DOI:
10.1039/C5AN00967G
(Paper)
Analyst, 2015,
140, 5055-5064
Recapitulation of in vivo-like neutrophil transendothelial migration using a microfluidic platform†
Received
14th May 2015
, Accepted 6th June 2015
First published on 9th June 2015
Abstract
Neutrophil transendothelial migration (TEM) is an essential physiological process that regulates the recruitment of neutrophils in response to inflammatory signals. Herein, a versatile hydrogel scaffold is embedded in a microfluidic platform that supports an endothelial cell layer cultured in the vertical direction and highly stable chemical gradients; this construct is employed to mimic the in vivo neutrophil TEM process. We found that the number of neutrophils migrating across the endothelial cell layer is dependent on the presented chemoattractant concentration and the spatial profile of the chemical gradient. Endothelial cells play a critical role in neutrophil TEM by promoting neutrophil morphological changes as well as expressing surface receptor molecules that are indispensable for inducing neutrophil attachment and migration. Furthermore, the microfluidic device also supports competing chemoattractant gradients to facilitate neutrophil TEM studies in complex microenvironments that more accurately model the in vivo system than simplified microenvironments without the complexity of chemical gradients. This work demonstrates that combinations of any two different chemoattractants induce more significant neutrophil migration than a single chemoattractant in the same total amount, indicating synergistic effects between distinct chemoattractants. The in vitro reconstitution of neutrophil TEM successfully translates planar neutrophil movement into in vivo-like neutrophil recruitment and accelerates understanding of cellular interactions between neutrophils and endothelial cells within the complicated physiological milieu.
Introduction
As the most abundant white blood cell type, neutrophils function as the primary immune cells in various relevant diseases and recruit to the sites of infection through the endothelial cell layer in response to the physiological signals generated from invading microorganisms or local macrophages.1,2 Neutrophil transendothelial migration (TEM) is a key multi-step process involved in inflammation since the activation of endothelial cells enables the capture of bypassing neutrophils and triggers the subsequent neutrophil inflammatory responses.3,4 The highly orchestrated interactions between endothelial cells and neutrophils include the initial neutrophil rolling on endothelium, firm adhesion mediated by receptor molecules on cell surfaces, transcellular or paracellular extravasation, and final migration towards the inflammation locus.1,5,6 Investigation of the neutrophil TEM process will shed light on the detailed mechanisms of cellular interactions between neutrophils and endothelial cells, also accelerating fundamental understanding of pathogenesis in neutrophil-related diseases.
Various traditional methods, such as the Boyden chamber7,8 and transwell assays,9 have been employed to recapitulate the in vivo leukocyte TEM process; however, these approaches are not able to accurately represent the characteristics based on two main limitations: (1) conventional methods cannot achieve stable long-lasting chemical gradients to support the quantitation of neutrophil TEM and (2) these methods build up endothelial cell layers on a two-dimensional (2D) substrate that only facilitates neutrophil TEM observation through the basement membrane while ignoring the recruitment in other directions. The chemical gradients generated by the chamber-based assays rely on the free diffusion of molecules between two separated chambers such that the shapes of gradients decay quickly and the results of neutrophil TEM cannot be interpreted in a quantitative and controllable fashion. In addition, the upright filter membrane set up for endothelial cell layer culture cannot reflect the whole picture of neutrophil TEM in different directions and introduces the contribution of gravity into neutrophil TEM, inspiring consideration of an improved platform to study the mechanisms of neutrophil migratory behaviors.
Microfluidic technology, devices that allow the manipulation of small volume fluids in microchannels,10 is promising for recapitulation of the in vivo neutrophil TEM process, especially with the inclusion of three-dimensional (3D) hydrogel matrices.11–14 The compact fibrous hydrogel structure, combined with the small dimensions of microfluidic devices, facilitate the creation of predictable, reproducible, and long-term stable chemical gradients with high spatiotemporal resolution so that the neutrophil TEM process can be characterized in a real-time and quantitative manner. More importantly, the inclusion of hydrogel materials not only provides mechanical support for the growth of an endothelial cell layer in the perpendicular direction, but also successfully models extracellular matrix (ECM) with realistic biophysical properties. With these efforts, a highly robust and accurate microfluidic model can be developed to study the neutrophil TEM process. Several previous examples have studied neutrophil migration through the endothelial cell layer using microfluidic platforms,15–18 but these efforts failed to account for the real configuration of blood vessels or the various cellular stimuli. One promising advantage of our device design compared to the existing microfluidic assays is the introduction of multiple chemical gradients in different directions relative to the endothelial cell layer. In an in vivo setting, the neutrophil TEM process occurring at one specific site is guided by an array of chemoattractants gradients in different directions released from various biological sources; however, the existing microfluidic assays cannot recapitulate this microenvironment and only characterize neutrophil TEM without the complexity of multiple chemical gradients. The goal of this work was to build on previous efforts to create a versatile microfluidic platform, more similar to the complex physiological milieu, to study the detailed mechanisms of neutrophil TEM.
Chemoattractants are the signaling molecules responsible for inducing neutrophil migration and activating endothelial cells in the neutrophil TEM process.19,20 Herein, we considered neutrophil TEM under the influence of three inflammatory chemoattractants: interleukin-8 (IL-8), N-formyl-methionyl-leucyl-phenylalanine (fMLP), and leukotriene B4 (LTB4). IL-8, one of the primary chemoattractants initiating in vivo neutrophil TEM, is known to enhance cell adherence to matrix proteins, endothelium and tissues to promote cell recruitment.21 Similar to IL-8, LTB4 is another type of host-derived chemoattractant that is known to induce cell adhesion, activation, and formation of reactive oxygen species.22,23 On the contrary, fMLP is a formylated short peptide of bacterial origin and functions as an intense chemoattractant for several cell types.24 As mentioned above, the introduction of various chemical gradients within our microfluidic device is able to establish the hierarchy among these three chemoattractants through developing competing chemical gradients in two opposing symmetric channels, which enables the mechanistic investigation of neutrophil migratory signaling cascades during decision-making process.
Experimental section
Device fabrication
Standard photolithography protocols were applied to fabricate microfluidic devices. The design of the device was printed on a film (CAD/Art Service Inc., Bandon, OR) with transparent channel patterns and a lightproof background. Through the exposure to UV light, channel patterns were transferred onto a chrome photomask plate coated with AZ1518 positive photoresist layer (Nanofilm, Westlake Village, CA), and then cross-linked photoresist in the channels was removed by placing the photomask in 351 developer solution (Rohm and Hass Electronic Materials LLC, Marlborough, MA). Then, the exposed chrome layer was etched down in the chrome etchant solution (Cyantek Corporation, Fremont, CA). To remove the residual photoresist, the photomask was immersed in piranha solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio of 30% hydrogen peroxide and 99.9% sulfuric acid, Avantor Performance Materials, Phillipsburg, NJ) and then washed using deionized (DI) water. After the preparation of the photomask, the microfluidic device mold was fabricated by spin-coating a 4 inch silicon wafer with 120 μm-thick negative SU-8 50 photoresist (MicroChem, Newton, MA). The channel patterns were imprinted on the SU-8 mold through the previously made photomask via UV exposure following an initial baking step. The silicon wafer was placed in SU-8 developer (MicroChem, Newton, MA) to remove the photoresist without exposure, and the channel patterns were left on the mold. A 10:1 mass ratio mixture of Sylgard 184 silicone elastomer base and curing agent (Ellsworth Adhesives, Germantown, WI) was poured on the SU-8 mold and kept on a hot plate at 95 °C overnight. Medium channel reservoirs and gel chamber inlets were punched at appropriate points in the polydimethylsiloxane (PDMS) layer using 3.5 mm and 1 mm disposable biopsy punches (Integra Miltex, Plainsboro, NJ), respectively. Finally, the PDMS layer was cut and then permanently attached to a glass slide by using oxygen plasma for 10 seconds at 100 L h−1 oxygen flow rate and 100 W.
