
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-instrumented DNA isolation, amplification and microarray-based hybridization for a rapid on-site detection of devastating Phytophthora kernoviae†
Lydia
Schwenkbier
abc,
Sibyll
Pollok
ad,
Anne
Rudloff
ac,
Sebastian
Sailer
a,
Dana
Cialla-May
abc,
Karina
Weber
*abc and
Jürgen
Popp
abc
aLeibniz Institute of Photonic Technology (IPHT Jena), Jenaer BioChip Initiative, Albert-Einstein-Straße 9, 07745 Jena, Germany
bFriedrich Schiller University Jena, Institute of Physical Chemistry and Abbe Center of Photonics, Helmholtzweg 4, 07743 Jena, Germany. E-mail: karina.weber@ipht-jena.de
cInfectoGnostics Forschungscampus Jena, Zentrum für Angewandte Forschung, Philosophenweg 7, 07743 Jena, Germany
dErnst-Abbe-Hochschule Jena, University of Applied Sciences, Carl-Zeiss-Promenade 2, 07745 Jena, Germany
First published on 18th August 2015
Abstract
A rapid and simple instrument-free detection system was developed for the identification of the plant pathogen Phytophthora kernoviae (P. kernoviae). The on-site operable analysis steps include magnetic particle based DNA isolation, helicase-dependent amplification (HDA) and chip-based DNA hybridization. The isothermal approach enabled the convenient amplification of the yeast GTP-binding protein (Ypt1) target gene in a miniaturized HDA-zeolite-heater (HZH) by an exothermic reaction. The amplicon detection on the chip was performed under room temperature conditions – either by successive hybridization and enzyme binding or by a combined step. A positive signal is displayed by enzymatically generated silver nanoparticle deposits, which serve as robust endpoint signals allowing an immediate visual readout. The hybridization assay enabled the reliable detection of 10 pg μL−1 target DNA. This is the first report of an entirely electricity-free, field applicable detection approach for devastating Phytophthora species, exemplarily shown for P. kernoviae.
Introduction
For more than a decade, invasive plant pathogens have been spreading across European and North American countries.1,2 This phenomenon was intensified by the increased globalization of the trade in plants. The most aggressive and important plant pathogens belong to the genus Phytophthora. They pose a serious threat to agriculture and food production worldwide, causing devastating diseases in hundreds of trees, shrubs and plants in natural and landscaped environments as well as in the nursery industry.2,3 The remarkably broad host range of these oomycetes plus their further geographical spread are major reasons for concern. Phytophthora kernoviae (P. kernoviae) is one of the emerging plant pathogens today. It was discovered in late autumn of 2003 as a phytopathogen of Rhododendron and beech trees in historic woodland gardens in Cornwall, Great Britain.4,5 The European and Mediterranean Plant Protection Organization (EPPO) recommended five Phytophthora species, including P. kernoviae, to be regulated as quarantine pests (http://www.eppo.int/QUARANTINE/listA2.htm). Common routine diagnoses of plant pathogens involve molecular biological, microbiological or immunological approaches.1,6,7 Most assays require expensive equipment, trained personnel and long sample-to-answer times. Thus, a rapid and reliable detection of Phytophthora kernoviae directly in the field is highly desirable in order to reduce the time delay between sampling and identification. Differentiation between the individual species is realizable mainly by nucleic acid based techniques.1,6 Polymerase chain reaction (PCR) is amongst the sensitive methods presently available for the detection of plant pathogens.7–9 Despite several efforts to adapt PCR approaches to energy-limited