
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single-molecule digital enzyme assay using alkaline phosphatase with a cumarin-based fluorogenic substrate
Yusuke
Obayashi
a,
Ryota
Iino
bc and
Hiroyuki
Noji
*ad
aDepartment of Applied Chemistry, The University of Tokyo, Japan. E-mail: hnoji@appchem.t.u-tokyo.ac.jp; Fax: +81 3 5841 1872; Tel: +81 3 5841 7252
bOkazaki Institute for Integrative Bioscience, Institute of Molecular Science, National Institutes of Natural Sciences, Japan
cSOKENDAI (The Graduate University for Advanced Studies), Japan
dCREST, Japan Science and Technology Agency, Japan
First published on 1st June 2015
Abstract
Digitalization of fluorogenic enzymatic assays through the use of femtoliter chamber array technology is an emerging approach to realizing highly quantitative bioassays with single-molecule sensitivity. However, only a few digital fluorogenic enzyme assays have been reported, and the variations of the digital enzyme assays are basically limited to fluorescein- and resorufin-based fluorogenic assays. This limitation hampers the realization of a multiplex digital enzyme assay such as a digital enzyme-linked immunosorbent assay (ELISA). In this study, after optimization of buffer conditions, we achieved a single-molecule digital enzyme alkaline phosphatase (ALP) assay with a cumarin-based fluorogenic substrate, 4-methylunbelliferyl phosphate (4-MUP). When ALP molecules were encapsulated in a 44-femtoliter chamber array at a low ratio of less than 1 molecule per chamber, each chamber showed a discrete fluorescence signal in an all-or-none manner, allowing the digital counting of the number of active enzyme molecules. The fraction of fluorescent chambers linearly decreased with the enzyme concentration, obeying the Poisson distribution as expected. We also demonstrated a dual-color digital enzyme assay with a ALP/4-MUP and β-galactosidase (β-gal)/resorufin-β-D-galactopyranoside combination. The activities of single ALP and β-gal molecules were clearly detected simultaneously. The method developed in this study will enable us to carry out a parallelized, multiplex digital ELISA.
Introduction
Miniaturization of bioassay systems provides many benefits to bioanalysis, such as massive parallelization, reductions in sample volumes, and more rapid responses due to the large surface-to-volume ratio.1,2 Another important benefit of the downsizing of reaction volumes is higher sensitivity. A particular case is the single-molecule fluorogenic enzyme assay using femtoliter chambers (hereafter referred to as the digital enzyme assay);3–6 individual enzyme molecules are stochastically encapsulated with the fluorogenic assay mixture in femtoliter-sized reactor chambers, and the catalytic activity is detected as the fluorescent signal from the reaction product molecules accumulate in the femtoliter chambers. The mean turnover rate of enzymes is around 10 turnovers per s.7 In a cube of 1 μm, which corresponds to 1 femtoliter, the concentration of reaction products reaches the micromolar range in a few minutes, allowing them to be readily detectable with a conventional fluorescent microscope.In a pioneering study of a single-molecule enzyme assay in femtoliter-scaled reactors,8 a diluted solution of β-galactosidase (β-gal) was partitioned in 7–30 μm water-in-oil (W/O) droplets with a fluorescein-based fluorogenic substrate at less than 1 enzyme molecule per droplet. The fluorescence signals from the droplets exhibited an all-or-none manner; while most droplets were not fluorescent, some showed clear fluorescence. The fraction of fluorescent droplets exhibited good linearity with the enzyme concentration, indicating that partitioning of the fluorogenic assay mixture into femtoliter reactors enabled the detection of the catalytic activity of a single β-gal molecule. However, due to the large inherent heterogeneity of the W/O droplets prepared by this technique (which was developed over 50 years ago), this approach did not become widespread as an analytical method until microfabrication technology allowed generation of the femtoliter chambers with identical shapes and volumes. In 2005, Rondelez et al. first reported the digital counting of active enzyme molecules through the use of a microfabricated chamber system.6 They prepared a microfabricated PDMS sheet with identically shaped micron-sized wells on the surface and encapsulated the enzyme solution between the fabricated PDMS sheet and a glass coverslip by mechanically pressing the PDMS sheet against the glass coverslip. When the enzyme solution was diluted to a ratio of less than 1 enzyme molecule per chamber, the individual chambers showed discrete fluorescence signals in the all-or-none fashion; while most of the chambers remained non-fluorescent, a few chambers showed fluorescence, and only a few showed fluorescence signals with double intensity, indicating the encapsulation of zero, one, or two molecules in each chamber, respectively. This work demonstrated that a very simple microdevice allows the formation of identically shaped femtoliter chambers and enables the single-molecule detection of enzyme molecules and quantification of the enzyme concentrations by directly counting the number of enzyme molecules (i.e., digital counting).
In recent years, several different formats for the femtoliter chamber system have been reported. For example, researchers have reported a femtoliter chamber array system formed from a plain PDMS sheet and a chemically etched optical fiber bundle.9 In addition, Sakakihara et al. developed an array system of W/O droplets that formed on micron-sized hydrophobic patterns on glass.10 Moreover, Ge et al. integrated a droplet chamber system in a microfluidic flow channel to form a gradient of the trapping probability of target molecules along the microchannel.3 This system enables the automatic preparation of a dilution series of specimens, allowing digital counting of enzyme molecules over a wide dynamic range. A microfluidic system for the generation and analysis of freestanding femtoliter droplets was also used for digital counting of enzyme molecules.11 Recently, an arrayed lipid bilayer chamber system (ALBiC) has been developed that allows digital counting and analysis of active transporter membrane proteins.12
Recently, the application of femtoliter chamber arrays has also been expanding. For example, PDMS-based femtoliter chamber arrays have been used for measurement of the chemomechanical coupling efficiency of a single rotary molecular motor protein13 and for detection of individual translation events in single bacterial cells.14 Many reports have described other applications of femtoliter chamber array systems that provide high sensitivity and/or high-throughput capacity, including DNA sequencing,15 single-cell drug efflux activity analysis,16in vitro translation,17 and single-enzyme analysis.4,18 Among them, one of the most important applications is the digital enzyme-linked immunosorbent assay (ELISA),3,11,19,20 in which antigen molecules recognized by enzyme-conjugated antibodies are individually entrapped in a chamber, and the number of antigen molecules is counted as the number of femtoliter chambers showing enzymatically produced fluorescence signals. Although the first report of a digital ELISA used the PDMS-etched optical fiber plate system,20 droplet-based array systems have been frequently used in recent studies.3,19,21 The digital ELISA has largely improved the limit of detection (LOD) down to the femto- or attomolar range, realizing the ultrasensitivity of diagnostic ELISAs.
