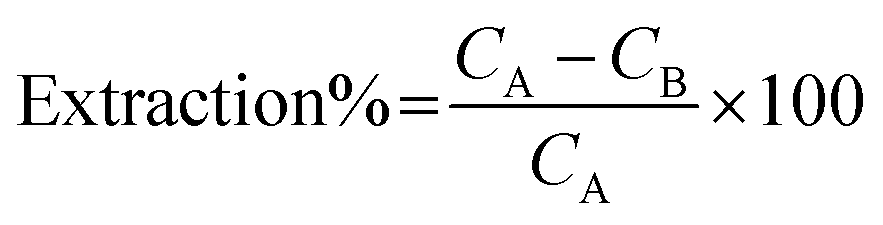DOI:
10.1039/C4AY01991A
(Paper)
Anal. Methods, 2015,
7, 313-320
An Fe3O4@SiO2@polypyrrole magnetic nanocomposite for the extraction and preconcentration of Cd(II) and Ni(II)†
Received
23rd August 2014
, Accepted 7th October 2014
First published on 18th November 2014
Abstract
This work describes a novel Fe3O4@SiO2@polypyrrole magnetic nanocomposite and its application in the preconcentration of Cd(II) and Ni(II) ions. The parameters affecting the preconcentration procedure were optimised by a Box–Behnken design using response surface methodology. Three variables (extraction time, magnetic sorbent amount, and pH) were selected as the main factors affecting the sorption step, while four variables (type, volume and concentration of the eluent, and elution time) were selected as the main factors in the optimisation study of the elution step. Following the sorption and elution of analytes, the ions were quantified by FAAS. The limits of detection were 0.3 and 1.2 ng mL−1 for Cd(II) and Ni(II) ions, respectively. All the relative standard deviations were less than 8.8%. The obtained sorption capacities (in mg g−1) of this new sorbent were 120 for Cd(II) and 98 for Ni(II). Ultimately, this nanocomposite was successfully applied to the rapid extraction of trace quantities of heavy metal ions from seafood samples, and satisfactory results were obtained.
1. Introduction
Heavy metal ions are toxic pollutants that exist in different samples and their presence concerns industries and environmental organisations all over the world. Most of these pollutants are very toxic and dangerous for human health. Thus the determination of trace amounts of heavy metals is often a major task for analytical chemists, as it is a good tool for the monitoring and identification of toxicants in environmental samples. Among heavy metals which exist in the environment, cadmium monitoring is very crucial due to the fact that cadmium concentration in the environment is increasing dramatically.1,2 Cadmium exposure can be linked to diseases associated with aging such as osteoporosis, prostate, and pancreatic cancer.3,4 Another heavy metal which is also toxic is nickel. Aspirating cadmium derivatives can lead to serious problems, including nasopharynx, lung and dermatological diseases and malignant tumours. Moreover, nickel can cause skin disorder, known as nickel-eczema which is a considerable health problem exclusively among women.5 It has been proven to be a carcinogenic agent. Various instrumental techniques, including electrothermal atomic absorption spectrometry (ETAAS),6,7 inductively coupled plasma-optical emission spectrometry (ICP-OES),8 flame atomic absorption spectrometry (FAAS),9 inductively coupled plasma-mass spectrometry (ICP-MS),10 and total reflection XRF-spectrometry,11 have been used for the determination of heavy metals. Since the heavy metals' concentration level in environmental samples is fairly low and the complexity of matrices is the main problem, thus preconcentration techniques are often required.12 Various procedures such as liquid–liquid extraction (LLE),13 cloud point extraction,14 chemical precipitation,15 ion exchange,16 and solid phase extraction (SPE) have been developed for the extraction and preconcentration of heavy metals in natural matrices.17–19
Among the aforementioned methods, the most commonly used technique for the preconcentration of heavy metal ions from environmental samples is solid phase extraction. Widespread application of SPE is due to its intrinsic simplicity, rapidity, minimal cost, and low consumption of reagents.20 By the advent of SPE, diverse sorbents such as carbon nanotubes,21–24 magnetic nanoparticles,25 solid sulfur,26 cotton,27 and modified porous materials28,29 have been utilized.
In recent years, much attention has been paid to the application of nano-structured materials, particularly magnetic nanoparticles, in analytical chemistry.30–32 Magnetic Fe3O4 nanoparticles (Fe3O4 NPs) are a special kind of nano-sized materials that exhibit magnetic properties in addition to the general features of nanometre-sized materials. However, naked Fe3O4 nanoparticles tend to aggregate, are prone to oxidation, and are not selective toward complex matrices. Hence, the surfaces of these magnetic nanoparticles have been modified with specific ligands to make them selective and appropriate sorbents. Another approach to this problem involves core–shell nanostructures. Fe3O4 NPs are one of the most generally used nanostructures as a core to synthesise core–shell systems due to their strong superparamagnetic properties. Silica is one of the most ideal shells due to its several unique advantages: (a) the silica shell prevents Fe3O4 NPs from aggregation at a wide range of pH values and improves their chemical stabilities; (b) the silica surface is often terminated by silanol groups that can react with silane coupling agents in order to conjugate with a variety of specific ligands; and (c) the SiO2 layers possess good hydrophilic behaviours.33 According to the hard and soft acid and base theory (HSAB), soft acids including most heavy metals react faster and form stronger bonds with ligands containing N and S moieties.34
In a previous work, yolk–shell composites with a movable Fe3O4 core inside the hollow polypyrrole (PPy) capsules were obtained through a template-assistant selective etching method. Yolk–shell Fe3O4@PPy composites were used as supports for the deposition of Pd NPs. Finally, Fe3O4@PPy/Pd catalysts were used in the reduction of methylene blue dye with sodium borohydride as reducing agent.35 In another work, a high-magnetic heavy metal ion adsorbent composed, from inside to outside, of an Fe3O4 polycrystal spherical cluster, an amorphous SiO2 protective layer and a polypyrrole adsorption outer layer was synthesised.36
In this work, an Fe3O4@SiO2@polypyrrole magnetic nanocomposite has been utilised as a novel sorbent for the fast separation and preconcentration of Cd(II) and Ni(II) ions in various matrices. In ref. 36, FeCl3 was used as an oxidising agent for the oxidative polymerisation of pyrrole; however, we used ammonium peroxydisulfate in 1.0 mol L−1 HCl solution for the polymerisation step, and the two Fe3O4 NPs synthetic procedures are completely different. In the previous work, a solvothermal method was used for the synthesis of Fe3O4 NPs, while we applied a co-precipitation route, which is easier and saves time compared to the solvothermal route. The sorbent was characterised by Fourier transform infrared spectroscopy (FT-IR), elemental analysis, transmission electron microscopy (TEM), X-ray diffraction (XRD) and scanning electron microscopy (SEM). The magnetic property of the sorbent provides a rapid and easy separation of the new solid phase from the solution. The modification of the sorbent helps it to show selectivity towards heavy metals. In addition, its nanosize characteristic enhances the surface area and sorption capacity of this new sorbent. A Box–Behnken design was used to explore the optimum conditions of this method through response surface methodology. Finally, the nanosorbent was used for the preconcentration and determination of Cd(II) and Ni(II) ions in different sea samples, and satisfactory results were obtained.