:1 volume ratio of 30% hydrogen peroxide and 99.9% sulfuric acid, Avantor Performance Materials, Phillipsburg, NJ) and then washed using deionized (DI) water. After the preparation of the photomask, the microfluidic device mold was fabricated by spin-coating a 4 inch silicon wafer with 120 μm-thick negative SU-8 50 photoresist (MicroChem, Newton, MA). The channel patterns were imprinted on the SU-8 mold through the previously made photomask via UV exposure following an initial baking step. The silicon wafer was placed in SU-8 developer (MicroChem, Newton, MA) to remove the photoresist without exposure, and the channel patterns were left on the mold. A 10:1 mass ratio mixture of Sylgard 184 silicone elastomer base and curing agent (Ellsworth Adhesives, Germantown, WI) was poured on the SU-8 mold and kept on a hot plate at 95 °C overnight. Medium channel reservoirs and gel chamber inlets were punched at appropriate points in the polydimethylsiloxane (PDMS) layer using 3.5 mm and 1 mm disposable biopsy punches (Integra Miltex, Plainsboro, NJ), respectively. Finally, the PDMS layer was cut and then permanently attached to a glass slide by using oxygen plasma for 10 seconds at 100 L h−1 oxygen flow rate and 100 W.
Endothelial cell culture
The human endothelial cell line hy926, a phenotype suitable for neutrophil–endothelial cell interaction studies,16,25,26 was purchased from American Type Culture Collection (ATCC, Manassas, VA) and stored in a liquid nitrogen storage container (MVE XC33/22, Select Genetics, Washington, PA). Upon thawing, endothelial cells were dispensed into a 75 cm2 flask containing 20 mL of Dulbecco's Modified Eagle Medium (DMEM, formula: 4 mM L-glutamine, 4.5 g L−1L-glucose, and 1.5 g L−1 sodium pyruvate, Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (Sigma-Aldrich, St. Louis, MO). Cells were fed every other day and, when necessary, cells were detached using 1× trypsin solution (Sigma-Aldrich, St. Louis, MO) for device injection. Endothelial cells were only used between the third and tenth passages.
Device preparation
First, microfluidic devices were filled with 30 μL of 1 mg mL−1 Poly-D-Lysine (PDL) solution (Sigma-Aldrich, St. Louis, MO) and incubated for 4 h under 5% CO2 at 37 °C. After the completion of surface coating, devices were rinsed with 30 μL of sterilized Milli-Q water (Millipore, Billerica, MA) twice to remove excess PDL solution that may cause damage to cells. Prior to introducing gel, devices were placed in the oven at 65 °C for 24 to 48 h so that the hydrophobicity of devices was restored. Collagen type I gel solution (BD Biosciences, San Jose, CA), one common hydrogel material used for simulating extracellular matrix,12,15,27 was diluted to a concentration of 2 mg mL−1 and injected into the gel chamber through the gel inlet. To avoid the evaporation of gel solution, all the devices were kept in humid pipette boxes after the gel injection and a thermally induced polymerization was carried out under 5% CO2 at 37 °C for 30 min. The porous fiber structure of the resulting collagen gel was visualized using a scanning electron microscope (SEM, see the details in ESI†). After the gel polymerization, 20 μL of cell culture medium was forcibly injected into each channel of the microfluidic device, and the medium in all six reservoirs was aspirated before loading endothelial cells. Endothelial cells were trypsinized and re-suspended in the cell culture medium at a proper density (1.5–2 × 106 cells per mL), and then 20 μL of endothelial cells were seeded into a reservoir of the bottom channel to enable the cell layer to attach on the side wall of gel because of the pressure difference between the bottom channel and side channels. Following an initial incubation under 5% CO2 at 37 °C for 30 min, the medium was aspirated from the bottom reservoirs, and 30 μL of fresh medium was added in each reservoir. Finally, all the devices were placed in the CO2 incubator (New Brunswick Scientific, Edison, NJ) overnight for confluent growth of the endothelial cell layer. For the conditions without an endothelial cell layer, the same procedure was used except for the addition of endothelial cells in the devices. The details about endothelial cell-conditioned medium experiments are included in ESI.†
Neutrophil isolation
Ethylenediaminetetraacetic acid (EDTA)-anticoagulated freshly drawn human blood samples were prepared by Memorial Blood Center (St. Paul, MN) according to IRB protocol E&I ID no. 07809. Samples were collected only from healthy donors following the guidelines that meet the standards of the Food and Drug Administration. Immediately after blood samples were collected, neutrophils were separated and purified using a previously reported isolation protocol.28 Carefully, 5 mL of blood sample was layered on the same volume of mono-poly resolving medium (Fisher Scientific, Waltham, MA) and promptly centrifuged to obtain a distinct neutrophil band. Neutrophils were washed using red blood cell lysis buffer (Miltenyi Biotec Inc., Auburn, CA) several times (2.5 mL for each time) until only white cells were left at the bottom of centrifuge tube. The final neutrophil pellet was re-suspended in Hank's buffered salt solution (HBSS, Fisher Scientific, Waltham, MA) containing 2% human serum albumin (HSA, Sigma-Aldrich, St. Louis, MO) at a cell density between 4–5 × 106 cells per mL.
Neutrophil transendothelial migration experiments
Before introducing neutrophils into the device, the medium in each of the reservoirs terminating the bottom and left channels was replaced with 30 μL of HBSS buffer while medium in each reservoir of the right channel was changed to 30 μL of chemoattractant solution (IL-8 or fMLP, Sigma-Aldrich, St. Louis, MO; LTB4, Cayman Chemical, Ann Arbor, MI). Different chemoattractant solutions were placed in the two opposing side channels for competing gradient conditions, and HBSS buffer was placed in the both channels in the chemoattractant-free condition. It took approximately 2 h to achieve completely stable diffusion of chemoattractant molecules in the gel scaffold. Then, 5 μL of neutrophils of the desired density were added into the bottom channel of the device. Neutrophil TEM was monitored using MetaMorph ver. 7.7.5 imaging software (images recorded every other hour for 5 h) on an inverted microscope equipped with a 10× objective (Nikon, Melville, NY) and a CCD camera (QuantEM, Photometrics, Tucson, AZ). Data from neutrophils collected from three different donors were measured in each condition.
