to selectively transmit NMP generated in the ESI ion source through the device for the determination of NMP in cefepime. Cefepime solutions were infused into the source 10 minutes after preparation to avoid degradation in solution and data acquired with an analysis time of 2 min per sample from the start of the infusion. The limit of detection for NMP was estimated to be 0.11 mg g−1 in the cefepime sample, giving a limit of quantitation 0.36 mg g−1, which is equivalent to 0.036%, well below the 0.3% threshold concentration for NMP in cefepime. The %RSD for replicate samples was 3.85% (n = 6, 0.37 μg ml−1) for NMP, with good linearity (R2 0.9979) for standard additions in the range 0.005–0.5 μg ml−1. These preliminary quantitative data demonstrate the potential of direct ESI-FAIMS-MS for the determination of NMP in cefepime at, and below, the threshold concentration, subject to full validation of the method.
4. Conclusions
The formation of NMP by solution degradation and CID of cefepime in the intermediate pressure region of the mass spectrometer interface presents an analytical challenge for the determination of NMP in cefepime. The use of a FAIMS separation of NMP and cefepime ions prior to mass spectrometric analysis allows NMP in a cefepime sample to be distinguished from NMP ions generated by CID in the mass spectrometer interface. Direct infusion of a cefepime solution using ESI-FAIMS-MS is shown to be a rapid, selective method for the quantitative determination of NMP in cefepime with an LOQ well below the required 0.3% threshold and good precision. The approach reported here for NMP in cefepime has potential for generic application to the direct determination of impurities present in other active pharmaceutical ingredients which fragment to yield the impurity ion by CID.Acknowledgements
The authors thank Owlstone Ltd. and Loughborough University for financial support. We thank Owlstone Ltd. and Agilent Technologies for the provision of instrumentation and technical support, and Quay Pharmaceuticals for provision of chemicals.References
- R. H. Barbhaiya, S. T. Forgue, C. R. Gleason, C. A. Knupp, K. A. Pittman, D. J. Weidler, H. Movahhed, J. Tenney and R. R. Martin, Antimicrob. Agents Chemother., 1992, 36, 552–557 CrossRef CAS.
- P. Van Der Auwera and P. Santella, J. Antimicrob. Chemother., 1993, 32, 103–115 CrossRef CAS PubMed.
- J. Fung-Tomc, E. Huczko, M. Pearce and R. E. Kessler, Antimicrob. Agents Chemother., 1988, 32, 1443–1445 CrossRef CAS.
- P. F. Sprauten, P. M. Beringer, S. G. Louie, T. W. Synold and M. A. Gill, Antimicrob. Agents Chemother., 2003, 47, 1991–1994 CrossRef CAS PubMed.
- N. Baririan, H. Chanteux, E. Viaene, H. Servais and P. M. Tulkens, J. Antimicrob. Chemother., 2003, 51, 651–658 CrossRef CAS PubMed.
- E. Viaene, H. Chanteux and P. M. Tulkens, Antimicrob. Agents Chemother., 2002, 46, 2327–2332 CrossRef CAS.
- Office of Prevention, Pesticides and Toxic Substances, United States Environmental Protection Agency, Washington, DC, http://www.epa.gov/opprd001/inerts/inert_nonfooduse.pdf, (accessed November 2014).
- S. J. Prasanna, H. K. Sharma, K. Mukkanti, V. J. Kumar, G. Raja and M. Sivakumaran, J. Chromatogr. Sci., 2010, 48, 830–834 CAS.
- M. A. Farajzadeh, L. Goushjuii and Y. Bashour, J. Sep. Sci., 2010, 33, 3767–3773 CrossRef CAS PubMed.
- N. H. Subramanian, S. Thyagarajan, P. Manigandan, R. G. Jeevan and G. Radhakrishnan, J. Chromatogr. Sci., 2009, 47, 549–552 CAS.
- N. Page, R. Stevenson and M. Powell, Anal. Methods, 2014, 6, 1248–1253 RSC.
- B. M. Kolakowski and Z. Mester, Analyst, 2007, 132, 842–864 RSC.
- R. Guevremont, Can. J. Anal. Sci. Spectrosc., 2004, 49, 105–113 CAS.
- C. S. Creaser, J. R. Griffiths, C. J. Bramwell, S. Noreen, C. a. Hill and C. L. P. Thomas, Analyst, 2004, 129, 984–994 RSC.
- R. W. Smith, L. J. Brown, D. E. Toutoungi, J. C. Reynolds, B. Boyle and C. S. Creaser, Int. Labmate, 2014, 39, 14–16 Search PubMed.
- R. W. Smith, D. E. Toutoungi, J. C. Reynolds, A. W. T. Bristow, A. Ray, A. Sage, I. D. Wilson, D. J. Weston, B. Boyle and C. S. Creaser, J. Chromatogr. A, 2013, 1278, 76–81 CrossRef CAS PubMed.
- L. J. Brown, R. W. Smith, D. E. Toutoungi, J. C. Reynolds, A. W. T. Bristow, A. Ray, A. Sage, I. D. Wilson, D. J. Weston, B. Boyle and C. S. Creaser, Anal. Chem., 2012, 84, 4095–4103 CrossRef CAS PubMed.
- R. W. Smith, J. C. Reynolds, S.-L. Lee and C. S. Creaser, Anal. Methods, 2013, 5, 3799–3802 RSC.
| This journal is © The Royal Society of Chemistry 2015 |