settings, a thermocycler for precise temperature control and a skilled operator for setting up the reaction are still required. Moreover, the on-site practicability is hampered due to power requirements of the instrumentation. To overcome these restrictions, a plethora of isothermal amplification techniques was developed as an attractive alternative for target gene amplification, omitting the use of such PCR thermocyclers.10–13 The corresponding reactions can be performed easily in a water bath or a conventional heating block. The potential for simplified instrumentation even promoted the implementation of isothermal amplification methods for in-field or point-of-care testing (POCT), enabling nucleic acid diagnostics without expensive equipment or even electricity.14–21 LaBarre and co-workers introduced a non-instrumented nucleic acid amplification (NINA) heater based on the exothermic reaction of calcium oxide with water for loop-mediated isothermal amplification (LAMP) of Plasmodium falciparum.17 Other isothermal amplification methods have been explored using this NINA heater, for example, the exponential amplification reaction (EXPAR),17 reverse transcription (RT)-LAMP,14 nicking enzyme amplification reaction (NEAR),17 or recombinase polymerase amplification (RPA).20 Recently, an improved NINA design incorporating magnesium iron alloy for heat production was reported for the amplification of viral nucleic acids.21,22 Additionally, a simplified DNA amplification platform based on helicase-dependent amplification (HDA) was presented for the specific detection of Clostridium difficile.15 In this case, the isothermal amplification was performed in a device consisting of a microfluidic chip and a toe warmer in a styrofoam cup. The respective exothermic reaction was initiated using sodium acetate as a latent heat storage system. Isothermal amplification techniques such as LAMP and HDA were also suitable for agriculture diagnostics.13,23 Though Phytophthora plant pathogens were reliably detected, power supply was needed for the experimental procedures. Nevertheless, an optimal assay would comprise an entirely non-laborious, instrument-free analysis of the plant sample.Due to the growing interest in electricity-free, user-friendly and robust agriculture diagnostic tools for pathogen identification, we established an in-field applicable approach for Phytophthora kernoviae as an example organism. By taking advantage of a naturally occurring exothermic reaction, we were able to perform isothermal amplification in a simple device, which is operating with the same accuracy as a thermocycler, but predestined for on-site handling. Furthermore, the presented assay combines electricity-free extraction and amplification of pathogenic DNA as well as chip-based detection. Thus, instructed personnel could easily perform the assay and interpret the robust endpoint signals, respectively.
Experimental
Detached leaf assay and DNA extraction
The isolate of P. kernoviae (JKI-080-09-00-00-00-03) was grown as Petri dish culture on carrot piece agar.24 Leaves of Rhododendron ponticum were inoculated with this oomycete. For this purpose the tissue was surface wounded in the middle of the lamina on the abaxial side, then inoculated with a plug of agar from active colonies and overlaid with 100 μl tap water. For 14 days it was maintained in a moist chamber at 20 °C and an 8 h day length. Agar debris around the infection site was removed.25 The genomic DNA (gDNA) of the species was extracted by homogenizing the leaf tissue with a mortar and pestle.26 The lysate was placed in a micro reaction tube and magnetic bead based DNA isolation was conducted using the innuPREP MP Basic kit (Analytik Jena AG, Germany). The extracted gDNA was adjusted to a concentration of 20 ng μL−1. The DNA concentration was determined by measuring OD260 with an Eppendorf Biophotometer plus (Eppendorf AG, Hamburg, Germany).Asymmetric helicase-dependent amplification