Compared with the active development of platforms and expansion of applications of femtoliter chamber-based digital bioassays, the variety of fluorogenic enzyme assays is still limited; however, researchers are hoping to develop parallelized digital counting assays, such as multiplex digital ELISAs, for improved analysis of multiple targets. To date, the chemistry of fluorogenic assays has mainly been based on three major fluorescent dyes: fluorescein, resorufin, and cumarin. Due to its high photostability and high fluorescent intensity, fluorescein is the first choice of probes; the first digital enzyme assay used a fluorescein derivative conjugated with galactose.6 Subsequently, digital enzyme assays with resorufin-based fluorogenic substrates were conducted for detection of β-gal,22 β-glucuronidase,4 and horseradish peroxidase.18 Although resorufin and fluorescein fluorescence signals are spectrally separable, the excitation and emission spectra overlap, causing fluorescence cross-talk. Therefore, simultaneous, dual-color digital enzyme assays using fluorescein- and resorufin-based fluorogenic assays have not been attempted. Since cumarin-based fluorogenic assays use excitation and emission wavelengths much shorter than those of fluorescein and resorufin, they will be suitable for dual digital enzyme assays with these dyes. However, digital enzyme assays using cumarin-based fluorogenic substrates have not been reported. In this study, we developed a digital enzyme assay with a cumarin-based fluorogenic substrate for detection of Escherichia coli alkaline phosphatase (ALP), which is widely used in diagnostic ELISAs.
Results
Before testing the digital enzyme assay with a cumarin-based fluorogenic substrate, we explored the optimal buffer conditions for the catalytic reaction of ALP in solution. To obtain a high fluorescent signal, we used a mutant ALP (D101S) from E. coli, for which the Vmax was 35-fold higher than that for the wild-type.23 We used 4-methylunbelliferyl phosphate (4-MUP) as the fluorogenic substrate for ALP; 4-MUP is a phosphorylated cumarin derivative and hydrolyzed into inorganic phosphate and 4-metylunbelliferon (4-MU) (Fig. 1). While 4-MUP is non-fluorescent, 4-MU emits fluorescence (excitation peak: 372 nm, emission peak: 445 nm). The enzymatic activity of ALP was monitored by determining the fluorescence of 4-MU. Fig. 2A shows typical time courses of the fluorogenic assay at different initial concentrations of 4-MUP. The concentrations of 4-MU increased linearly for 300 s, indicating the constant turnover rates at this time scale under the conditions used. Furthermore, the slope of the time course increased as the 4-MUP concentration increased, indicating increased activity of ALP.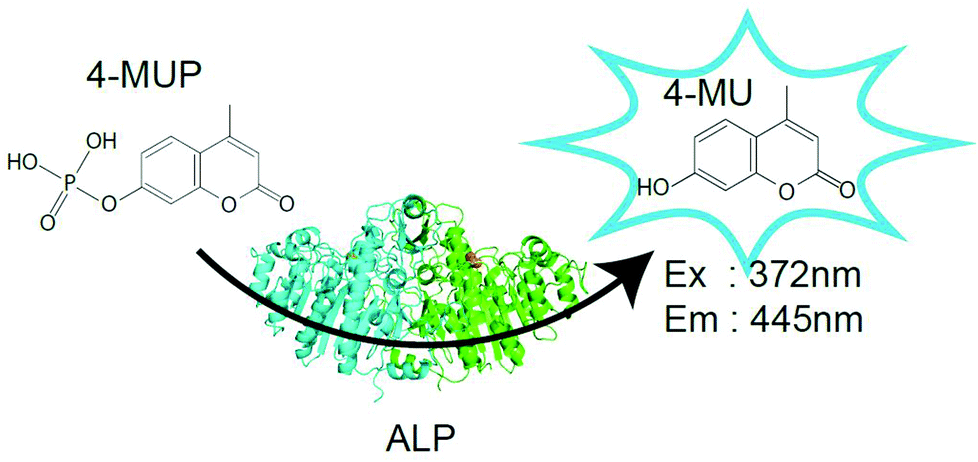 | ||