Results and discussion
Characterization of neutrophil TEM system
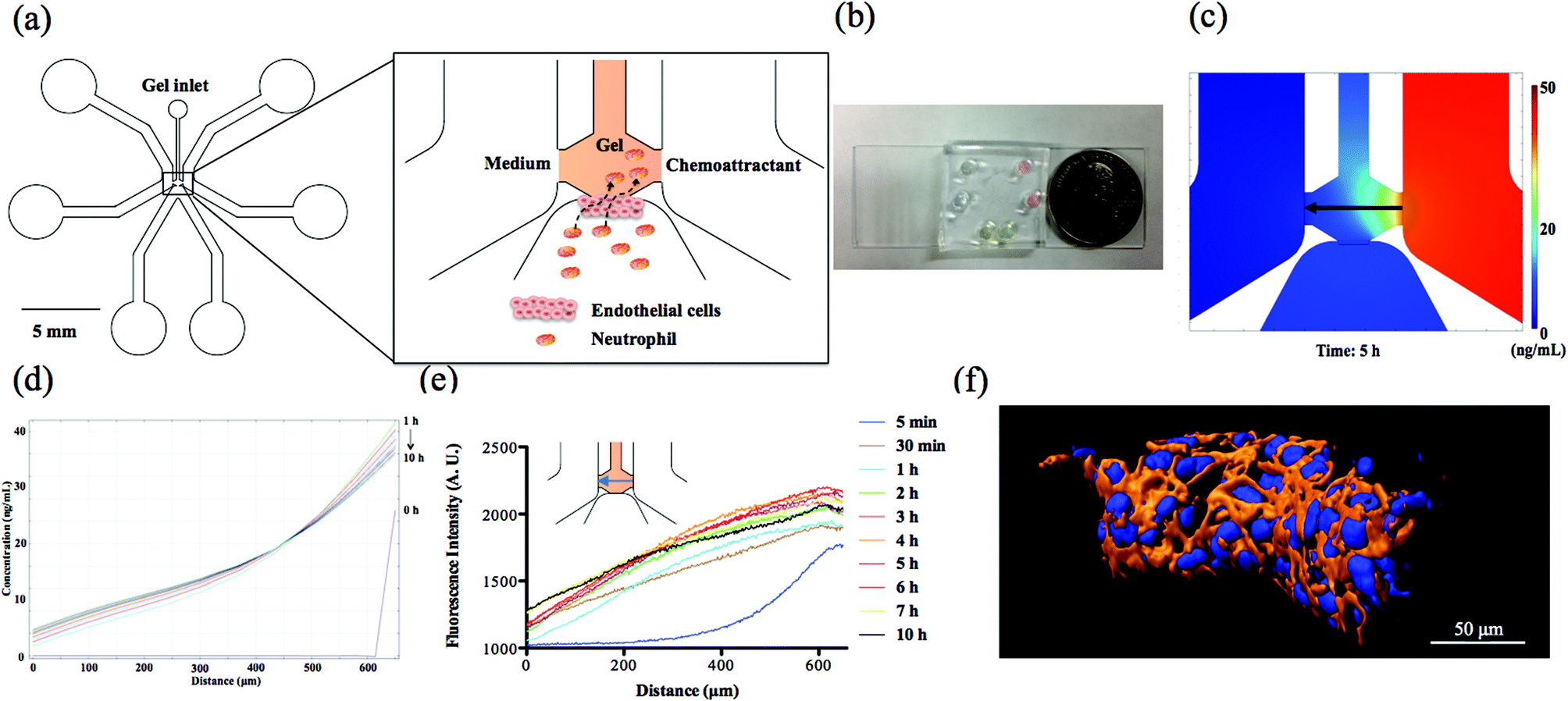
The microfluidic device consists of two side channels, one bottom channel and the central gel chamber that separates these three channels (Fig. 1(a) and (b)). In our design, endothelial cells attached to the side wall of collagen gel are activated by the chemoattractants originating from the side channels; meanwhile, neutrophils in the bottom channel receive the biological signals from endothelial cells and complete the TEM process along the direction of the chemical gradients. The prerequisite for establishing chemical gradients in the collagen gel is the stable diffusion of chemoattractant molecules between two symmetrical side channels. Due to the solid 3D cross-linked network of collagen gel, molecular diffusion is confined at a slow and uniform rate that promotes the long-term stabilization of chemical gradient. To verify the diffusion characteristics of chemoattractant molecules, theoretical simulation and experimental fluorescence imaging have been employed to observe the chemical gradient in the collagen gel. The simulation result (Fig. 1(d)) using finite element method software COMSOL 4.3b reveals that the chemical gradient across the center line in the gel chamber produced by 50 ng mL−1 fMLP solution is linear and stable from 1 h to 10 h diffusion (Fig. 1(c) shows the diffusion of fMLP molecules at 5 h), which is suitable for examining neutrophil TEM with reproducible spatiotemporal resolution. Also, the chemical gradient was visualized at different time points by placing Rhodamine 6G solution in the right side channel and monitoring the fluorescence gradient across the center line in the gel chamber; this experiment demonstrated that a stable fluorescent gradient can be achieved after 2 h diffusion, and there is no apparent decay until 10 h (Fig. 1(e)). The profiles of chemical gradients are similar between those apparent in the COMSOL simulation and the empirical fluorescence imaging results, but it takes longer than expected (2 h vs. 1 h as predicted by COMSOL) to reach stable diffusion for fluorescence imaging; as a result, 2 h was used as the wait time for gradient formation before neutrophil injection. Since the diffusion coefficient is directly related to the molecular weight, the established Rhodamine 6G (∼479 Da) gradient will be very similar to fMLP (∼437 Da) and LTB4 (∼340 Da) conditions. Although IL-8 has a much higher molecular weight (∼8.4 kDa) than the other two chemoattractants, this difference is likely compromised in the highly compact gel structure such that all of the chemoattractants have similar diffusive behaviors.15 In addition to examining the center line of gel chamber, we found that the fluorescence gradient becomes steady after 2 h at other positions, including the gel-endothelial cell interface and the vertical direction across cell layer that characterizes the gradient from top to bottom part (Fig. S1†), suggesting that gradients in various parts of the microfluidic devices reach stabilization after the first 2 h and can be maintained for a long time. Furthermore, the results of confocal and dark-field imaging confirm the confluency of the whole endothelial cell layer structure on the side wall of the gel scaffold (Fig. 1(f) and S2†) and the confocal images of three different devices clearly indicate the good reproducibility of cell layer configuration from device to device (Fig. S3†). The permeability of the endothelial cell layer was measured by analyzing fluorescence images of the device after 2 h diffusion of fluorescein isothiocyanate (FITC)-dextran solution across the endothelial cell layer from the bottom channel to the gel scaffold (Fig. S4†); the measured permeability was 5.73 × 10−7 m s−1, a value similar to those reported in the other in vitro systems with a non-permeable endothelial cell layers,15,29 indicating a good seal between the endothelial cell layer and PDMS substrate. Together, these device characterizations suggest that this microfluidic platform will be a good model for the in vivo neutrophil TEM system.
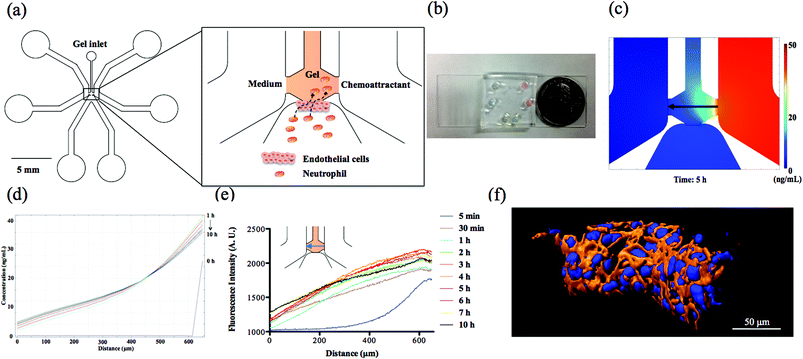 |
| | Fig. 1 Characterization of neutrophil TEM microfluidic device. (a) Schematic of neutrophil TEM microfluidic device design. Endothelial cells (not to scale) are cultured on the side wall of the collagen gel, and the chemoattractant solution or medium is placed in the side channels for developing the chemical gradients. The black arrow line indicates the migration route of neutrophils across the endothelial cell layer towards the chemoattractant source. (b) Photograph of a real device from the top view. (c) COMSOL simulation of a chemical gradient after 5 h diffusion using 50 ng mL−1 fMLP in the right side channel. The black arrow indicates the direction of gradient from high concentration to low concentration. (d) The COMSOL simulation results of the chemical gradient induced by 50 ng mL−1 fMLP. (e) The visualization of the fluorescence gradient at the center line of the gel chamber at different time points. (f) Deconvoluted confocal imaging of endothelial cell layer cultured on the side wall of the gel (blue indicates cell nucleus stained by DAPI and orange represents cytoskeletal F-actin labeled by rhodamine phalloidin). | |