In the present study an asymmetric, thermophilic HDA (tHDA) approach was applied. The IsoAmp® II Universal tHDA kit (BioHelix Corporation, Beverly, MA, USA) was utilized according to the manufacturer's recommendations to amplify a fragment within the yeast GTP-binding protein (Ypt1) target gene region.9,27 The primers for amplification were designed using the program Sequencher 5.1 and fulfil the following criteria: (i) a length of 29–34 bp and (ii) an optimized melting temperature of 64 °C (±2 °C) and a G/C base content between 45 and 55%.13 The reaction mixture contained 1× annealing buffer II, 4 mM MgSO4, 40 mM NaCl, 1 μM bovine serum albumin, 3.5 μl dNTPs, 75 nM biotinylated reverse and 25 nM forward primers (Table 1), 3.5 μl of IsoAmp enzyme mixture and 2.5 μl gDNA in a final volume of 50 μl.| DNA | Sequence 5′–3′ | Modification |
|---|---|---|
| *M = adenine (A)/cytosine (C). | ||
| HDA_ker.F | GGC TGC ACG AGA TCG ATA GGT GAG TTC TAC | |
| HDA_ker.R | TCT CM*C AGG CGT ATC TGA TTT AAC ACG TGT TCC | 5′-Biotin |
| Process control | AGA ATC AAG GAG CAG ATG CTG AAA AAA | 5′-NH2, 3′-biotin |
| 1 Negative control I | TTA GAC CTT TTT GAA GAA GGT TCT GTT ACT AAC ATG | 5′-NH2-C6 |
| 2 Negative control II | ATC GAG CTG GAC GGC AAG ACC ATC AAG CT | 5′-NH2-C6 |
| 3 P. lateralis | CGG GAG ATT TTT TCC CGC TTT CCT TGG GGT AAG | 5′-NH2-C6 |
| 4 P. ramorum | CCC CCC ACT TTC CGT GGG TGA GTT TCC TTT | 5′-NH2-C6 |
| 5 P. pinifolia | CCG CGG ACG AAA ACT AAC TCT CTT GTG TAG TG | 5′-NH2-C6 |
| 6 P. fragariae | CTA GCC TTG CCA TTT CTA GGT CCA AAA AGG C | 5′-NH2-C6 |
| 7 P. kernoviae | CAC CAC ATG AAT ACC TGC CAG GCG AGA TGC | 5′-NH2-C6 |
| 8 P. austrocedrae | CCT CCG TGG TTC ATG TAC AAA ACG TGC AGC | 5′-NH2-C6 |
| 9 P. cinnamomi | CTG TCT GCC CCA TTC AAC AGA CGC TAA CGT C | 5′-NH2-C6 |
The tHDA reaction was conducted asymmetrically whereas the ratio between forward and reverse primers was set at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The reaction mixture was filled into 0.2 ml micro reaction tubes and incubated for up to 90 min at 60–70 °C in a conventional heating block (Analytik Jena AG, Germany) or an in-house constructed miniaturized HDA-zeolite-heater.
:4. The reaction mixture was filled into 0.2 ml micro reaction tubes and incubated for up to 90 min at 60–70 °C in a conventional heating block (Analytik Jena AG, Germany) or an in-house constructed miniaturized HDA-zeolite-heater.
HDA-zeolite-heater
The in-house constructed HDA-zeolite-heater (HZH) consists of an aluminum device filled with paraffin wax as a phase change material (PCM) (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) and a polyoxymethylene (POM) cap. This device is placed within zeolite beads 13× (aluminosilicate, Zeo-Tech GmbH, Unterschleissheim, Germany) in a stainless steel container (Henkelmann-Lunchtime, Baesweiler, Germany). The POM cap was chosen for its resistance to hot wax and its good processability. Moreover, the low thermal conductivity of the POM material isolates the implemented aluminum cavities from excessive heat transfer from the container wall. The PCM used had a melting point of around 65 °C. In the above-mentioned cavities, six micro reaction tubes (0.2 ml) can be placed.For the approach, 120 g zeolites were filled in the container and the aluminum device was placed in the middle of the material. The exothermal reaction was started by pouring 30 ml of water onto the zeolite beads. Afterwards the container was sealed with the lid.