2. Experimental
2.1. Reagents and solutions
All reagents (pyrrole (Py), FeCl2, FeCl3, HCl, HNO3, NaOH, KCl, ammonium hydroxide (25%), and tetraethyl orthosilicate (TEOS), ammonium peroxydisulfate (APS), ethanol, methanol ethanol, and acetone) were of analytical grade, were purchased from Merck (Darmstadt, Germany) or Fluka (Seelze, Germany) and were used without further purification. Standard solutions of Cd(II) and Ni(II) (1000 mg L−1) were purchased from Merck. All solutions were prepared using doubly distilled water.
2.2. Instrumentation
An AA-680 Shimadzu (Kyoto, Japan) flame atomic absorption spectrometer with a deuterium background corrector was used for the determination of Cd(II) and Ni(II) ions. Cadmium and nickel hollow cathode lamps (HCL) were used as the radiation sources with wavelengths of 228.8 and 232.0 nm, respectively. All measurements were carried out in an air/acetylene flame. The pH of the solutions were measured at 25 ± 1 °C with a digital WTW Metrohm 827 Ion analyser (Herisau, Switzerland) equipped with a combined glass-calomel electrode. CHN analysis was performed by a Thermo Finnigan Flash EA112 elemental analyser (Okehampton, UK). IR spectra were recorded by a Bruker IFS-66 FT-IR spectrophotometer (Bruker). SEM (KYKY-3200, Beijing, China) was performed by gently distributing the sample powder on stainless steel stubs. TEM analyses were performed using a LEO 912AB electron microscope (Leo Ltd., Germany). XRD patterns were obtained with a Philips-PW 12C diffractometer (Amsterdam, the Netherlands) using Cu Kα radiation.
2.3. Preparation of standard solutions
The standard reference material was digested with 6 mL of HCl (37% (v/v)) and 2 mL of HNO3 (65% (v/v)) in a microwave digestion system. Digestion was carried out for 2 min at 250 W, 2 min at 0 W, 6 min at 250 W, 5 min at 400 W and 8 min at 550 W followed by venting for 8 min. The digestion residue was then diluted with doubly distilled water.37 Standard stock solutions (1000 mg L−1) of K+, Na+, Ca(II), Mn(II), Mg(II), Hg(II), Co(II), Zn(II), Ni(II), Fe(III), Cr(III), Cu(II), Al(III), AsO43− and CrO42− were prepared in 2% (v/v) HNO3 solution. The mixed working standard solutions were prepared by diluting an appropriate amount of the stock solution with doubly distilled water. All of these solutions were stored at ambient temperature.
2.4. Synthesis of Fe3O4@SiO2@polypyrrole magnetic nanocomposite
2.4.1. Synthesis of Fe3O4@SiO2 core–shell nanoparticles.
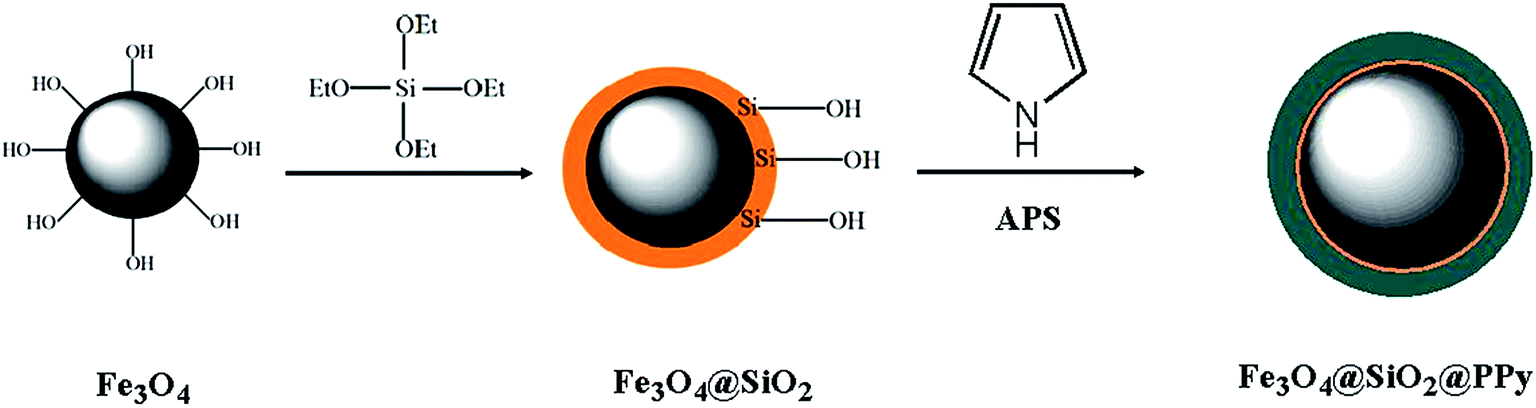
Fe3O4 NPs were synthesised according to our previously reported procedure.29 Subsequently, 1.0 g of the synthesised Fe3O4 NPs were dispersed in a solution of 200 mL deionised water, 50 mL ethanol and 3.0 mL NH4OH (25%). In the next step, a total of 2.5 mL TEOS was added dropwise to the mixture under vigorous stirring in order to obtain the Fe3O4@SiO2 core–shell NPs.38 After 12 h of stirring at 40 °C, the Fe3O4@SiO2 NPs were separated by a strong magnet, washed with ethanol and then dried at room temperature (Fig. 1).