Neutrophil TEM under single chemoattractant gradients
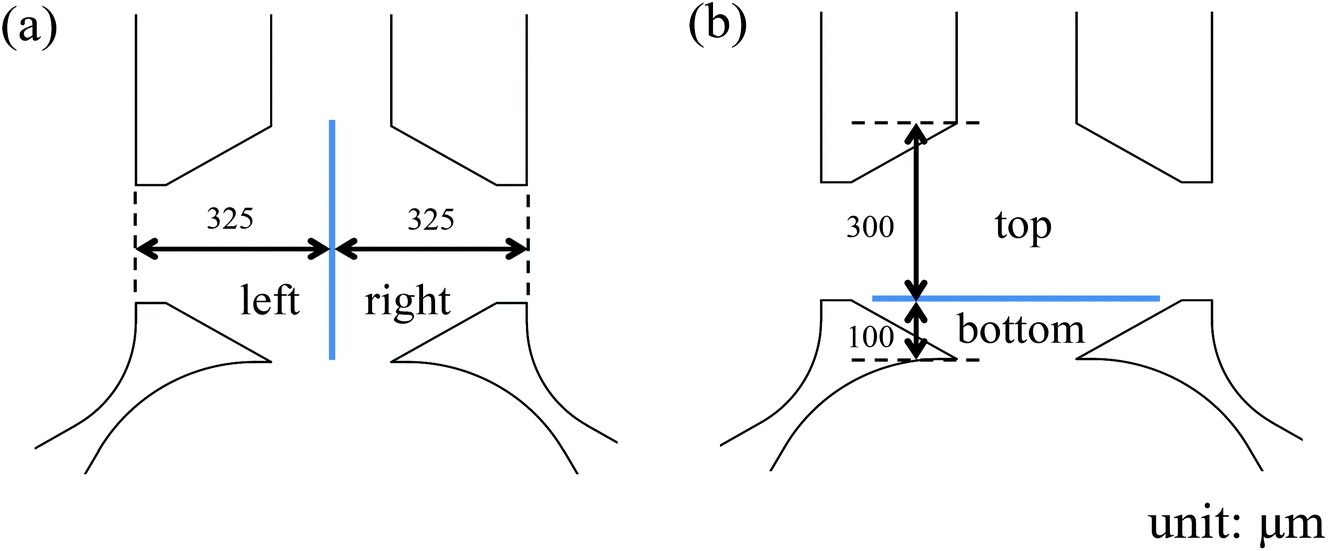
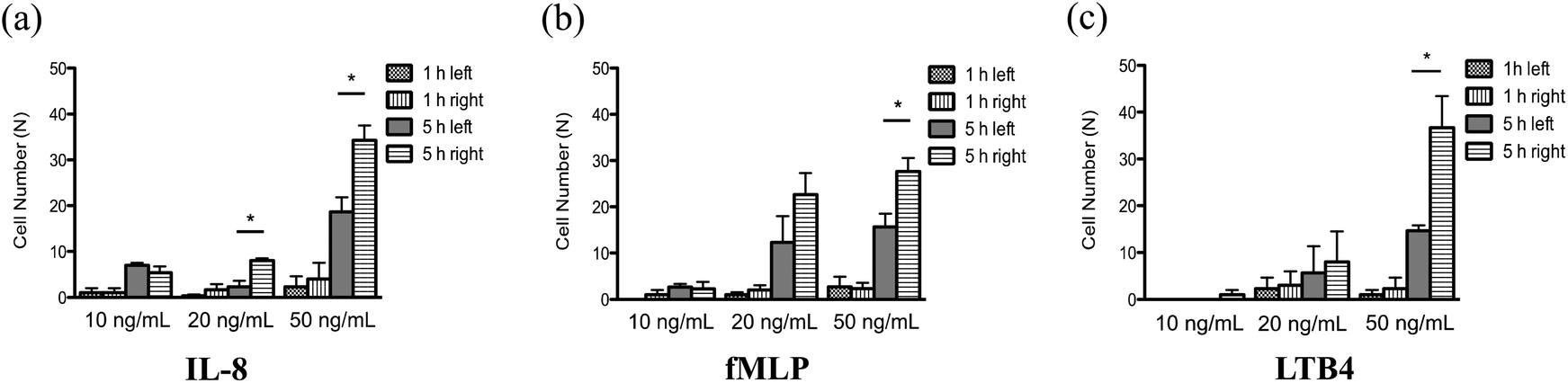
With a well-characterized device, the neutrophil TEM process was first examined under single chemoattractant gradients. For each chemoattractant, three different concentrations (10 ng mL−1, 20 ng mL−1, and 50 ng mL−1) were employed to build up chemical gradients from the right channel to the left channel; neutrophil migration in the gel chamber was monitored every other hour after neutrophil injection into the device. The images at 5 h (Fig. 2(a)–(c)) clearly show that a number of neutrophils migrate into the gel region across the endothelial cell layer in each condition. To quantify the results of neutrophil TEM, we simply counted the number of cells in different regions of the gel chamber (Fig. 3) with linear and continuous gradients so that the effects of localized chemoattractant concentrations on neutrophil polarization could be determined. The largest concentrations (50 ng mL−1) for all the chemoattractants (fMLP: 114 nM; LTB4: 147 nM; IL-8: 5.95 nM) lead to significant differences between the neutrophils present in the left and right portions of the devices; however, there is no significant difference found with lower chemoattractant concentrations (10 ng mL−1 and 20 ng mL−1) except for 20 ng mL−1 IL-8 (Fig. 4). It is worth mentioning that different gel interface shapes caused by various surface tensions do not lead to the considerable deviation in average values of three replicates since the flat gradient shape in the vertical direction across the cell layer is not sensitive to small changes in the gel interface position (Fig. S1(b)†). Additionally, the arc-shaped interfaces do not influence the number of neutrophils interacting with the endothelial cell layer due to the significantly larger dimension of bottom channel for neutrophil injection. The amount of chemoattractants in each part of the gel chamber is proportional to the total chemoattractant concentration presented in the side channel, so the concentration differences between the left and right gel region become larger as the total chemoattractant concentrations increase. Neutrophils sense a steeper gradient in the condition of 50 ng mL−1 chemoattractant compared to the lower concentrations, and many more neutrophils prefer moving towards the chemoattractant sources (i.e. the steeper portion of the chemoattractant gradient) on the right side. On the contrary, no statistical difference is observed between the cell numbers located in the top and bottom portion of the gel-filled chamber for any of the three chemoattractants, even at the highest concentrations (Fig. S5†). The flat slope of the fluorescence gradient in the vertical direction (Fig. S1(b)†) suggests that chemoattractant molecules distribute evenly between the top and bottom regions, and the small concentration difference is not enough to induce significant neutrophil migration. To examine neutrophil TEM without chemical gradients in the horizontal direction, 25 ng mL−1 of each chemoattractant was placed in both side channels such that the average concentration is the same as the single chemical gradient condition (50 ng mL−1). The simulation results using fMLP as a model chemoattractant indicate that the gradient profile is symmetric along the horizontal direction in the “no gradient” condition while the gradient profiles in the vertical direction are the same for single chemoattractant gradient and “no gradient” conditions, which means these two conditions both have the steepest gradients in the perpendicular direction and the same total amounts of chemoattractant molecules in the gel scaffold because of the identical average concentration (Fig. S6†). The neutrophil migration results reveal no significant difference in cell numbers between the left and right device regions in the “no gradient” condition, and there are still not statistically more neutrophils moving into the top device region (Fig. S7†). Thus, we can conclude that the symmetric gradient profile diminishes the polarization of neutrophil TEM in the horizontal direction, and the migration of cells along the vertical direction is determined by the average concentration of chemoattractants in the side channels. Based on the results above, this work reveals that high chemoattractant concentrations with steep chemical gradients are able to cause distinguishable neutrophil TEM processes, which is consistent with disease models where excessive amounts of neutrophils accumulate around infection sites, likely induced by the high level of chemoattractants in the context of diseases.