| Fig. 1 Fluorogenic substrates and enzymes used in this study. Schematic images of the fluorogenic assay of alkaline phosphatase (ALP) which hydrolyzes 4-methylunbelliferyl phosphate (4-MUP) to 4-metylunbelliferon (4-MU) and inorganic phosphate. While 4-MUP is non-fluorescent, 4-MU is fluorescent (excitation peak: 372 nm, emission peak: 445 nm). | ||
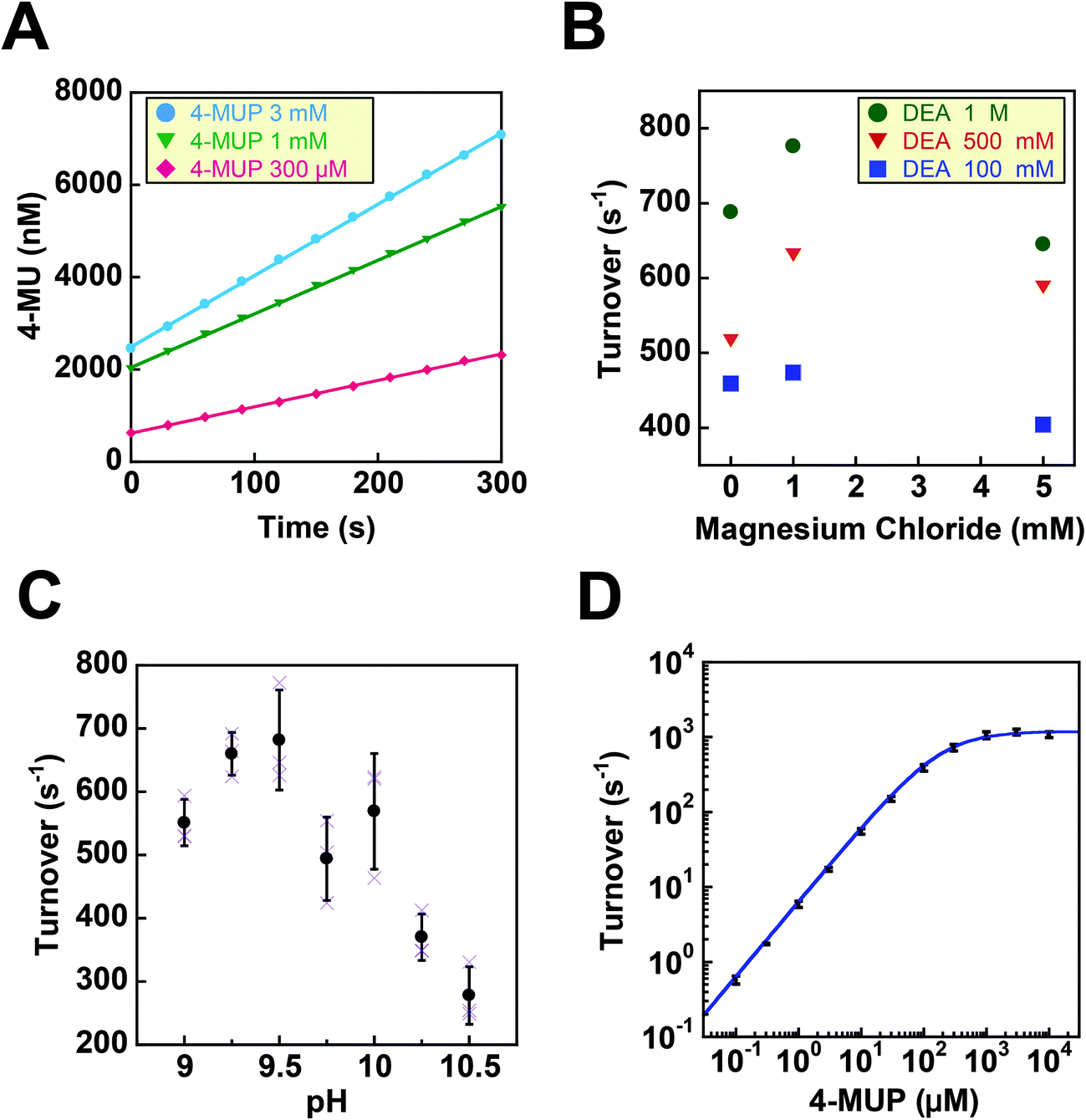 | ||
| Fig. 2 Fluorogenic assays of ALP in bulk solution. (A) Time courses of fluorogenic assays of ALP with 4-MUP at 300 μM (red), 1 mM (green), 3 mM (blue). The reaction was monitored by the fluorescence of the reaction product, 4-MU. The turnover rate was determined from the linear fitting of a time-course. (B) Effects of diethanolamine (DEA) and magnesium chloride on the turnover rate. (C) pH dependence of the turnover rate. The activity was measured in the presence of 1 M DEA and 1 mM MgCl2. Error bars represent the standard deviations of three independent measurements. (D) Dependence of the turnover rate on 4-MUP concentration. The activity was measured in the presence of 1 M DEA and 1 mM MgCl2 at pH 9.25. Data points were fitted to the Michaelis–Menten equation to give a Km of 183 μM and a Vmax of 1.19 × 103 s−1. | ||
Next, we examined the effects of diethanolamine (DEA), which enhances the transphosphorylation activity of ALP as an acceptor of inorganic phosphate.24 We also tested the effects of magnesium ions (Fig. 2B). Among the tested conditions, 1 M DEA and 1 mM MgCl2 yielded the highest ALP activity. Thus, we then tested various pH conditions with 1 M DEA and 1 mM MgCl2 (Fig. 2C). We found that pH 9.25 was the optimal pH showing the highest activity. Finally, we investigated the ALP activity at different 4-MUP concentrations to determine the basic kinetic parameters of ALP (Fig. 2D). By fitting the data points with the Michaelis–Menten equation, the maximum turnover rate (kcat) and the Michaelis–Menten constant (KM) were determined to be 1.19 × 103 s−1 and 183 μM, respectively. These values were consistent with a previous report on the mutant ALP.23
Next, digital enzyme assays for detection of the ALP activity in W/O-type femtoliter chambers were conducted at 2 mM 4-MUP in the presence of 1 M DEA and 1 mM MgCl2 at pH 9.25, at which ALP hydrolyzes 4-MUP at 1090 s−1 according to the Michaelis–Menten analysis in Fig. 2D. Note that at the estimated consumption of 4-MUP in a 44 fL chamber containing a single ALP molecule for 20 min reaction, is only 2.5%, so that the substrate consumption as well as the product inhibition would be negligible. After adding the enzyme solution to the 4-MUP solution, the reaction mixture was immediately introduced into the device and sealed with fluorinated oil. When the calculated mean number of ALP molecules per chamber (λ) was well below 1, the chambers showed a discrete fluorescent intensity. Fig. 