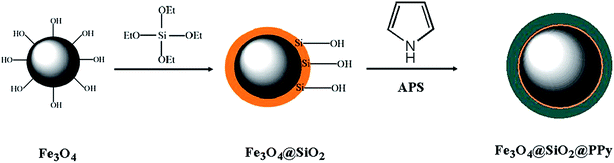 |
| | Fig. 1 A schematic diagram for the synthesis of Fe3O4@SiO2@PPy magnetic nanocomposite. | |
2.4.2. Modification of Fe3O4@SiO2 nanoparticles by polypyrrole.
The procedure for modifying Fe3O4@SiO2 core–shell nanoparticles includes two steps as follows. In the first step, 1.0 g of Fe3O4@SiO2 NPs was suspended in 150 mL of HCl solution (1 mol L−1) and sonicated for 15 min. Subsequently, 1.0 mL of pyrrole was added into the reaction mixture and stirred for 30 min. The solution was then cooled to 0 °C, and a stoichiometric amount of APS in 1 mol L−1 HCl solution was added dropwise to the reaction mixture. Polymerisation continued for 10 h, and a precipitate was finally obtained.39 Afterward, the Fe3O4@SiO2@PPy nanocomposites were separated by a strong magnet, washed several times with methanol and water to remove any impurities and dried at room temperature. A schematic of the synthetic procedure for the Fe3O4@SiO2@PPy nanocomposite is depicted in Fig. 1.
2.5. Sorption and elution steps
The extraction of Cd(II) and Ni(II) ions from aqueous solutions was investigated by batch mode analysis. The sorption step was performed in test tubes containing 25 μg of Cd(II) and Ni(II) ions in 50 mL of doubly distilled water. According to the preliminary experimental design, the pH of the solutions was adjusted by the dropwise addition of 1.0 mol L−1 NH3 and 1.0 mol L−1 HCl. The magnetic sorbent was then added into the solutions, and the mixture was stirred for an appropriate amount of time to completely extract these heavy metal ions from solution. Finally, the test tubes were exposed to a strong magnet (15 cm × 12 cm × 5 cm, 1.4 T), and the permanent magnet in the wall caused the sorbent to aggregate on one side of the test tube. The sorbed amounts of Cd(II) and Ni(II) ions were determined using FAAS based on their concentration change in solution after the sorption procedure was completed. The instrument's response was periodically checked with known Cd(II) and Ni(II) standard solutions. The extraction percentage for each ion was calculated using the following equation:
where CA and CB are the initial and final concentrations (mg L−1) of each ion in solution, respectively. In the elution step, 7.5 mL of a 1.5 mol L−l HNO3 solution was added to the magnetic sorbent as an eluent, and the solution was shaken. This mixture was again exposed to the strong magnet, and the clear solution containing the eluted heavy metal ions was introduced to FAAS to determine the amount of each ion.
2.6. Real sample pre-treatment
2.6.1. Fish and shrimp samples.
Fish samples were collected from Caspian Sea, Siahroud River and two different sites in Moosa creek. Moosa creek is located in the northwest of the Persian Gulf (southern Iran) and has several subsidiary creeks. Among them, Jafar creek was chosen as a polluted site, and Behad creek was chosen as a reference site since it is far from petrochemical industries. Shrimp and canned tuna were purchased from local supermarkets in Tehran, Iran. The river fish were collected from Siahroud River (Gilan Province in northern Iran). The fish samples and shrimp were placed in ice, transferred to the laboratory and stored at −20 °C prior to analysis. The fish (Platycephalus indicus) and shrimp samples were dissected with a clean plastic knife, and a part of the muscle was quickly removed and dried in an oven at 70 °C for 48 h. Analyses were carried out according to the procedure reported by Yilmaz (2003).37 A porcelain mortar was used to grind the dried tissues, and 0.5 g of each sample was digested with 5 mL of concentrated HNO3 in Teflon beakers for 4 h at 100 °C. The content was filtered into a 25 mL standard volumetric flask and diluted to 25 mL with doubly distilled water. Sediment samples were collected using a Peterson grab sampler, stored in ice in a plastic bag, transferred to the laboratory and kept at −20 °C before analysis. Samples (1 g) were digested with 6 mL of HCl (37%) and 2 mL of HNO3 (65%) in a microwave digestion system. Digestion was carried out for 2 min at 250 W, 2 min at 0 W, 6 min at 250 W, 5 min at 400 W and 8 min at 550 W followed by venting for 8 min. After acid digestion was completed, the acid digests were diluted to 100 mL with doubly distilled water.
2.6.2. Reference material.
The concentrations of Cd(II) and Ni(II) ions in a standard reference material (seafood mix 02-2932) were determined at optimum conditions. The standard material was digested according to the abovementioned procedure for fish and blank digestion. The pH of the solution was adjusted to 6.4 for the separation and preconcentration of Cd(II) and Ni(II) ions from solution. Ultimately, the above preconcentration procedure was applied to the resulting solutions.
2.7. Experimental design methodology
In order to thoroughly investigate the effects of the experimental variables that can significantly influence the extraction procedure, individual factors must be considered along with nonlinear and interaction effects. A rational experimental design is a type of chemometric approach that allows the simultaneous variation of all experimental factors and reduces the required time and number of trials, which results in the reduction of the overall required cost. The Box–Behnken design (BBD) is probably the most widely used experimental design applied for fitting second-order response surfaces. This cubic design is characterised by a set of points lying at the midpoint of each edge of a multidimensional cube; a centre point can be replicated, whereas the ‘missing corners’ help the experimenter avoid using the combined factor extremes. This property prevents a potential loss of data.40
In this study, the StatGraphics plus 5.1 package was used to analyse the experimental design data and calculate the predicted responses.