Analytical agarose gel electrophoresis
Successful DNA amplification was verified on a 2% (w/v) agarose gel (Carl Roth, Karlsruhe, Germany) stained with GelRed (VWR International GmbH, Darmstadt, Germany). The molecular weight marker used was GeneRuler 100 bp DNA Ladder (Fisher Scientific, Germany).Chip-based DNA hybridization and signal detection
White planar polypropylene (PP) sheets (Modulor GmbH, Berlin, Germany) were cut into chip size (17 × 17 mm), and successively cleaned in an ultrasonic bath with acetone, ethanol and water for 10 min each.The Ypt1 region was chosen to design species-specific capture probes to achieve (i) the highest discrimination of target sequences in relation to non-corresponding sequences, (ii) a length of 30–35 bp and (iii) a melting temperature between 62 and 65 °C. The low tendency for sequence secondary structure formation is expressed in delta G values between −1 and 1.5.13 The probes (Eurofins MWG Operon, Ebersberg, Germany) were dissolved in 1× Micro Spotting Solution (ArrayIt Corporation, Sunnyvale, USA) to a final concentration of 20 μM and spotted (Nanoplotter 2.1, GeSim, Grosserkmannsdorf, Germany) in an array format on the PP chips. A biotin-labeled non-complementary probe was immobilized as a process control to verify the binding of streptavidin–enzyme and the subsequent silver deposition.
The specific biomolecule interaction on the chip was realized using either a long or a combined mode. The long protocol encompassed the incubation of an 80 μl hybridization mixture (30 μl HDA product in 3 × SSC/0.5% SDS) for one hour at room temperature, followed by 30 minutes of incubation of the streptavidin–horseradish peroxidase (Sigma Aldrich Chemie GmbH, Taufkirchen, Germany; 1:1000 diluted in 1× phosphate buffered saline with Tween® 20, PBST). For the short approach, DNA hybridization and enzyme binding were done by incubating an 80 μl reaction mixture (30 μl HDA product, 0.5 μl enzyme in 5 × SSC/0.1% SDS) for one hour at room temperature. Afterwards the chips were washed five times with solution 1 (2 × SSC/0.1% SDS) and solution 2 (water). Finally, the enzymatic silver deposition was performed by applying the EnzMet™ HRP detection kit (Nanoprobes Inc., Yaphank, USA; components A–C). The generated silver nanoparticles in the case of a positive reaction served as robust endpoint signals allowing an immediate visual readout by the naked eye. In addition, the amount of silver deposits was quantified by the grey values. The respective spots were scanned (ProScan 7200, reflecta GmbH, Rottenburg, Germany) and analyzed using the software ImageJ (National Institutes of Health, USA). The grey value was calculated by mean grey value calculation, subtracting the measured background values from the sample values. The mean grey values of the negative controls of all experiments were used to set the threshold, which equals three times the standard deviation.
Results
Our work has focused on the development of an entirely electricity-free analysis system usable for the on-site detection of Phytophthora kernoviae as an example organism (Fig. 1). Thus, we not only performed the isothermal DNA amplification without any energy-dependent device, but also conducted the obligatory pre- and post-amplification steps electricity-free. Using recyclable zeolites and disposable materials, the system offers great potential for direct application in the field.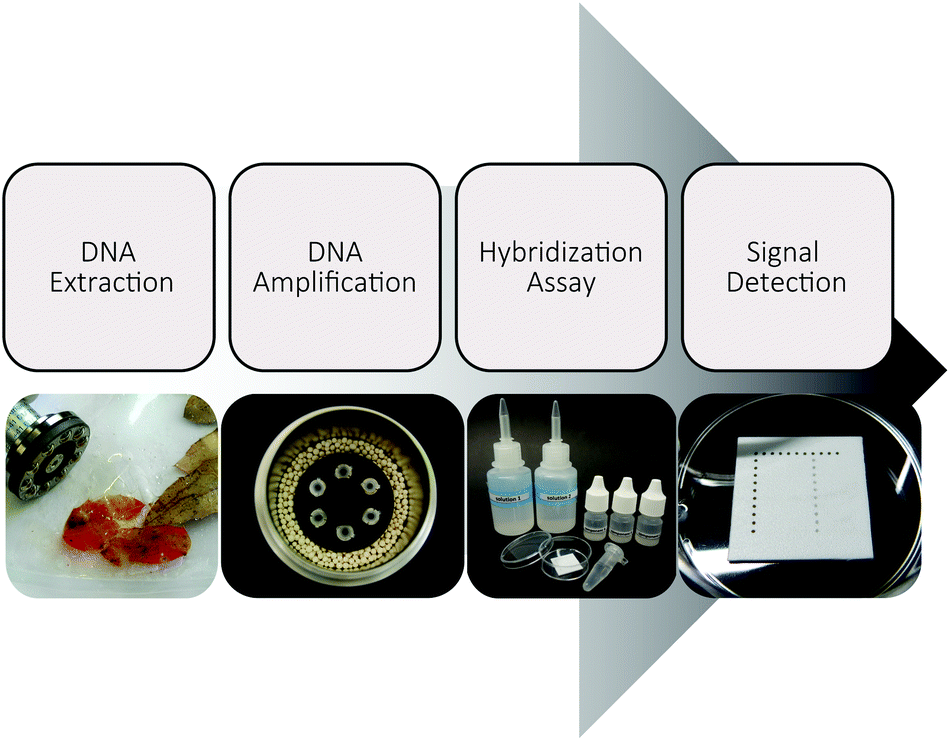 | ||