3A shows the fluorescence image at λ = 0.52 after a 4 min incubation. Although more than 50% of the chambers remained non-fluorescent, some chambers showed weak or strong fluorescence (Fig. 3B). The fractions of non-fluorescent, weak and strong fluorescent chambers were consistent with expectations based on the Poisson distribution. When ALP molecules are randomly encapsulated, the probability of encapsulation obeys the Poisson distribution:
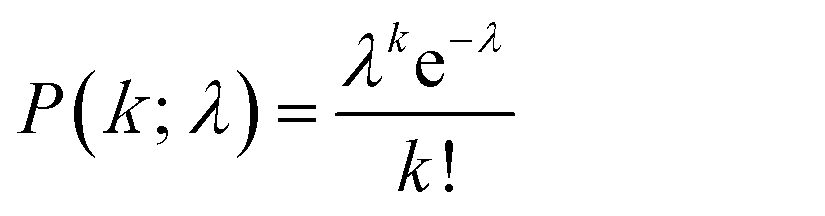
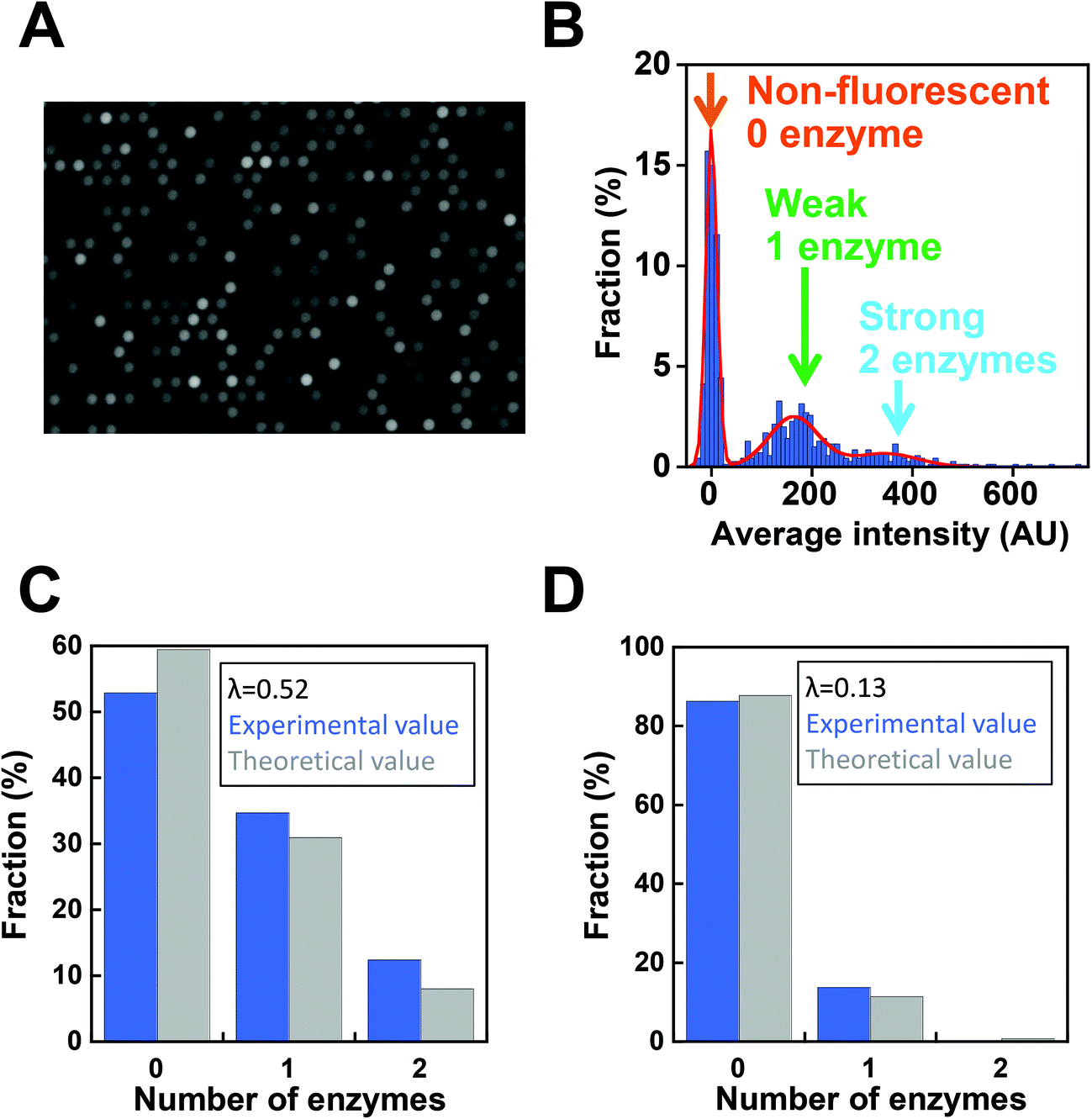 | ||
| Fig. 3 Digital enzyme assays of ALP. (A) Fluorescence images of digital enzyme assays of ALP with a 44 fL chamber array device at the mean number of ALP molecules per chamber (λ) of 0.52. Images were obtained after a 10 min incubation. The exposure time was 100 ms. (B) Distribution of the fluorescence intensity of the chambers. The data points were fit to the sum of the three Gaussians. Chambers were assigned to having 0, 1, or 2 enzymes according to the fluorescence intensity. (C) The fractions of chambers with 0, 1, and 2 enzymes experimentally determined by the Gaussian fitting in (B) (blue), and the theoretical value estimated assuming the Poisson distribution with λ = 0.52. (D) The fractions of chambers with 0, 1, or 2 enzymes when λ = 0.13. | ||
Next, we conducted a time-course analysis of digital enzyme assays of ALP at λ = 0.13. To minimize the photobleaching of 4-MU, images were taken every 5 min with a 100 ms exposure time (Fig. 4A). The rate of increase in fluorescence intensity was determined for each chamber by linear fitting of the time course (Fig. 4A, right). Fig. 4B shows the distribution of the rate. The leftmost peak represents the non-fluorescent chamber (0 molecule). The second peak, magnified in the inset of Fig. 4B represents the catalytic activity of a single ALP molecule at 2 mM 4-MUP. The Gaussian fit yielded an average turnover rate of 891 s−1. This value was slightly lower than the expected value from the measurement shown in Fig. 2D (1090 s−1). We do not attribute it to slow leakage of the fluorescent reaction product 4-MU into the oil phase, because the chambers enclosed with 4-MU retained almost constant fluorescence over 30 min in the control experiments (data not shown). The frequent interaction of the enzyme with surfaces might interfere with the catalysis to some extent. As expected from the Poisson distribution with λ = 0.13, a few chambers (0.7%) showed double activity by encapsulation of two enzyme molecules in a chamber (Fig. 4A and B).
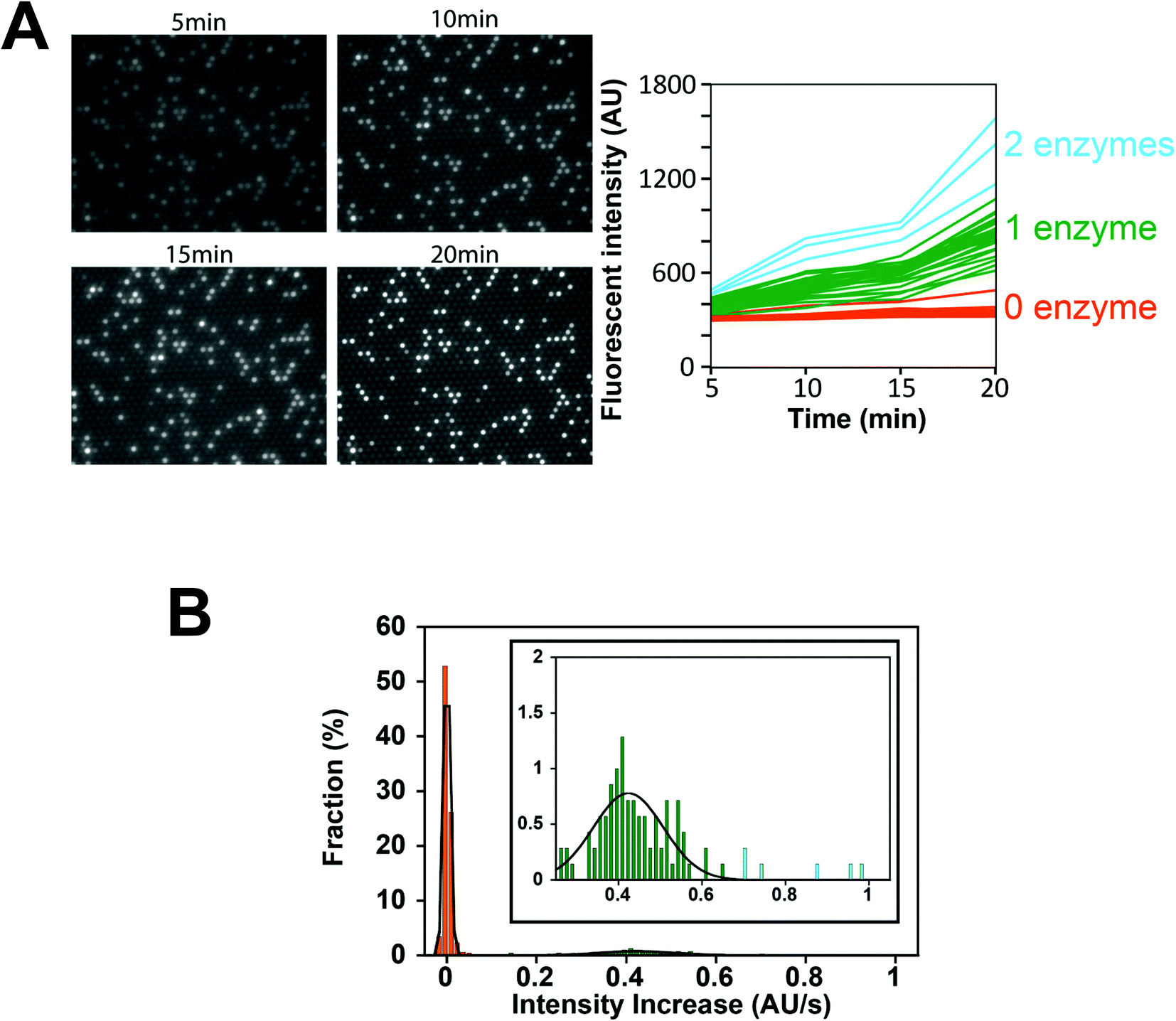 | ||