3. Results and discussion
3.1. Characterisation studies
3.1.1. FT-IR spectra and elemental analysis.
The FT-IR spectrum of the modified Fe3O4@SiO2 NPs was recorded using the KBr pellet method. The presence of absorption peaks at 3412 cm−1 (N–H), 1583 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and 1479 cm−1 (CC) indicated the existence of PPy on the surface of the Fe3O4@SiO2 NPs (Fig. 1S, ESI†). Moreover, elemental analysis revealed the presence of 0.52% N in the structures of the Fe3O4@SiO2 NPs, indicating that the Fe3O4@SiO2 NPs were successfully modified with PPy.
N) and 1479 cm−1 (CC) indicated the existence of PPy on the surface of the Fe3O4@SiO2 NPs (Fig. 1S, ESI†). Moreover, elemental analysis revealed the presence of 0.52% N in the structures of the Fe3O4@SiO2 NPs, indicating that the Fe3O4@SiO2 NPs were successfully modified with PPy.
3.1.2. Transmission and scanning electron microscopies.
The morphologies of the Fe3O4@SiO2@PPy NPs were studied by SEM. As illustrated in Fig. 2a, the spherical Fe3O4 NP structures were approximately preserved after modification with PPy. The SEM micrograph also confirmed that the magnetic sorbents are nano-sized with an average particle size of 50 nm. The morphologies of the Fe3O4@SiO2@PPy NPs were also characterised by TEM (Fig. 2b); Fig. 2b demonstrates that Fe3O4 nanoparticles were successfully coated by a thin layer of SiO2. In this figure, the formation of the core–shell structure is confirmed by the presence of two regions with different electron densities:33 an electron dense region corresponding to Fe3O4 cores with uniform sizes of about 15–20 nm and a less dense and more translucent region surrounding these cores composed of SiO2 and PPy coating shells with a thickness of about 10–15 nm.
 |
| | Fig. 2 (a) SEM and (b) TEM images of the Fe3O4@SiO2@PPy magnetic nanocomposite. | |
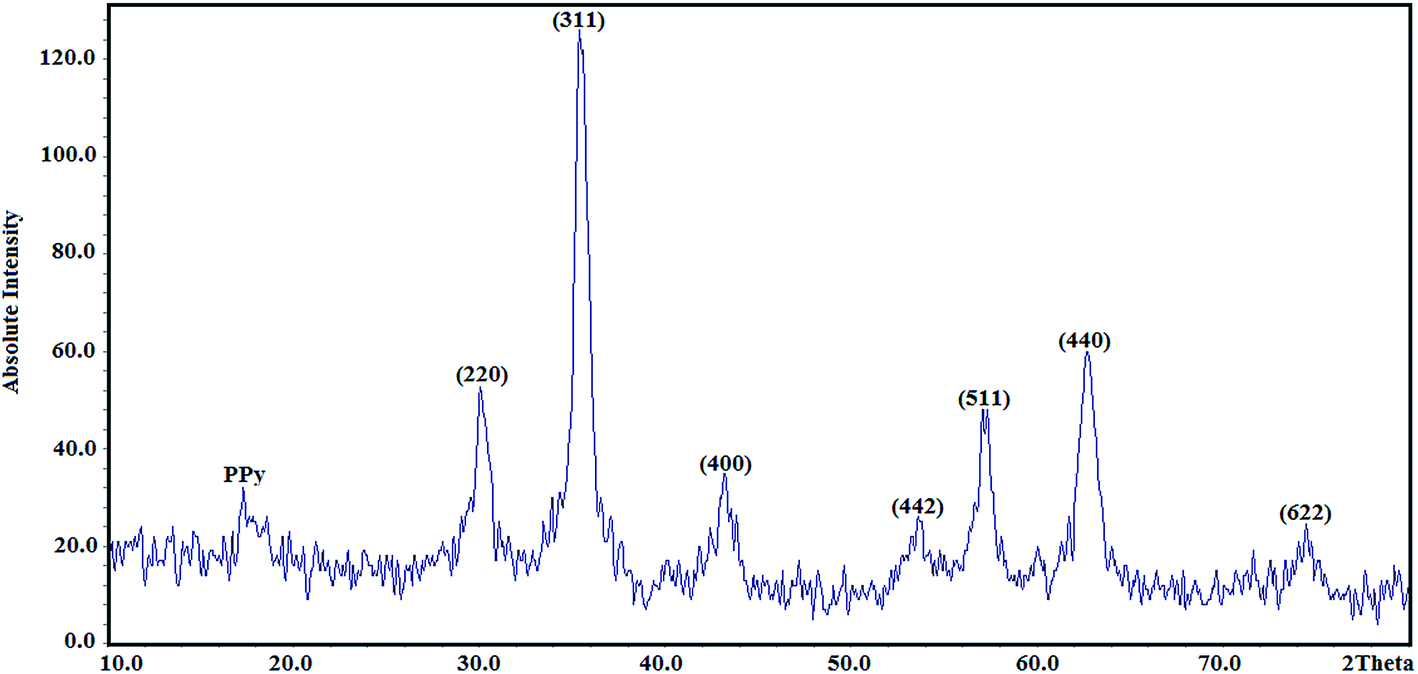
3.1.3. X-ray diffraction.
The structures of the Fe3O4@SiO2@PPy NPs were analysed by XRD, and the results are shown in Fig. 3. All of the Fe3O4@SiO2@PPy diffraction peaks show that the Fe3O4 crystal is well retained even after modification with PPy. In addition, seven characteristic diffraction peaks appeared at 2θ = 30.1°, 35.5°, 43.2°, 53.8°, 57.1°, 62.6° and 74.7°, which correspond to the (220), (311), (400), (422), (511), (440) and (622) Bragg diffractions of the face-centred cubic Fe3O4 core. Moreover, the diffraction peak appearing at 2θ = 17.3° is related to PPy.35 The average crystallite size of the Fe3O4@SiO2@PPy NPs was estimated from the XRD pattern using the Scherrer formula:
where D is average crystallite size, λ is the X-ray wavelength, β is the full-width at half maximum (FWHM) and θ is the diffraction angle. Here, K = 0.9 is chosen for a spherical shape. Thus, the crystallite size of Fe3O4@SiO2@PPy NPs was computed from the XRD pattern and found to be about 60 nm.