 |
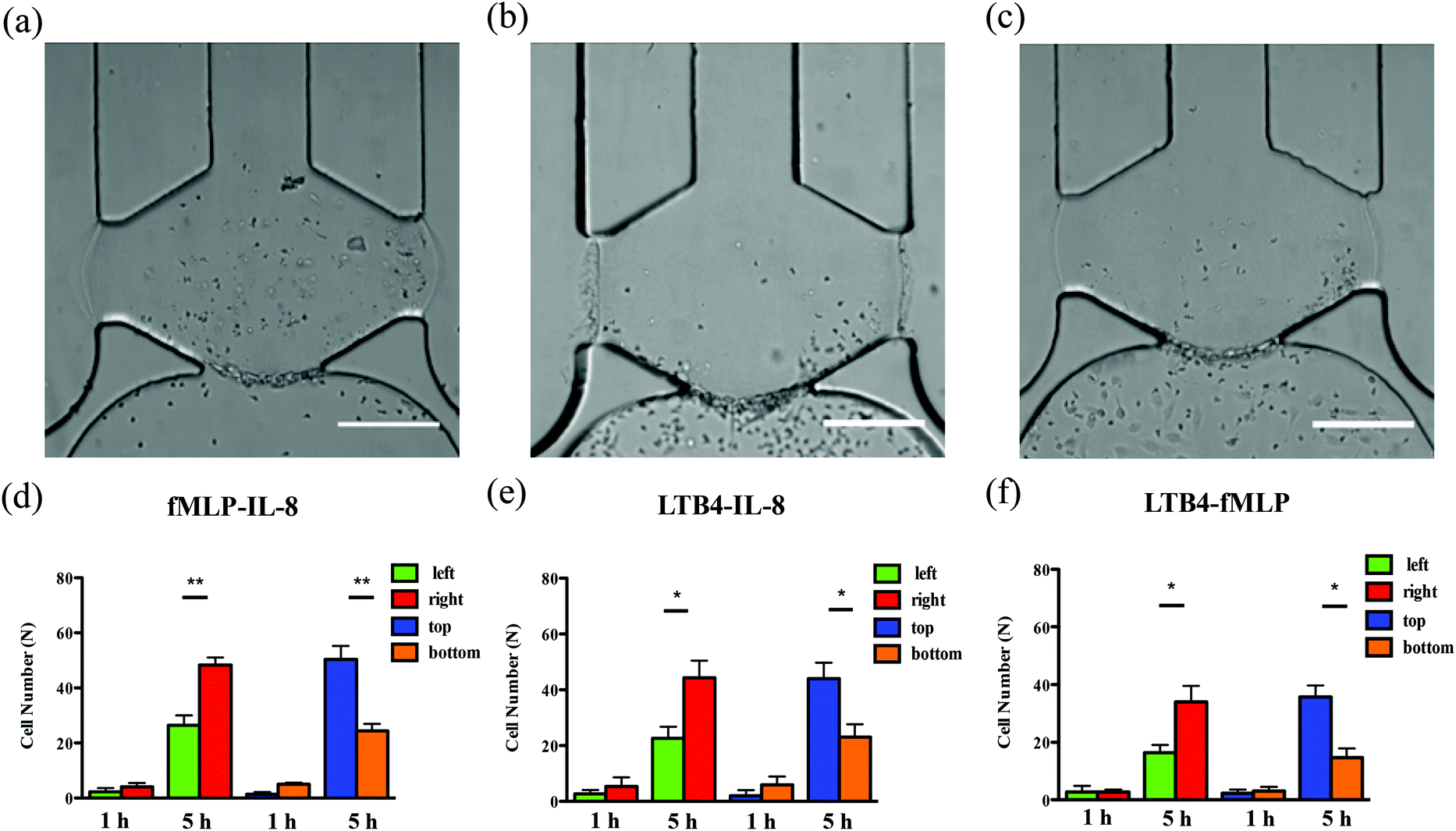
| | Fig. 2 Bright-field images of neutrophil TEM 5 h after neutrophil injection under single chemoattractant gradients (gradient direction is indicated in (a)): (a) 50 ng mL−1 of IL-8; (b) 50 ng mL−1 of fMLP; (c) 50 ng mL−1 of LTB4. Bright-field images of neutrophil migration without an endothelial cell layer 5 h after neutrophil injection under single chemoattractant gradients: (d) 50 ng mL−1 of IL-8; (e) 50 ng mL−1 of fMLP; (f) 50 ng mL−1 of LTB4. Endothelial cells cultured in the bottom channel appear to be in elongated shape (30–40 μm) and much larger than the surrounding round neutrophils (∼10 μm). (scale bar: 200 μm). | |
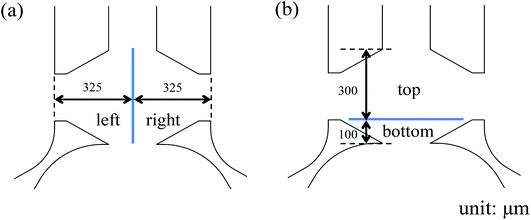 |
| | Fig. 3 Division of collagen gel chamber into two different parts: (a) left and right parts; (b) top and bottom parts. | |
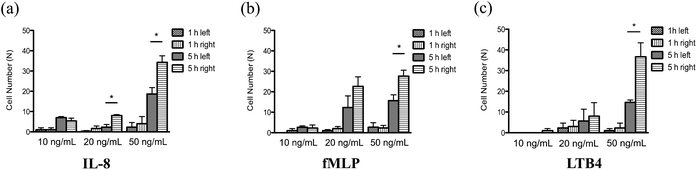 |
| | Fig. 4 Quantitative analysis of neutrophil TEM 1 h and 5 h after neutrophil injection under various single chemoattractant gradients (*, p < 0.05, using a two-tailed unpaired t-test): (a) the number of neutrophils in the left and right parts under (a) 10 ng mL−1, 20 ng mL−1, and 50 ng mL−1 of IL-8 gradient; (b) 10 ng mL−1, 20 ng mL−1, and 50 ng mL−1 of fMLP gradient; (c) 10 ng mL−1, 20 ng mL−1, and 50 ng mL−1 of LTB4 gradient. | |
The role of endothelial cell layer in neutrophil transmigration
One interesting phenomenon revealed in this study is that neutrophils do not migrate into the gel chamber without an endothelial cell layer in any of the presented conditions; without the endothelial cell layer, neutrophils only gather at the interface between the gel chamber and the bottom channel after 5 h migration (Fig. 2(d)–(f)). The collagen gel with a small pore size (Fig. S8†) functions as a physical barrier to prevent the infiltration of neutrophils and endothelial cells into the gel, and neutrophils must undergo morphological changes before entering the gel due to the comparatively large diameter of a single neutrophil (∼10 μm). A morphological difference is clear between the spherical neutrophils without an endothelial cell layer and the stretched neutrophils that are moving through the gel chamber in the presence of an endothelial cell layer (Fig. S9†). To assess the possibility that biological molecules secreted by the endothelial cells promote the neutrophil morphological changes, all the molecules in the endothelial cell conditioned-medium after overnight (∼12 h) chemoattractant-activation were collected and placed in the aforementioned microfluidic devices (see the details in ESI†) without adding the actual endothelial cells. The timescale used herein is suitable to maintain the activity of the endothelial cell secreted species.30–32 Neutrophils were introduced as previously described, images were captured, and cells were counted. Even in the presence of the endothelial cell-secreted soluble molecules, neutrophils stayed in the bottom channel instead of penetrating into the collagen gel (Fig. S10†), signifying that the biological secretion alone is not strong enough to induce neutrophil deformation. Some previous work33–35 indicates that the mechanical interactions between neutrophils and endothelia cells initiate the disruption of cell–cell junctions and enable neutrophils to undergo morphological changes to complete the extravasation step. The biological molecules regulating this process, such as intercellular adhesion molecule-1 (ICAM-1), guanine exchange factor (GEF), and myosin light chain kinase (MLCK),33 are either intracellular species or molecules expressed on the surfaces of cells, and thus not secreted as soluble factors into the free medium, meaning that secreted molecules cannot promote neutrophil migration in the absence of the actual endothelial cell layer; however, more experiments will be pursued in the future to further explore mechanical effects in a biologically relevant environment. In addition to inducing shape change in neutrophils, another important role of endothelial cells in neutrophil TEM is to express surface receptors for triggering neutrophil attachment. The result of chemoattractant-free control conditions (Fig. S11(a)†) shows that no neutrophil attachment or migration is detected without chemoattractant signals, even in the presence of an endothelial cell layer. To examine the effects of chemoattractants on endothelial cell activation, the expression of two major adhesion molecules known to regulate neutrophil–endothelial cell interaction, p-selectin and intercellular adhesion molecule-1 (ICAM-1),5,36 were visualized using antibody fluorescence imaging (Fig. S11(b) and (c)†). After the activation by 50 ng mL−1 of IL-8 gradient (fMLP and LTB4 data not shown), endothelial cells display a much higher level of adhesion molecules than the condition in the absence of chemoattractant, suggesting that chemoattractant activation is the main driving force for receptor expression and non-activated endothelial cells are not able to induce the neutrophil TEM process. Further evidence was obtained by examining neutrophil TEM under the same IL-8 gradient but with the antibodies for adhesion molecules, and no neutrophil TEM was detected 5 h after cell addition (Fig. S11(d)†). Based on the observations above, the role of endothelial cells in neutrophil TEM must: (1) promote the morphological changes of neutrophils to enable cell extravasation into the ECM and (2) present the surface receptor molecules for initiating neutrophil attachment and migration.