| Fig. 4 Time-course analysis of the digital enzyme assay of ALP. (A, left) Fluorescence images after 5, 10, 15, and 20 min. λ = 0.13. (A, right) Time course of the fluorescence intensity from (A, left). Orange, green, and light blue lines were assigned to chambers with 0, 1, and 2 enzymes, respectively. (B) Distribution of the rate of fluorescence increase determined by linear fitting of (A, right). The data points were fit to the sum of two Gaussians for chambers having 0 (orange) or 1 (green) enzyme molecule. Blue data points were assigned to chambers having 2 enzyme molecules. Inset indicates an enlarged distribution of chambers having 1 or 2 enzymes. | ||
In order to determine the LOD of digital counting of ALP, a 10-fold dilution series of the ALP solution from λ = 0.13 to λ = 0.00013 was subjected to digital enzyme assays. After a 10 min incubation, 120 fluorescence images of the different field of view were taken for each dilution sample. Fig. 5A shows typical fluorescence images. Each field of view contains 7600 chambers, and the total number of chambers analyzed was over 0.9 million. Because the fraction of encapsulation of two enzyme molecules in a chamber was essentially negligible at λ ≤ 0.13, each fluorescent chamber was counted as one enzyme molecule. The threshold level for fluorescent chambers was set as the mean background level plus 10 times the standard deviation (SD) of the background fluorescence. As shown in Fig. 5B, the number of fluorescent chambers was proportional to the λ and essentially consistent with the theoretical values, although the experimental values were slightly higher than the theoretical values, presumably due to the inaccuracy of protein quantification. The background count (false-positive fluorescent chambers detected in the absence of ALP) was much higher than that for the fluorogenic digital enzyme assay of β-gal with fluorescein-di-β-D-galactopyranoside (about 0.0001%).19 This difference could be explained by contaminating impurities or photoresists remaining on the device (see the Discussion section). The high background count resulted in the LOD of 7.0 fM (λ = 1.9 × 10−4) (Fig. 5C).
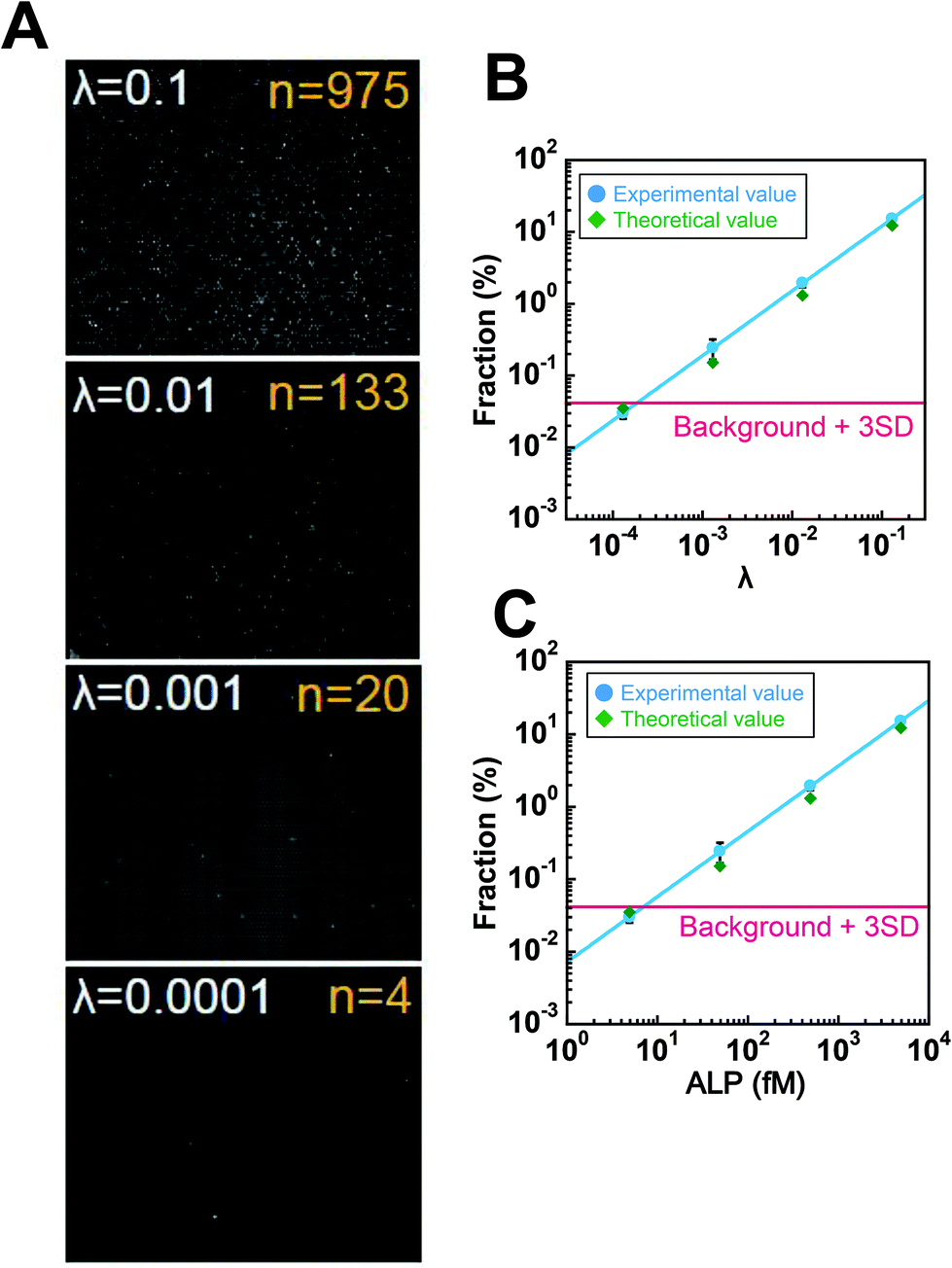 | ||
| Fig. 5 Digital counting of ALP. (A) Fluorescence images of digital enzyme assays of ALP at λ = 0.13, 0.013, 0.0013, and 0.00013 after a 10 min incubation. Typical examples of the single field of view obtained at each λ are shown. The numbers of fluorescent chambers (n) at each λ is also shown. (B and C) The fractions of fluorescent chambers versus λ (B) or ALP concentration (C). Error bars are the standard deviations of three independent measurements. The red line represents the fraction of the background count plus 3 times the SD of the background count determined under the ALP-free conditions. | ||