| Fig. 1 Electricity-free DNA-based detection scheme for Phytophthora kernoviae. The convenient DNA-based detection of Phytophthora kernoviae is realized by electricity- and instrument-free process steps. DNA extraction from infected plant leaves was realized by manual homogenizing and magnetic particle based nucleic acid extraction. DNA amplification was done isothermally and the detection was performed by hybridization and signal detection on white polymer chips. | ||
Instrument-free DNA extraction
The isolation of P. kernoviae gDNA from infected Rhododendron leaves was performed according to Julich et al.26 by combining a handy homogenizer and extraction bags for an effective cell disruption in conjunction with nucleic acid release. This manually operated homogenization is applicable for softer tissues such as Rhododendron leaves and provided an incredibly facile way to release DNA from plant material. A subsequent magnetic particle based DNA extraction enabled easy handling as well as short processing times.Instrument-free isothermal DNA amplification in a miniaturized HDA-zeolite-heater
The helicase-dependent amplification allowed the amplification of the target Ypt1 region of P. kernoviae. Only recently, a novel DNA-chip hybridization assay, which focused on the Ras-related GTP-binding protein 1 (Ypt1) gene, was examined for the identification of several devastating Phytophthora species.25,28 In a further study this region was explored to establish the helicase-dependent amplification for P. ramorum, P. fragariae and P. kernoviae.13The optimal temperature range for the thermophilic HDA process is considered to be set between 60–65 °C.29 In order to examine the capability of the isothermal amplification variant for higher temperature tolerance, various temperature increments (60, 62.5, 65, 67.5, and 70 °C) were tested. The respective tHDA reactions were conducted in a conventional heating block. As shown in Fig. 2, amplification was also successfully realizable at 67.5 °C and 70 °C and only slight differences in the signal intensities of the bands were observed. Thus, the tHDA process is adaptable to higher temperatures, which can emerge by using exothermic reactions for tempering.14–17,21
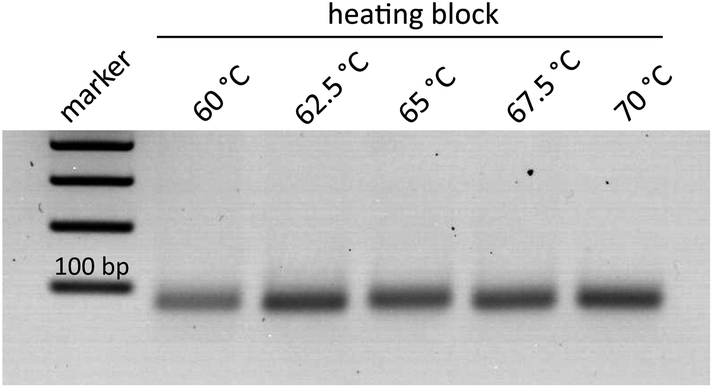 | ||
| Fig. 2 Comparison of amplification temperatures and product yield for P. kernoviae; HDA conducted in a heating block. | ||
In order to test the usability of such an alternative heating source, an in-house constructed HZH (Fig. 3) was used for the HDA reaction. This HZH, which does not require an energy supply for heat production, consists of a stainless steel container filled with zeolite beads. An aluminum device, allowing for the insertion of six micro reaction tubes, is placed in the zeolite granulate by adjusting its edge in line with the beads. As soon as water is added to the zeolites, the heat generation starts. Thermal energy is simply produced by zeolite hydration, resulting in an exothermal reaction.