Neutrophil TEM under competing chemoattractants gradients
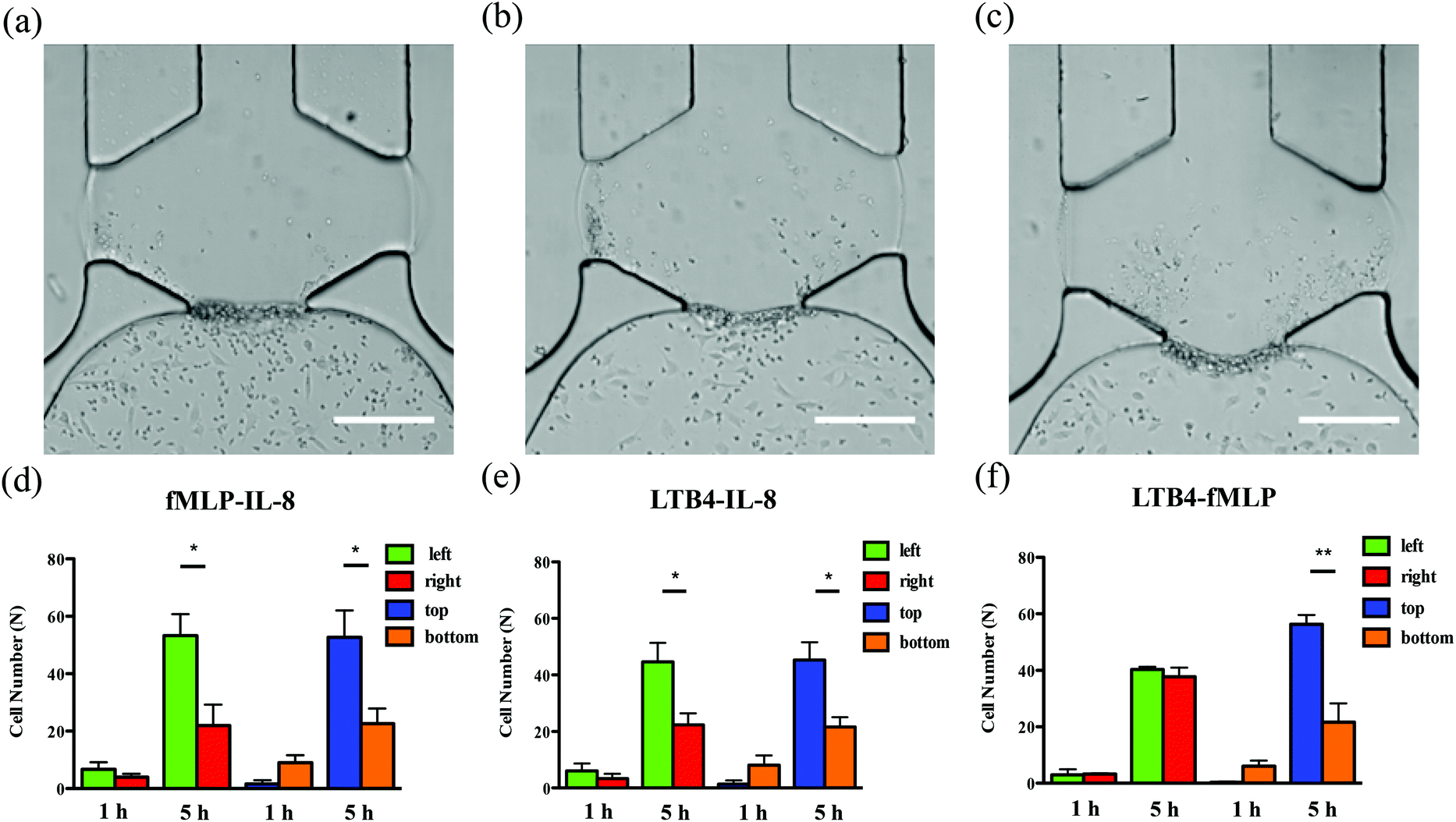
To examine neutrophil TEM under competing gradients, different types of chemoattractants at 50 ng mL−1 were placed in two opposing side channels. The results of single chemoattractant gradients conditions reveal that 50 ng mL−1 of each chemoattractant is capable of inducing similar numbers of neutrophils to transmigrate across the endothelial cell layer, so 50 ng mL−1 was used as the concentration for developing competing gradients. Of the three chemoattractant pairs, significant differences in cell numbers between the left and right regions of the gel chamber, thus indicating a neutrophil preference for one chemoattractant or the other, are observed in the conditions of fMLP vs. IL-8 and LTB4 vs. IL-8 (the former chemoattractant is in the left channel) while there is no significant difference found in the condition of LTB4 vs. fMLP (Fig. 5). Statistically, more neutrophils migrate towards the other type of chemoattractant in IL-8-containing competing gradients, which means both fMLP and LTB4 are dominant chemoattractants over IL-8 during the neutrophil TEM process. The comparison between LTB4 and fMLP indicates that these two chemoattractants have similar abilities to mediate the polarization of neutrophil TEM. Considering the results above, the hierarchy among these chemoattractants is fMLP = LTB4 > IL-8. Although neutrophil migration under competing gradients has been studied in previous research using microfluidic platforms,37–39 these studies did not incorporate the endothelial cell layer into the devices, and the hierarchy among multiple chemoattractants was only obtained in simplified microenvironments. The hierarchy reported herein agrees with the previous conclusion that the p38 mitogen-activated protein (MAP) pathway related to fMLP overwhelms the phosphatidylinositol 3-kinase (PI3K) signaling pathway activated by IL-8.40,41 As another PI3K pathway-controlled chemoattractant, the competence of LTB4 in attracting neutrophil migration is enhanced in the presence of the endothelial cell layer; better understanding of the molecular mechanisms behind this behavior will be the focus of future work. Unlike the single chemoattractant gradients, all the competing gradient conditions demonstrate significant differences between the number of cells in the top and bottom regions of the gel chamber, confirming the observation that the increase in the total amount of chemoattractants (from 50 ng mL−1 to 100 ng mL−1) enables the production of steeper gradients and thus, statistically distinct neutrophil TEM.