Finally, we tested the feasibility of dual-color digital enzyme assays of ALP and β-gal. In the fluorogenic assay for β-gal, the enzyme cleaved resorufin-β-D-galactopyranoside (RGP) to galactose and resorufin, yielding red fluorescence. The 4-MU and resorufin have different fluorescent emission peaks, 445 nm and 585 nm, respectively, and were spectrally separable in the fluorescence images. Because the optimal pH values for ALP and β-gal are different, the assays were conducted under near alkaline conditions (pH 8.25), at which ALP retains 67% of the maximum activity. After mixing well-diluted enzyme solutions with fluorogenic substrates, the reaction mixture was introduced into a flow cell and observed under a fluorescence microscope. Fig. 6 shows a pseudo-colored overlay image of 4-MU and resorufin. Green and red represent the fluorescence of 4-MU and resorufin, respectively. As expected, the green and red fluorescent chambers were distributed randomly, with some showing both green and red fluorescence (Fig. 6, yellow arrow), indicating the simultaneous encapsulation of ALP and β-gal. Thus, the feasibility of dual-color digital enzyme assays with cumarin- and resorufin-based fluorogenic assays was successfully demonstrated.
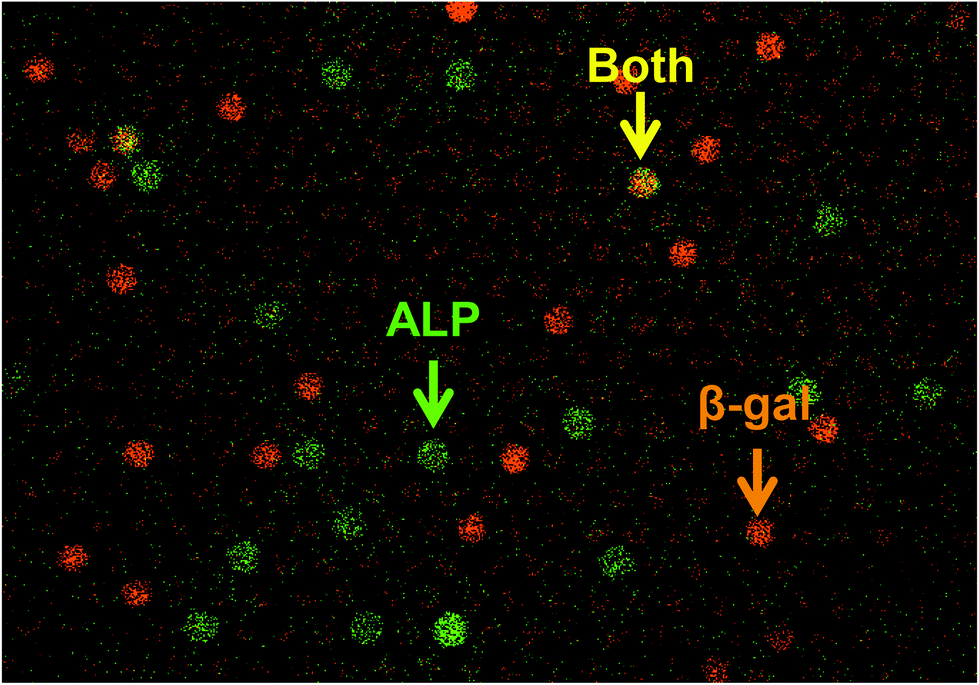 | ||
| Fig. 6 Dual-color digital enzyme assays for ALP and β-gal. Dual-color fluorogenic assays for ALP and β-gal were conducted by 45 min incubation. ALP and β-gal were encapsulated at λ = 0.033 and 0.05, respectively. ALP produced 4-MU, and β-gal produced resorufin. A pseudo-colored overlay image of 4-MU (green) and resorufin (red) is shown. The yellow color indicates a mixture of red and green signals, representing the coexistence of ALP and β-gal. | ||
Discussion
This study presented a cumarin-based fluorogenic assay that can be applied as a digital enzyme assay. Because many cumarin-based fluorogenic assays have been developed for analysis and detection of enzymes, digital enzyme assays using cumarin-based probes are expected to have a variety of applications. In addition, the present study also demonstrated a dual-color digital enzyme assay using cumarin and resorufin for the first time.Although fluorescein- and resorufin-based digital enzyme assays have been reported independently, dual-color digital enzyme assays using these two probes have not been achieved due to overlap of their excitation and fluorescence spectra. However, cumarin emits fluorescence signals of distinctively shorter wavelengths. In this study, we verified that the dual-color digital enzyme assays for ALP and β-gal were feasible using resorufin and cumarin. Dual-color digital enzyme assays are expected to enable multiplex digital ELISAs; while such multiplex digital ELISAs have been reported using differently colored plastic beads to identify the captured antigen molecules,21 the expansion of color variations in digital enzyme assays will provide an alternative approach for multiplex digital ELISAs or further expand the multiplicity of digital ELISAs by combination with the multi-colored bead method.