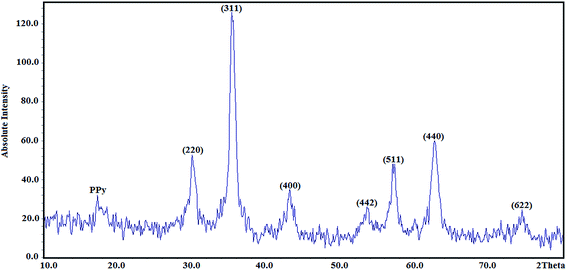 |
| | Fig. 3 High-angle X-ray diffraction pattern of the Fe3O4@SiO2@PPy magnetic nanocomposite. | |
3.2. Optimisation of the preconcentration procedure
3.2.1. Sorption step.
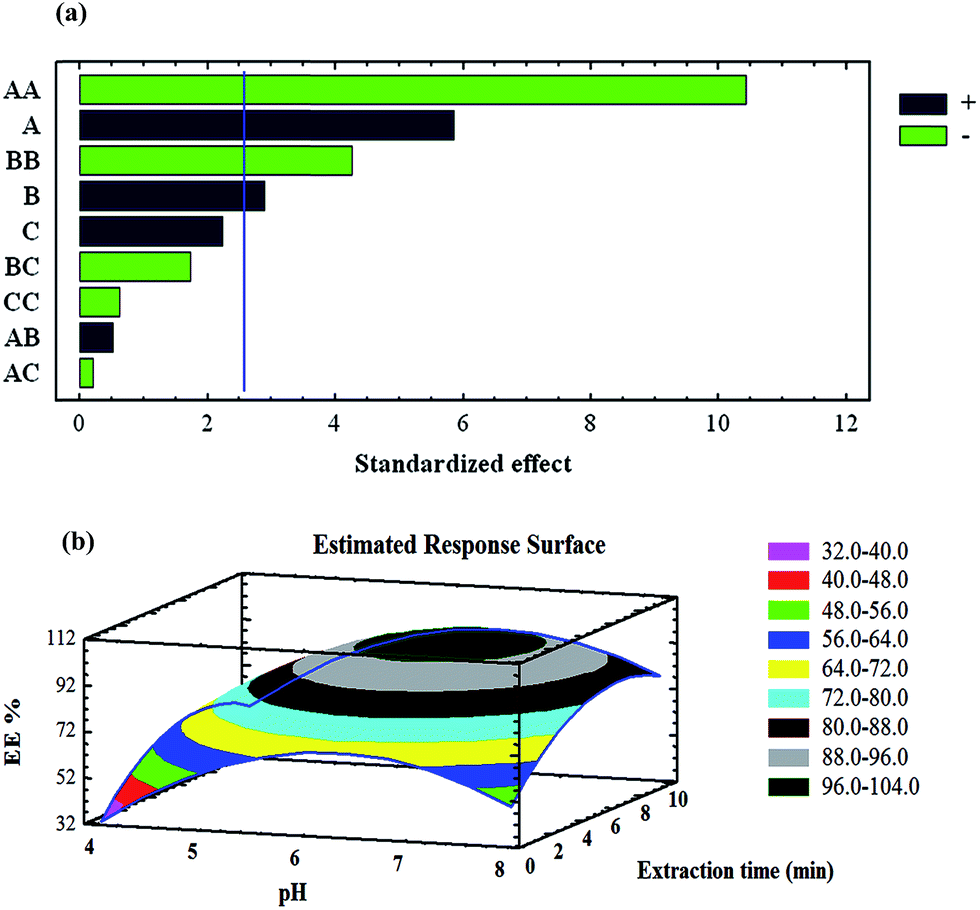
The optimisation step for the uptake of metal ions on the magnetic nanosorbent was carried out using BBD. Variables affecting the extraction efficiency were chosen to be pH, amount of magnetic nanosorbent, and extraction time. Other parameters involved in the extraction were kept constant, particularly the concentration of heavy metal ions (0.5 mg L−1). This design permitted the responses to be modelled by fitting a second-order polynomial, which can be expressed as follows:
| Y = β0 + β1x1 + β2x2 + β3x3 + β12x1x2 + β13x1x3 + β23x2x3 + β11x12 + β22x22 + β33x132 |
where x1, x2, and x3 are the independent variables, β0 is the intercept, β1–β33 are the regression coefficients, and Y is the response (removal% or recovery%). The number of experiments (N) is then defined by the expression below:
where K is the number of variables and Co is the number of centre points.28 In this study, K and Co were both set at 3, which meant that 15 experiments had to be done. The levels of each factor are listed in Table 1. The results of the analysis of variance (ANOVA) produced the Pareto chart of main and interaction effects (Fig. 3a). The standard effect was estimated for computing the t-statistic for each effect. The vertical line on the plot indicates statistically significant effects. The bar extracted beyond the line corresponds to the effects that are statistically significant at the 95% confidence level.40,41 Furthermore, the positive or negative sign (corresponding to a coloured or colourless response) can enhance or reduce the extraction efficiency, respectively, while increasing from the lowest to the highest level set for that specific factor. According to the Pareto chart, the pH of the solution has the most significant positive effect on the extraction efficiency; the uptake of heavy metal ions increases as pH increases. In less acidic solutions, the uptake is quite low. This observation is due to the protonation of the magnetic nanosorbent active sites, especially the N atoms of PPy. As pH increases, the protonation of these active sites decreases, making complex formation and the sorption of heavy metal ions to the magnetic nanosorbent more favourable. Response surface methodology (RSM; Fig. 3b) was applied to analyse the simultaneous effects of extraction time and pH on the response plot displaying the interaction between these independent variables. The sorption efficiency of target metal ions increased with increasing pH. Moreover, the extraction time and sorbent amount both showed positive effects on the extraction efficiency and were the second and third most important factors, respectively. According to the overall results of the optimisation study, the following experimental conditions were chosen: pH, 6.4; extraction time, 6.0 min; amount of magnetic sorbent, 30 mg.