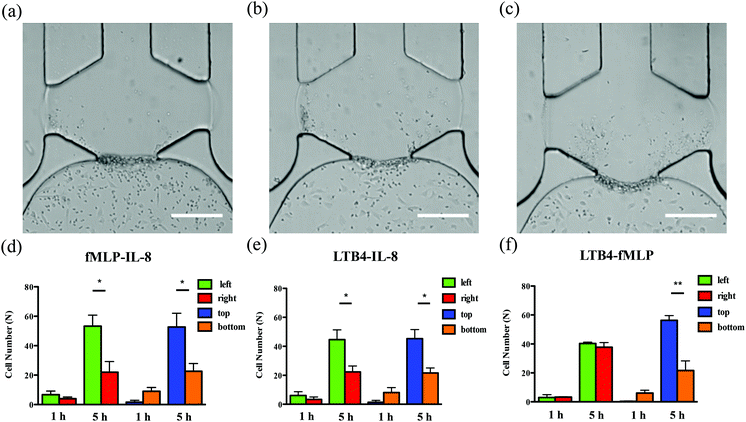 |
| | Fig. 5 Characterization of neutrophil TEM under competing gradients. Bright-field images of neutrophil TEM after 5 h neutrophil injection (left channel vs. right channel): (a) 50 ng mL−1 fMLP vs. 50 ng mL−1 IL-8; (b) 50 ng mL−1 LTB4 vs. 50 ng mL−1 IL-8; (c) 50 ng mL−1 LTB4 vs. 50 ng mL−1 fMLP. Quantitative analysis of neutrophil numbers in different parts of gel chamber after 1 h and 5 h neutrophil injection: (d) 50 ng mL−1 fMLP vs. 50 ng mL−1 IL-8; (e) 50 ng mL−1 LTB4 vs. 50 ng mL−1 IL-8; (f) 50 ng mL−1 LTB4 vs. 50 ng mL−1 fMLP (*, p < 0.05, **, p < 0.005, using a two-tailed unpaired t-test). Endothelial cells cultured in the bottom channel appear to be in elongated shape (30–40 μm) and much larger than the surrounding round neutrophils (∼10 μm). (scale bar: 200 μm). | |
Synergistic chemoattractant effects on neutrophil TEM
In the conditions of single chemoattractant gradients, 50 ng mL−1 of each chemoattractant was not able to induce significant differences between the number of neutrophils migrating into the top and bottom portions of the gel-filled chamber due to the flat gradient shape in the vertical direction. The previous study suggested the cooperative interplay taking place between two different chemoattractants to promote neutrophil migratory responses.37 With this neutrophil transendothelial migration model, we also hypothesized that various chemoattractants coexisting in the channel, at the same total chemoattractant concentration as the single chemoattractant gradients, would influence neutrophil TEM in the vertical direction. The mixture of any two chemoattractants, with 25 ng mL−1 concentration for each one, was employed to replace single chemoattractant solutions in this experiment. For all three conditions, significant differences are observed between the number of cells in the top and bottom portions of the chamber (Fig. 6), which means that these chemoattractants function through synergistic effects to mediate the neutrophil TEM process. This is likely attributable to the fact that multiple chemoattractants trigger downstream signaling pathways using different surface receptors cooperatively, thus speeding up the responses of neutrophil migration. On the other hand, a single chemoattractant only binds to the corresponding receptor in a competitive manner that reduces the efficiency of initiating neutrophil TEM. In addition, the combination of any two chemoattractants does not alter the chemoattractant concentration gradients from right to left, and the significant differences between the number of cells in the left and right portions of the chamber remain unchanged compared to the single chemoattractant gradients. We also evaluated the synergistic effects within the competing gradients by introducing 25 ng mL−1 of two different chemoattractants in the side channels separately. Compared to integrating two chemoattractants in one side channel, competing gradients provide a symmetric distribution of different chemoattractant molecules across the gel chamber but maintain the same total amount of chemoattractants in the top and bottom regions. All three competing gradients still indicate the synergistic effects in the vertical direction compared to placing 25 ng mL−1 of the same chemoattractant in both side channels (Fig. S7†) while the hierarchy among these three chemoattractants is disrupted, and the significant difference between the cell numbers in the left and right portions of the chamber is only found in the LTB4-IL-8 pair (Fig. S12†). Another interesting discovery is that the total cell numbers completing transmigration across the endothelial cell layer after 5 h observation is not statistically different for 25 ng mL−1 and 50 ng mL−1 competing gradients conditions although the total concentration of chemoattractants for 50 ng mL−1 competing gradients is twice as that in 25 ng mL−1 competing gradients conditions. Further examination of adhesion molecule expression (p-selectin and ICAM-1) reveals that there is no significant difference in the levels of receptor molecule expression for these two conditions (Fig. S13†), and thus, the activation of endothelial cells by chemoattractant must be saturated with 25 ng mL−1 competing gradients. Accordingly, the increase in chemoattractant concentration does not enhance adhesion molecule expression significantly. Based on the results of competing gradients and the consideration of synergistic effects, it is clear that neutrophils prioritize and integrate different chemoattractant signals simultaneously during the neutrophil TEM process.
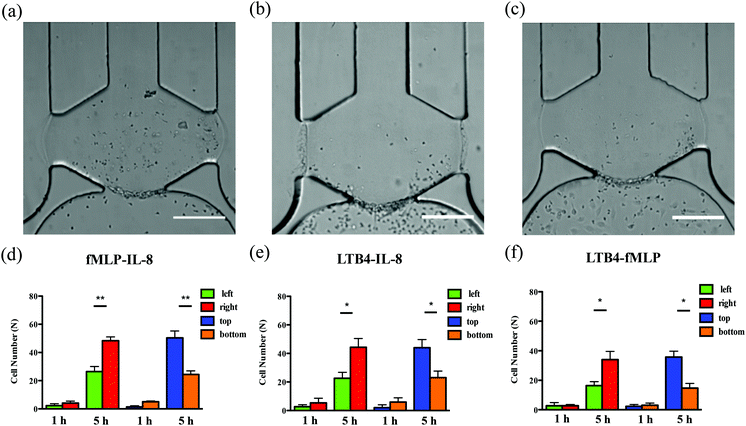 |
| | Fig. 6 Examination of synergistic effects by mixing two different chemoattractants in the right side channel. Bright-field images of neutrophil TEM after 5 h neutrophil injection: (a) 25 ng mL−1 fMLP and 25 ng mL−1 IL-8 in the right channel; (b) 25 ng mL−1 LTB4 and 25 ng mL−1 IL-8 in the right channel; (c) 25 ng mL−1 LTB4 and 25 ng mL−1 fMLP in the right channel. Quantitative analysis of neutrophil numbers in different parts of the gel chamber after 1 h and 5 h neutrophil injection: (d) 25 ng mL−1 fMLP and 25 ng mL−1 IL-8 in the right channel; (e) 25 ng mL−1 LTB4 and 25 ng mL−1 IL-8 in the right channel; (f) 25 ng mL−1 LTB4 and 25 ng mL−1 fMLP in the right channel (*, p < 0.05, **, p < 0.005, using a two-tailed unpaired t-test). (scale bar: 200 μm). | |
Conclusions
An in vivo-like neutrophil TEM model was fabricated as a microfluidic platform incorporating a biomimetic hydrogel matrix and a vertical endothelial cell layer to examine neutrophil migratory responses in various complex microenvironments. We found that the profiles of single chemical gradients are heavily dependent on the total amounts of each chemoattractant, and only the largest concentration (50 ng mL−1) was able to induce significantly more neutrophils moving towards chemoattractant sources with all considered chemoattractants due to the steepest gradient shapes. In addition, the single chemoattractant gradients experiments without the cultured endothelial cell layer reveal that endothelial cells play a crucial role in promoting neutrophil morphological changes and the expression of relevant adhesion molecules. The creation of competing chemoattractant gradients across the hydrogel matrix reveals the hierarchy among three common neutrophil chemoattractants (fMLP = LTB4 > IL-8), and this order confirms the previous conclusion that the p38 MAP pathway is dominant over the PI3K pathway for neutrophil migration, but the introduction of an endothelial cell layer enhances the ability of LTB4 to promote neutrophil migration. Compared to the conditions of single chemoattractant gradients, the coexistence of two different chemoattractants in the same total amount indicates a statistically higher number of cells migrating into the collagen gel, implying synergistic effects between any two neutrophil chemoattractants. This is the first report of competing and synergistic effects among various chemoattractants in the neutrophil TEM process. In conclusion, this research describes a promising candidate for neutrophil immunology studies and provides new insights on the mechanisms of cellular interactions that can be used to predict in vivo neutrophil behaviors during the migration process.