In addition to the above, multi-color digital enzyme assays are expected to exhibit an improved background count compared with digital ELISAs; a high background count may impair the potential sensitivity of digital ELISAs. The main factor affecting the background count in digital ELISAs is non-specific binding of the detection antibody–enzyme conjugate to the bead surface on which the captured antibody is immobilized. Plastic beads are the most frequently used surface for antigen capture. When the target antigen molecule is marked with two different detection antibody–enzyme conjugates simultaneously, we can distinguish the true signal from the false-positive signal with high efficiency. This is because the simultaneous non-specific binding of two detection conjugates on the same bead should be much more infrequent than the single nonspecific binding events. Thus, by increasing the color variations of fluorogenic assays, the background count of digital ELISAs will be dramatically reduced. This strategy is expected to be highly effective, particularly for digital ELISAs targeting multi-epitope antigens such as infectious viruses.
However, several drawbacks of this method were also found when compared with a fluorescein-based digital enzyme assay. The first is the leakage of cumarin to the fluorinated oil phase under neutral pH conditions. Although 4-MU has an additional hydroxyl moiety on the cumarin structure which enhances water solubility, the hydroxyl group of 4-MU has to be deprotonated to prevent the leakage into the fluorinated oil phase. Actually, slow leakage was found at neutral pH (pH 7.0). This phenomenon is consistent with the pKa of the hydroxyl group of 4-MU (pH 7.8). Thus, 4-MU-based fluorogenic assays are currently limited to the conditions under which the pH is alkaline or near alkaline. Several cumarin derivatives carrying dissociative groups with different pKa values have been reported. To expand the cumarin-based fluorogenic assay to the conditions under which the pH is neutral or acidic, cumarin derivatives with lower pKa, such as a fluorinated 4-MU, should be tested.
Another drawback of cumarin-based digital enzyme assays is their relatively high background count. As shown in Fig. 5B, 0.03–0.04% of chambers showed apparent fluorescence signals under ALP-free conditions. Thus, because of this high background count, the digital counting of ALP with 4-MUP has not achieved an LOD below the fM level. We tested the possible contamination of ALP enzymes by the bacteria grown in buffers by using freshly prepared chemicals and buffers. However, the background count was not reduced. However, when observed with an optical setup for fluorescein imaging, such background counts were not observed. The background count was observed even when pure water was introduced into the device. These results suggest that the background count was attributable to the dissolution of unknown impurities from CYTOP or photoresist polymers. Thus, to reduce or eliminate the background count, microfabrication procedures may have to be improved.
Experimental
Materials
The D101S mutant of alkali phosphatase (ALP) from E. coli was a kind gift from Abott Japan.23 Powder of ALP was dissolved in buffer A (20 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 150 mM NaCl, 0.1% sodium azide) and stored at −30 °C. The ALP stock was diluted in buffer A before use. Enzymatic activity was measured in assay buffer containing the indicated concentrations of diethanolamine (DEA)–HCl at pH 9.25 and magnesium chloride. The fluorogenic substrate for ALP, 4-methylunbelliferyl phosphate (4-MUP) and the reaction product, 4-metylunbelliferon (4-MU) were purchased from Sigma Aldrich (St. Louis, MO, USA). Stock solutions of 4-MUP and 4-MU were dissolved in dimethyl sulfoxide (DMSO) and stored at −30 °C.ALP assay in bulk solution
The ALP activity in bulk solution was measured in 96-well black plates (Greiner, Germany). Stocks of 4-MUP and ALP solutions were diluted in 200 μL assay buffer. The time course of the fluorescence intensity (excitation: 372 nm, emission: 445 nm) was measured at 28 °C with 30 s intervals for 5 min with a microplate reader (Flex Station 3; Molecular Devices, USA). The turnover rate was estimated from the linear fitting of the time-course and the calibration curve between the fluorescence intensity and the 4-MU concentration.Microfabrication of the femtoliter chamber array
A chamber array device was prepared as previously reported.19 A glass coverslip (24 × 32 mm) was sonicated in acetone and isopropanol, and deionized in water for 10 min. After sonication treatment, the coverslip was immersed in 10 M KOH for several hours and rinsed with deionized water. The coverslip was then spin-coated with an amorphous fluorocarbon polymer (CYTOP 816AP; Asahi Glass, Japan) at 3000 rpm for 30 s and baked for 1 h on a hotplate at 180 °C. The thickness of the CYTOP layer was 3 μm. The CYTOP-coated coverslip was spin-coated with a positive photoresist (AZ-4903; AZ Electronic Materials, USA) at 4000 rpm for 60 s and baked at 55 °C for 3 min and then at 110 °C for 5 min. Subsequently, photolithography was carried out with a mask structure with 3 μm holes, which were each separated by 3 μm. The resist-patterned coverslip was dry-etched with O2 plasma in a reactive ion etching system (RIE-10NR; Samco, Japan) to remove the exposed CYTOP. The substrate was then cleaned and rinsed with acetone and ethanol to remove the photoresist layer remaining on the substrate. The resulting CYTOP-on-coverslip device had an array of exposed SiO2 patterns with a diameter of 4.3 μm, which each held a water droplet in the digital enzyme assay. The device had 120 square areas each having 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 223 (167 × 169) patterns within an area of 10 × 10 mm2.
223 (167 × 169) patterns within an area of 10 × 10 mm2.
Digital enzyme assay for ALP in the chamber array
The flow cell was constructed from a CYTOP-on-coverslip device and a non-fabricated coverslip, which were bound via a paper spacer with silicone grease. The ALP stock solution was diluted with the assay buffer containing 1.1 mg mL−1 Tween20 (Sigma Aldrich) and 2 mM 4-MUP. Next, 40 μL of reaction mixture was introduced into the flow cell by manual pipetting. Then, 200 μL of fluorinated oil (Fluorinert FC-40; Sigma) was introduced into the flow cell to flush out an excess amount of the reaction mixture and form W/O droplets on the 4.3 μm wells of the device. The enzymatic activity of ALP molecules in the chambers was measured from the fluorescence signal of the catalytically produced 4-MU under a fluorescence microscope.Fluorescence image analysis
Fluorescent images were observed with a CMOS camera (Neo sCMOS camera; Andor, UK) using an inverted microscope (IX81; OLYMPUS, Japan) equipped with a 20× objective lens (UPlanSApo 20×/0.75; OLYMPUS) and a 1.6× image extender lens (in total 32× image magnification). The 120 fluorescence images of the different field of view (each contains 7600 chambers) in a device were taken with 100 ms exposure time for each, and analyzed with image analysis software (MetaMorph; Molecular Devices). The fluorescence intensity of each chamber was determined as the averaged intensity of 7 × 7 pixels (1.4 × 1.4 μm2) containing a single chamber.Dual digital enzyme assay
The indicated amount of ALP and β-gal from E. coli (Roche Applied Science, USA) was mixed in buffer B (1 M diethanolamine–HCl, pH 8.25, 1 M MgCl2) containing 250 μM 4-MUP and 250 μM resorufin-β-D-galactopyranoside (RGP) (Life Technologies, USA). After infusion into the flow cell and sealing with FC 40 oil, the chambers were imaged with a confocal microscope (Nikon Eclipse Ti microscope; Nikon, Japan) equipped with a CMOS camera (NIKON A1R MP; Nikon). The objective lens used was PlanApo 60×/1.40 oil (Nikon), and 401 nm and 561 nm lasers were used as the excitation light sources for 4-MU and resorufin, respectively.Acknowledgements
We would like to thank Dr Toru Yoshimura, Dr Eisaku Yoshida, and Dr Ryotaro Chiba from Abbott Japan for the expression vector of the mutant ALP and for technical supports. This research was supported by the Japan Science and Technology Agency for Core Research for Evolutional Science and Technology (CREST).References
- T. Schneider, J. Kreutz and D. T. Chiu, The potential impact of droplet microfluidics in biology, Anal. Chem., 2013, 85, 3476–3482 CrossRef CAS PubMed.