Table 1 Experimental variables and levels of the Box Behnken design (BBD)
|
|
|
Level |
| Lower |
Central |
Upper |
| Sorption step |
A: pH |
4.0 |
6.0 |
8.0 |
| B: uptake time (min) |
2.0 |
6.0 |
10 |
| C: magnetic sorbent amount (mg) |
20 |
30 |
40 |
| Elution step |
A: elution time (min) |
10 |
15 |
20 |
| B: eluent concentration (mol L−1) |
0.1 |
1.05 |
2.0 |
| C: eluent volume (mL) |
2.0 |
6.0 |
10 |
3.2.2. Selection of eluent.
In this work, several acidic eluents including HCl, HNO3 and HClO4 solutions were examined as the elution solvent. Other factors were kept constant during the optimisation (pH, 6.4; extraction time, 6.0 min; amount of magnetic sorbent, 30 mg; eluent volume, 4 mL; elution time, 20 min). The results showed that HNO3 can recover the target metal ions. In the next step, the effect of HNO3 volume and concentration were optimised.
3.2.3. Elution step.
Three factors were studied in the elution step using the experimental design: eluent volume (mL), elution time (min), and eluent concentration (mol L−l). In this condition, a response surface design could be performed without having previously performed a screening design. The BBD was chosen because it requires the least number of experiments (15). The obtained data were evaluated by ANOVA. The results of the experimental design were evaluated at 5% of significance and analysed by standardised Pareto chart (Fig. 4a). Based on BBD, both the concentration and volume of the eluent showed significant positive effects on the recovery of the target metal ions, while elution time had an insignificant positive effect. These observations are likely due to the increased protonation of the sorbent heteroatoms as the concentration of the eluent increased; the fast kinetics of the elution process is also of great importance. As shown in Fig. 4a, eluent volume has the greatest influence on extraction recovery. RSM (Fig. 4b) was applied to analyse the simultaneous effects of elution time and eluent concentration on the responses. The extraction efficiency of target ions increased along with increasing eluent concentration and elution time. According to the overall results of the optimisation study, the following experimental conditions were chosen as the optimal ones: eluent volume, 7.5 mL; elution time, 14.5 min; and eluent concentration, 1.5 mol L−l HNO3 solution (Fig. 5).
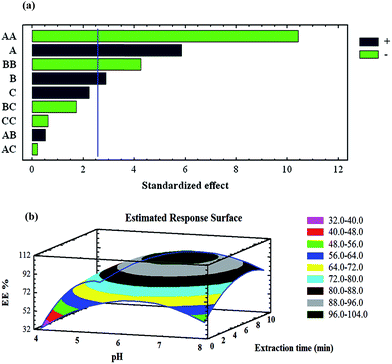 |
| | Fig. 4 (a) Pareto chart of the main effects in the BBD (sorption step). AA, BB and CC are the quadratic effects of pH, the extraction time and the amount of the magnetic sorbent, respectively. AB, AC and BC are the interaction effects between pH and extraction time, pH and the amount of sorbent, and the extraction time and amount of magnetic sorbent, respectively. (b) RSM obtained by plotting pH vs. extraction time using the BBD. | |
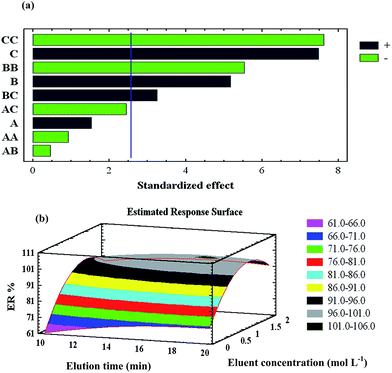 |
| | Fig. 5 (a) Pareto chart of the main effects in the BBD (elution step). AA, BB and CC are the quadratic effects of elution time, eluent concentration and eluent volume, respectively. AB, AC and BC are the interaction effects between elution time and eluent concentration, elution time and eluent volume, and eluent concentration and eluent volume, respectively. (b) RSM obtained by plotting elution concentration vs. eluent volume. | |
3.3. Effect of breakthrough volume
For the analysis of real samples, sample volume is one of the important parameters affecting the preconcentration factor. Hence, the breakthrough volume of sample solutions was investigated by dissolving 1 mg of each Cd(II) and Ni(II) ions in 250, 500, 750, 1000, 1250, 1500, and 1750 mL distilled water. The SPE protocol was then carried out, and the results demonstrated that the dilution effect for both of these ions was not significant for sample volumes of 1500 mL on the magnetic nanosorbent. It was concluded that the simultaneous quantitative recovery of Cd(II) and Ni(II) ions on the magnetic sorbent can be obtained for sample volumes up to 1500 mL. Thus, the new nanosorbent enables an enrichment factor of 200 for Cd(II) and Ni(II) ions.
3.4. Effect of potential interfering ions
To investigate the effect of potential interfering ions found in natural samples, various metal ions were added to 100 mL of a solution containing 0.01 mg Cd(II) and 0.01 mg Ni(II) ions. The degrees of tolerance for potential interfering ions are presented in Table 1S (ESI†) The tolerance results show that even high levels of potential interfering ions have no effect on the preconcentration of Cd(II) and Ni(II) ions at pH 6.4. Thus, the proposed method can be applied to determine the target ions in complicated matrix samples.