 | ||
| Fig. 3 HZH for thermophilic helicase-dependent DNA amplification. (A) Stainless steel container, (B) weighted zeolite beads and pouring flask with water, (C) zeolite beads filled into the container, (D) aluminum device with PCM and cavities for six micro reaction tubes, (E) device with micro reaction tubes placed within the zeolites, (F) starting of exothermal reaction by pouring water on the zeolite beads. | ||
The pouring of water onto the zeolite granulate has to be uniform, so a small spray bottle was used to guarantee easy handling. The optimal amount of zeolites and water was empirically elucidated (data not shown).
For the characterization of the thermal profile (Fig. 4) within different HZH compartments, four individual thermal sensors were inserted. One sensor was placed in the PCM, one inside the zeolite beads and two sensors were placed into micro reaction tubes. Once the reaction started, the temperature was measured every 2 seconds. By varying the amounts of zeolites and water the best choice to provide good long-lasting temperature stability was by combining 120 g zeolites and 30 ml water. Thus, an optimal reaction temperature range between 60–70 °C for tHDA inside the sample tubes could be maintained for up to 90 min (Fig. 4B). After a short adjustment phase of approximately 15 min, the desired temperatures were reached. Note that the initial high temperature of the zeolite beads (Fig. 4A), which can peak over 100 °C, declined within a few minutes. Additionally we want to emphasize that the temperature in the sample tubes did not overshoot the desired optimal tHDA range. The paraffin wax temperature (Fig. 4A) ensured the optimal reaction conditions for isothermal DNA amplification. Furthermore, sample-to-sample variations of the reaction temperature were in the expected span.
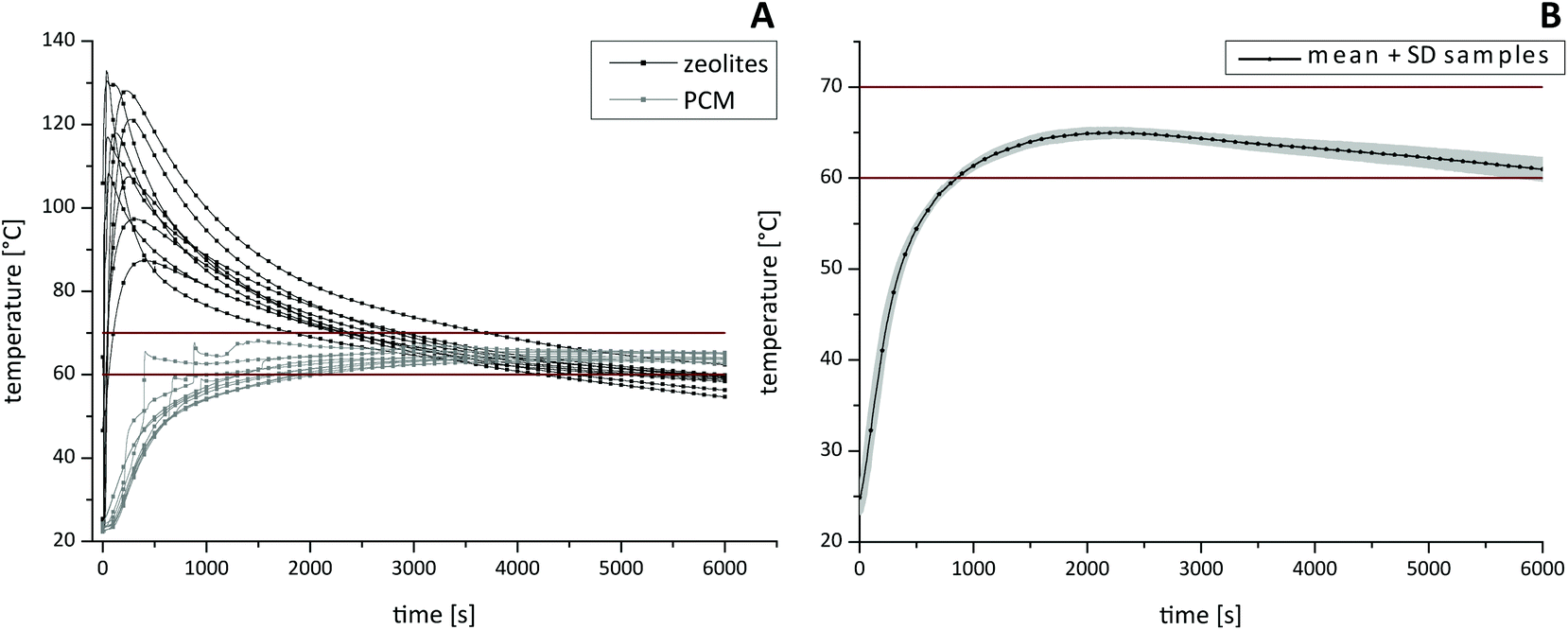 | ||
| Fig. 4 Temperature profiles of the HDA-zeolite-heater. (A) Temperature profiles of the zeolites (black lines) and PCM (grey lines), (B) temperature profiles measured in the samples, n = 10 independent measurements, the tHDA reaction temperature range is set as 60–70 °C, SD = standard deviation. | ||