- D. Witters, B. Sun, S. Begolo, J. Rodriguez-Manzano, W. Robles and R. F. Ismagilov, Digital biology and chemistry, Lab Chip, 2014, 14, 3225–3232 RSC.
- S. Ge, W. Liu, T. Schlappi and R. F. Ismagilov, Digital, ultrasensitive, end-point protein measurements with large dynamic range via Brownian trapping with drift, J. Am. Chem. Soc., 2014, 136, 14662–14665 CrossRef CAS PubMed.
- R. B. Liebherr, M. Renner and H. H. Gorris, A single molecule perspective on the functional diversity of in vitro evolved beta-glucuronidase, J. Am. Chem. Soc., 2014, 136, 5949–5955 CrossRef CAS PubMed.
- D. M. Rissin, H. H. Gorris and D. R. Walt, Distinct and long-lived activity states of single enzyme molecules, J. Am. Chem. Soc., 2008, 130, 5349–5353 CrossRef CAS PubMed.
- Y. Rondelez, G. Tresset, K. V. Tabata, H. Arata, H. Fujita, S. Takeuchi and H. Noji, Microfabricated arrays of femtoliter chambers allow single molecule enzymology, Nat. Biotechnol., 2005, 23, 361–365 CrossRef CAS PubMed.
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi, D. S. Tawfik and R. Milo, The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters, Biochemistry, 2011, 50, 4402–4410 CrossRef CAS PubMed.
- B. Rotman, Measurement of activity of single molecules of beta-D-galactosidase, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1981–1991 CrossRef CAS.
- D. M. Rissin and D. R. Walt, Digital concentration readout of single enzyme molecules using femtoliter arrays and Poisson statistics, Nano Lett., 2006, 6, 520–523 CrossRef CAS PubMed.
- S. Sakakihara, S. Araki, R. Iino and H. Noji, A single-molecule enzymatic assay in a directly accessible femtoliter droplet array, Lab Chip, 2010, 10, 3355–3362 RSC.
- J. U. Shim, R. T. Ranasinghe, C. A. Smith, S. M. Ibrahim, F. Hollfelder, W. T. Huck, D. Klenerman and C. Abell, Ultrarapid generation of femtoliter microfluidic droplets for single-molecule-counting immunoassays, ACS Nano, 2013, 7, 5955–5964 CrossRef CAS PubMed.
- R. Watanabe, N. Soga, D. Fujita, K. V. Tabata, L. Yamauchi, S. Hyeon Kim, D. Asanuma, M. Kamiya, Y. Urano, H. Suga and H. Noji, Arrayed lipid bilayer chambers allow single-molecule analysis of membrane transporter activity, Nat. Commun., 2014, 5, 4519 CAS.
- Y. Rondelez, G. Tresset, T. Nakashima, Y. Kato-Yamada, H. Fujita, S. Takeuchi and H. Noji, Highly coupled ATP synthesis by F1-ATPase single molecules, Nature, 2005, 433, 773–777 CrossRef CAS PubMed.
- L. Cai, N. Friedman and X. S. Xie, Stochastic protein expression in individual cells at the single molecule level, Nature, 2006, 440, 358–362 CrossRef CAS PubMed.
- P. A. Sims, W. J. Greenleaf, H. Duan and X. S. Xie, Fluorogenic DNA sequencing in PDMS microreactors, Nat. Methods, 2011, 8, 575–580 CrossRef CAS PubMed.
- R. Iino, K. Hayama, H. Amezawa, S. Sakakihara, S. H. Kim, Y. Matsumono, K. Nishino, A. Yamaguchi and H. Noji, A single-cell drug efflux assay in bacteria by using a directly accessible femtoliter droplet array, Lab Chip, 2012, 12, 3923–3929 RSC.
- S. H. Kim, S. Yoshizawa, S. Takeuchi, T. Fujii and D. Fourmy, Ultra-high density protein spots achieved by on chip digitalized protein synthesis, Analyst, 2013, 138, 4663–4669 RSC.
- B. N. Ehrl, R. B. Liebherr and H. H. Gorris, Single molecule kinetics of horseradish peroxidase exposed in large arrays of femtoliter-sized fused silica chambers, Analyst, 2013, 138, 4260–4265 RSC.
- S. H. Kim, S. Iwai, S. Araki, S. Sakakihara, R. Iino and H. Noji, Large-scale femtoliter droplet array for digital counting of single biomolecules, Lab Chip, 2012, 12, 4986–4991 RSC.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- D. M. Rissin, C. W. Kan, L. Song, A. J. Rivnak, M. W. Fishburn, Q. Shao, T. Piech, E. P. Ferrell, R. E. Meyer, T. G. Campbell, D. R. Fournier and D. C. Duffy, Multiplexed single molecule immunoassays, Lab Chip, 2013, 13, 2902–2911 RSC.
- H. H. Gorris, D. M. Rissin and D. R. Walt, Stochastic inhibitor release and binding from single-enzyme molecules, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17680–17685 CrossRef CAS PubMed.
- W. Mandecki, M. A. Shallcross, J. Sowadski and S. Tomazic-Allen, Mutagenesis of conserved residues within the active site of Escherichia coli alkaline phosphatase yields enzymes with increased kcat, Protein Eng., 1991, 4, 801–804 CrossRef CAS PubMed.
- R. B. McComb and G. N. Bowers Jr., Study of optimum buffer conditions for measuring alkaline phosphatase activity in human serum, Clin. Chem., 1972, 18, 97–104 CAS.
| This journal is © The Royal Society of Chemistry 2015 |