3.5. Sorption capacity study
To investigate the sorption capacity of the magnetic sorbent, a standard solution containing 10 mg L−1 of Cd(II) and Ni(II) was used. In order to evaluate the maximum sorption capacity, the initial and equilibrium amounts of the target metal ions were determined by FAAS. The maximum sorption capacity is defined as the total amount of heavy metal ions sorbed per gram of magnetic nanosorbent. The obtained capacities of the magnetic nanosorbent were found to be 120 and 98 mg g−1 for Cd(II) and Ni(II) ions, respectively.
3.6. Analytical performance of the method
Under the optimised conditions, calibration curves were obtained for the determination of Cd(II) and Ni(II) ions according to the mentioned procedure. Linearity was within the initial concentration range of 1–100 ng mL−1 for Cd(II) and 4–150 ng mL−1 for Ni(II). The correlation of determination (r2) was 0.996 for Cd(II) and 0.994 for Ni(II) ions. The limit of detection is defined as LOD = 3Sb/m, where Sb is the standard deviation of 10 replicate blank signals and m is the slope of the calibration curve after preconcentration. For a sample volume of 750 mL, r2 was found to be 0.3 ng mL−1 for Cd(II) and 1.2 ng mL−1 for Ni(II). The precision of the method for a standard solution containing 25 ng mL−1 of Cd(II) and Ni(II) ions (n = 3) was evaluated as the relative standard deviation (RSD%) and was 6.9 and 8.8%, for Cd(II) and Ni(II), respectively.
3.7. Validation of the method
The concentrations of Cd(II) and Ni(II) ions obtained by the current method were compared to the exact concentrations of the target ions in standard reference materials. For this reason, the concentrations of the heavy metal ions were determined at optimum conditions in the standard reference material (seafood mix 02-2932). As seen in Table 2, good correlations were obtained between the contents estimated using the present method and the real amounts of the studied ions in the reference material. Therefore, the magnetic nanosorbent can be used as a reliable solid phase for the extraction and determination of Cd(II) and Ni(II) ions in various samples.
Table 2 Determination of Cd(II) and Ni(II) in a certified reference material
| Sample |
Concentration (mg kg−1) |
Relative error % |
| Element |
Certified |
Found |
| Seafood mix |
Cd |
4.764 |
4.63 |
−2.8 |
| Ni |
3.071 |
2.90 |
−5.6 |
3.8. Determination of Cd(II) and Ni(II) ions in various real samples
Since natural samples have complex matrices, non-specific background absorption is always caused by interfering species of the sample matrix. To reduce this undesirable effect, the magnetic nanosorbent was applied for the selective extraction of Cd(II) and Ni(II) ions at pH 6.4. Table 3 shows the recoveries of Cd(II) and Ni(II) ions in various real samples; in all cases, the recoveries were almost quantitative.
Table 3 Determination of Cd(II) and Ni(II) ions in different real samples
| Sample |
Element |
Real sample (ng g−1) |
Added (ng g−1) |
Found (ng g−1) |
Recovery (%) |
| Fish (Behad creek) |
Cd |
6.7 |
10 |
15.6 |
89.0 |
| Ni |
21.3 |
10 |
30.5 |
92.0 |
| Fish (Jafare creek) |
Cd |
8.8 |
10 |
18.7 |
99.0 |
| Ni |
60.5 |
10 |
71.4 |
109 |
| Shrimp |
Cd |
15.0 |
10 |
23.7 |
87.0 |
| Ni |
49.3 |
10 |
57.6 |
83.0 |
| Canned tuna |
Cd |
5.1 |
10 |
14.6 |
95.0 |
| Ni |
40.1 |
10 |
48.5 |
84.0 |
| Fish (Caspian sea) |
Cd |
19.6 |
10 |
29.0 |
94.0 |
| Ni |
98.4 |
10 |
107 |
86.0 |
| Fish (Siahroud river) |
Cd |
32.2 |
10 |
41.5 |
93.0 |
| Ni |
25.3 |
10 |
36.0 |
107 |
4. Conclusion
A simple, fast, reproducible, and selective magnetic solid-phase extraction procedure based on Fe3O4@SiO2@PPy nanocomposites has been developed for the determination of Cd(II) and Ni(II) ions. The modification of the sorbent helps it to show selectivity towards these heavy metals. In comparison with other solid-phases, the magnetic sorbent exhibits the advantages of excellent sorption capacity, low limit of detection, and high enrichment factor (Table 2S, ESI†). In addition, it has been shown that the RSDs of the present method are similar to those of other methods. Hence, the analytical performance of this method is acceptable. Another advantage of this method is its low time-consumption due to the magnetically-assisted separation of the sorbent and its high surface area; therefore, satisfactory results can be achieved using smaller amounts of the sorbent. Due to the relatively high preconcentration factor, trace amounts of heavy metals in high-volume samples can be quantified by the magnetic nanosorbent.
Acknowledgements
The authors would like to thank Young Researchers and Elite Club, Tabriz Branch, Islamic Azad University, Tabriz, Iran, for the financial support for this research.
References
- U. Farooq, M. A. Khan, M. Athar and J. A. Kozinski, Chem. Eng. J., 2011, 171, 400–410 CrossRef CAS PubMed.
- J. N. Bianchin, E. Martendal, R. Mior, V. N. Alves, C. S. T. Araújo, N. M. M. Coelho and E. Carasek, Talanta, 2009, 78, 333–336 CrossRef CAS PubMed.
- N. M. Kalariya, B. Nair, D. K. Kalariya, N. K. Wills and F. J. G. M. V. Kuijk, Toxicol. Lett., 2010, 198, 56–62 CrossRef CAS PubMed.
- W. Guo, S. Hu, Y. Xiao, H. Zhang and X. Xie, Chemosphere, 2010, 81, 1463–1468 CrossRef CAS PubMed.
- J. Kristiansen, J. M. Christensen, T. Henriksen, N. H. Nielsen and T. Menne, Anal. Chim. Acta, 2000, 403, 265–272 CrossRef CAS.
- D. Bohrer, P. Cícero do Nascimento, M. Guterres, T. Trevisan and E. Seibert, Analyst, 1999, 124, 1345–1350 RSC.