In order to demonstrate the comparability of the HZH performance regarding tHDA-dependent duplication of the Ypt1 fragment with a common heating block, different amplification times were tested. According to the manufacturer's recommendations, ideal conditions for the tHDA approach are accomplished by incubating for 90 min at 65 °C. Thus, the temperature of the heating block was kept constant at 65 °C. For the HZH we assumed the recorded temperature profiles (Fig. 4). The amplification products were analyzed on an agarose gel as a reference method (Fig. 5). An incubation time of 60 min led to the appearance of distinctive bands of the precise length for both the conventional heating block and the HZH, indicating an appropriate reproducibility. The product yield did not increase after a reaction time of 90 min. Thus, for all further investigations we kept the amplification time constant at 60 min, which favors a reliable DNA amplification.
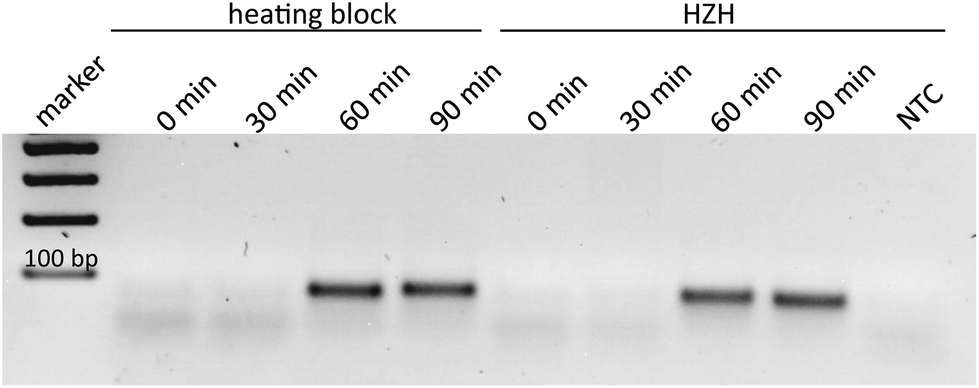 | ||
| Fig. 5 Side-by-side comparison of HDA amplification times and product yield for P. kernoviae Ypt1 fragment, conducted either in a heating block or the HZH. The amplification temperature of the heating block was kept constant at 65 °C. The amplification times for the HZH were displayed as times the desired temperature range between 60–70 °C, which was reached 15 min after the induction of the exothermal reaction (15 min warm-up time). | ||
As the prototype HZH provides cavities for six micro reaction tubes, the homogeneous heat distribution in combination with amplification yield had to be examined for parallel placement of six samples. Therefore, the amplification products of six parallel and at once independent reactions in both devices were analyzed on an agarose gel (Fig. 6). Prominent bands with almost the same intensity verify a suitable reproducibility due to a homogeneous heat distribution in the HZH. Thus with one single HZH run different samples could be placed and successfully amplified.