Acknowledgements
This work was partially supported by a Defense Advanced Research Projects Agency (DARPA) funding (DARPA 00026142). Device fabrication was done in the Minnesota Nano Center at University of Minnesota. We would like to thank Dr Zhe Gao for taking SEM images of the collagen gel. We also thank the help from Guillermo Marques and Thomas Pengo in University Imaging Center (University of Minnesota) for recording and processing confocal images.
References
- B. Amulic, C. Cazalet, G. L. Hayes, K. D. Metzler and A. Zychlinsky, Annu. Rev. Immunol., 2012, 30, 459–489 CrossRef CAS PubMed.
- E. Kolaczkowska and P. Kubes, Nat. Rev. Immunol., 2013, 13, 159–175 CrossRef CAS PubMed.
- N. D. Burg and M. H. Pillinger, Clin. Immunol., 2001, 99, 7–17 CrossRef CAS PubMed.
- H. L. Wright, R. J. Moots, R. C. Bucknall and S. W. Edwards, Rheumatology, 2010, 49, 1618–1631 CrossRef CAS PubMed.
- N. Borregaard, Immunity, 2010, 33, 657–670 CrossRef CAS PubMed.
- J. D. van Buul and P. L. Hordijk, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 824–833 CrossRef CAS PubMed.
- S. J. Roth, M. W. Carr, S. S. Rose and T. A. Springer, J. Immunol. Methods, 1995, 188, 97–116 CrossRef CAS.
- Z. Ding, K. Xiong and T. B. Issekutz, J. Leukocyte Biol., 2001, 69, 458–466 CAS.
- J. Xu, X.-P. Gao, R. Ramchandran, Y.-Y. Zhao, S. M. Vogel and A. B. Malik, Nat. Immunol., 2008, 9, 880–886 CrossRef CAS PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- S. Chung, R. Sudo, P. J. Mack, C.-R. Wan, V. Vickerman and R. D. Kamm, Lab Chip, 2009, 9, 269–275 RSC.
- G. S. Jeong, S. Han, Y. Shin, G. H. Kwon, R. D. Kamm, S.-H. Lee and S. Chung, Anal. Chem., 2011, 83, 8454–8459 CrossRef CAS PubMed.
- W. J. Polacheck, J. L. Charest and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11115–11120 CrossRef CAS PubMed.
- M. B. Chen, S. Srigunapalan, A. R. Wheeler and C. A. Simmons, Lab Chip, 2013, 13, 2591–2598 RSC.
- S. Han, J.-J. Yan, Y. Shin, J. J. Jeon, J. Won, H. Eun Jeong, R. D. Kamm, Y.-J. Kim and S. Chung, Lab Chip, 2012, 12, 3861–3865 RSC.
- D. Kim and C. L. Haynes, Anal. Chem., 2013, 85, 10787–10796 CrossRef CAS PubMed.
- E. K. Sackmann, E. Berthier, E. W. K. Young, M. A. Shelef, S. A. Wernimont, A. Huttenlocher and D. J. Beebe, Blood, 2012, 120, e45–e53 CrossRef CAS PubMed.
- U. Y. Schaff, M. M. Q. Xing, K. K. Lin, N. Pan, N. L. Jeon and S. I. Simon, Lab Chip, 2007, 7, 448–456 RSC.
- R. Snyderman and M. C. Pike, Annu. Rev. Immunol., 1984, 2, 257–281 CrossRef CAS PubMed.
- C. D. Sadik, N. D. Kim and A. D. Luster, Trends Immunol., 2011, 32, 452–460 CrossRef CAS PubMed.
- S. E. Van Eeden and T. Terashima, Leuk. Lymphoma, 2000, 37, 259–271 CAS.
- I. A. Reilly, H. R. Knapp and G. A. Fitzgerald, J. Clin. Pathol., 1988, 41, 1163–1167 CrossRef CAS.
- S. P. Mathis, V. R. Jala, D. M. Lee and B. Haribabu, J. Immunol., 2010, 185, 3049–3056 CrossRef CAS PubMed.
- G. Cavicchioni, A. Fraulini, S. Falzarano and S. Spisani, Eur. J. Med. Chem., 2009, 44, 4926–4930 CrossRef CAS PubMed.
- F. Pellegatta, Y. Lu, A. Radaelli, M. R. Zocchi, E. Ferrero, S. Chierchia, G. Gaja and M. E. Ferrero, Br. J. Pharmacol., 1996, 118, 471–476 CrossRef CAS PubMed.
- F. Pellegatta, S. L. Chierchia and M. R. Zocchi, J. Biol. Chem., 1998, 273, 27768–27771 CrossRef CAS PubMed.
- Y. Zheng, J. Chen, M. Craven, N. W. Choi, S. Totorica, A. Diaz-Santana, P. Kermani, B. Hempstead, C. Fischbach-Teschl, J. A. López and A. D. Stroock, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9342–9347 CrossRef CAS PubMed.
- H. Oh, B. Siano and S. Diamond, J. Visualized Exp., 2008, 17, e745, DOI:10.3791/745.
- B. M. Eaton, V. J. Toothill, H. A. Davies, J. D. Pearson and G. E. Mann, J. Cell. Physiol., 1991, 149, 88–99 CrossRef CAS PubMed.
- M. Dhanabal, R. Ramchandran, M. J. F. Waterman, H. Lu, B. Knebelmann, M. Segal and V. P. Sukhatme, J. Biol. Chem., 1999, 274, 11721–11726 CrossRef CAS PubMed.
- J. Heidemann, H. Ogawa, M. B. Dwinell, P. Rafiee, C. Maaser, H. R. Gockel, M. F. Otterson, D. M. Ota, N. Lügering, W. Domschke and D. G. Binion, J. Biol. Chem., 2003, 278, 8508–8515 CrossRef CAS PubMed.
- A. Li, S. Dubey, M. L. Varney, B. J. Dave and R. K. Singh, J. Immunol., 2003, 170, 3369–3376 CrossRef CAS.
- K. M. Stroka, H. N. Hayenga and H. Aranda-Espinoza, PLoS One, 2013, 8, e61377 CAS.
- A. R. Burns, R. A. Bowden, S. D. MacDonell, D. C. Walker, T. O. Odebunmi, E. M. Donnachie, S. I. Simon, M. L. Entman and C. W. J. Smith, Cell Sci., 2000, 113, 45–57 CAS.
- S. K. Shaw, P. S. Bamba, B. N. Perkins and F. W. Luscinskas, J. Immunol., 2001, 167, 2323–2330 CrossRef CAS.
- M. W. J. Boehme, U. Raeth, W. A. Scherbaum, P. R. Galle and W. Stremmel, Clin. Exp. Immunol., 2000, 119, 250–254 CrossRef CAS.
- H. Cho, B. Hamza, E. A. Wong and D. Irimia, Lab Chip, 2014, 14, 972–978 RSC.
- D. Kim and C. L. Haynes, Anal. Chem., 2012, 84, 6070–6078 CrossRef CAS PubMed.
- F. Lin, C.-C. Nguyen, S.-J. Wang, W. Saadi, S. Gross and N. Jeon, Ann. Biomed. Eng., 2005, 33, 475–482 CrossRef.
- B. Heit, L. Liu, P. Colarusso, K. D. Puri and P. Kubes, J. Cell Sci., 2008, 121, 205–214 CrossRef CAS PubMed.
- B. Heit, S. M. Robbins, C. M. Downey, Z. Guan, P. Colarusso, B. J. Miller, F. R. Jirik and P. Kubes, Nat. Immunol., 2008, 9, 743–752 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an00967g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a