- Y.-H. Sung and S.-D. Huang, Anal. Chim. Acta, 2003, 495, 165–176 CrossRef CAS PubMed.
- E. L. Silva, P. dos Santos Roldan and M. F. Giné, J. Hazard. Mater., 2009, 171, 1133–1138 CrossRef CAS PubMed.
- C. Duran, H. B. Senturk, L. Elci, M. Soylak and M. Tufekci, J. Hazard. Mater., 2009, 162, 292–299 CrossRef CAS PubMed.
- J. Yin, Z. Jiang, G. Chang and B. Hu, Anal. Chim. Acta, 2005, 540, 333–339 CrossRef CAS PubMed.
- B. Zawisza and R. Sitko, Spectrochim. Acta, Part B, 2007, 62, 1147–1152 CrossRef PubMed.
- P. Bruno, M. Caselli, G. Gennaro, P. Ielpo, T. Ladisa and C. M. Placentino, Chromatographia, 2006, 64, 537–542 CAS.
- S. Abe, K. Fuji and T. Sono, Anal. Chim. Acta, 1994, 293, 325–330 CrossRef CAS.
- J. Chen and K. C. Teo, Anal. Chim. Acta, 2011, 450, 215–222 CrossRef.
- M. M. Matlock, B. S. Howerton and D. A. Atwood, Water Res., 2002, 36, 4757–4764 CrossRef CAS.
- M. C. Yebra-Biurrun, A. Bermejo-Barrera, M. P. Bermejo-Barrera and M. C. Barciela-Alonso, Anal. Chim. Acta, 1995, 303, 341–345 CrossRef CAS.
- M. Faraji, Y. Yamini, A. Saleh, M. Rezaee, M. Ghambarian and R. Hassani, Anal. Chim. Acta, 2010, 659, 172–177 CrossRef CAS PubMed.
- A. Duran, M. Tuzen and M. Soylak, J. Hazard. Mater., 2009, 169, 466–471 CrossRef CAS PubMed.
- M. Tuzen, M. Soylak and L. Elci, Anal. Chim. Acta, 2005, 548, 101–108 CrossRef CAS PubMed.
- M. Tuzen, K. O. Saygi and M. Soylak, J. Hazard. Mater., 2008, 156, 591–595 CrossRef CAS PubMed.
- A. A. ALOthman, M. Habila, E. Yilmaz and M. Soylak, Microchim. Acta, 2012, 177, 397–403 CrossRef.
- M. Tuzen, K. O. Saygi, C. Usta and M. Soylak, Bioresour. Technol., 2008, 99, 1563–1570 CrossRef CAS PubMed.
- M. Tuzen, K. O. Saygi and M. Soylak, J. Hazard. Mater., 2008, 152, 632–639 CrossRef CAS PubMed.
- M. Taghizadeh, A. A. Asgharinezhad, N. Samkhaniany, A. Tadjarodi, A. Abbaszadeh and M. Pooladi, Microchim. Acta, 2014, 181, 597–605 CrossRef CAS.
- J. S. Suleiman, B. Hu, H. Peng and C. Huang, Talanta, 2009, 77, 1579–1583 CrossRef CAS PubMed.
- H. Parham, N. Pourreza and N. Rahbar, J. Hazard. Mater., 2009, 163, 588–592 CrossRef CAS PubMed.
- M. Faraji, Y. Yamini and S. Shariati, J. Hazard. Mater., 2009, 166, 1383–1388 CrossRef CAS PubMed.
- M. R. Sohrabi, Z. Matbouie, A. A. Asgharinezhad and A. Dehghani, Microchim. Acta, 2013, 180, 589–597 CrossRef CAS.
- M. Taghizadeh, A. A. Asgharinezhad, M. Pooladi, M. Barzin, A. Abbaszadeh and A. Tadjarodi, Microchim. Acta, 2013, 180, 1073–1084 CrossRef CAS.
- M. Ghaemi and G. Absalan, Microchim. Acta, 2014, 181, 45–53 CrossRef CAS.
- M. Faraji, Y. Yamini and M. Rezaee, Talanta, 2011, 81, 831–836 CrossRef PubMed.
- H. Bagheri, A. Afkhami, M. Saber-Tehrani and H. Khoshsafar, Talanta, 2012, 97, 87–95 CrossRef CAS PubMed.
- M. Helmi, R. Farimani, N. Shahtahmassebi, M. R. Roknabadi and N. Ghows, Nanomed. J., 2014, 1, 71–78 Search PubMed.
- A. A. Asgharinezhad, H. Ebrahimzadeh, M. Rezvani, N. Shekari and M. Loni, Food Addit. Contam., 2014, 31, 1196–1204 CAS.
- T. Yao, T. Cui, X. Fang, J. Yu, F. Cui and J. Wu, Chem. Eng. J., 2013, 225, 230–236 CrossRef CAS PubMed.
-
S. Weijie, C. Ping, Y. Ye, C. Yang, T. Qin and Z. Shan, high-magnetic heavy-metal ion adsorbent carrying conductive high molecules and preparation method thereof, CN 101708463 B, 2010.
- A. B. Yilmaz, Environ. Res., 2003, 92, 277–281 CrossRef CAS.
- S. Sadeghi and E. Aboobakri, Microchim. Acta, 2012, 178, 89–97 CrossRef CAS.
- E. Sahmetlioglu, E. Yilmaz, E. Aktas and M. Soylak, Talanta, 2014, 119, 447–451 CrossRef CAS PubMed.
-
G. E. P. Box and N. R. Draper, Empirical Model Building and Response Surfaces, John Wiley and Sons, New York, 1987 Search PubMed.
-
StatGraphics Plus 5.1 for Windows, Statistical Graphic Crop., online manuals, 2001 Search PubMed.
- M. Faraji, Y. Yamini, A. Saleh, M. Rezaee, M. Ghambarian and R. Hassani, Anal. Chim. Acta, 2010, 659, 172–177 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ay01991a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.