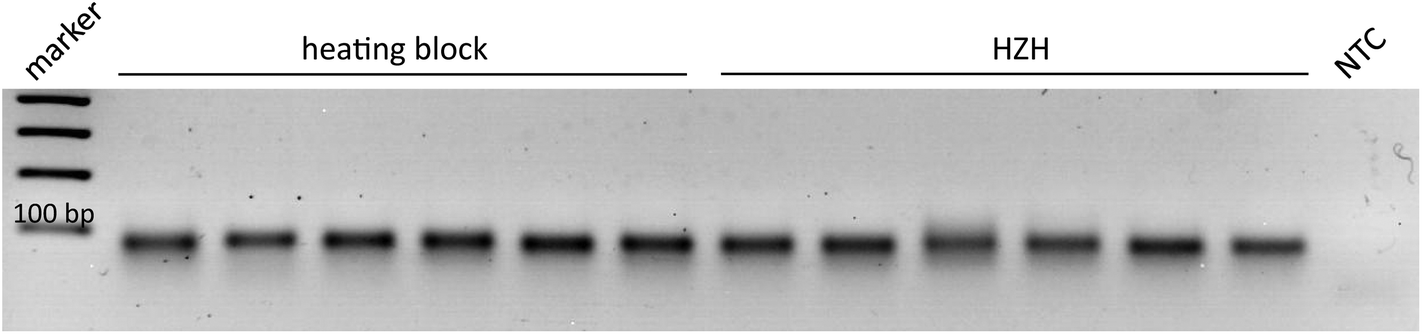 | ||
| Fig. 6 Homogeneous heat distribution leads to successful amplification of the P. kernoviae Ypt1 fragment in the six cavities of the HZH. The amplification temperature of the heating block was kept constant at 65 °C. Six parallel amplifications in the heating block or the HZH were conducted for 60 min to verify the homogeneous heat distribution and thus amplification consistency when all the six available cavities of the HZH were used. | ||
Instrument-free microarray-based DNA detection
Recently a novel DNA chip hybridization assay based on the Ypt1 region was examined.25 It allows for parallel and specific detection of nine Phytophthora species due to immobilized capture probes in an array format. Based on these capture probes, on-chip hybridization experiments were performed to prove the functionality of the amplified products. We chose the asymmetric tHDA approach to generate single-stranded target DNA within the amplification reaction for a subsequent hybridization. Thus, the identification of P. kernoviae is realized by analyzing the appearance of black spots at the respective chip positions, where nine various capture probes were immobilized in a defined pattern. Each specific probe was spotted with at least eight replicates in a column (Fig. 7). The usage of white PP material as a chip substrate allowed an appropriate capture probe spotting without the need for prior chemical surface modification. Furthermore, we applied a suitable and rapid washing mode by only pouring the respective solutions onto the chip several times. The enzymatically generated silver nanoparticle deposits served as robust endpoint signals allowing for signal readout by the naked eye.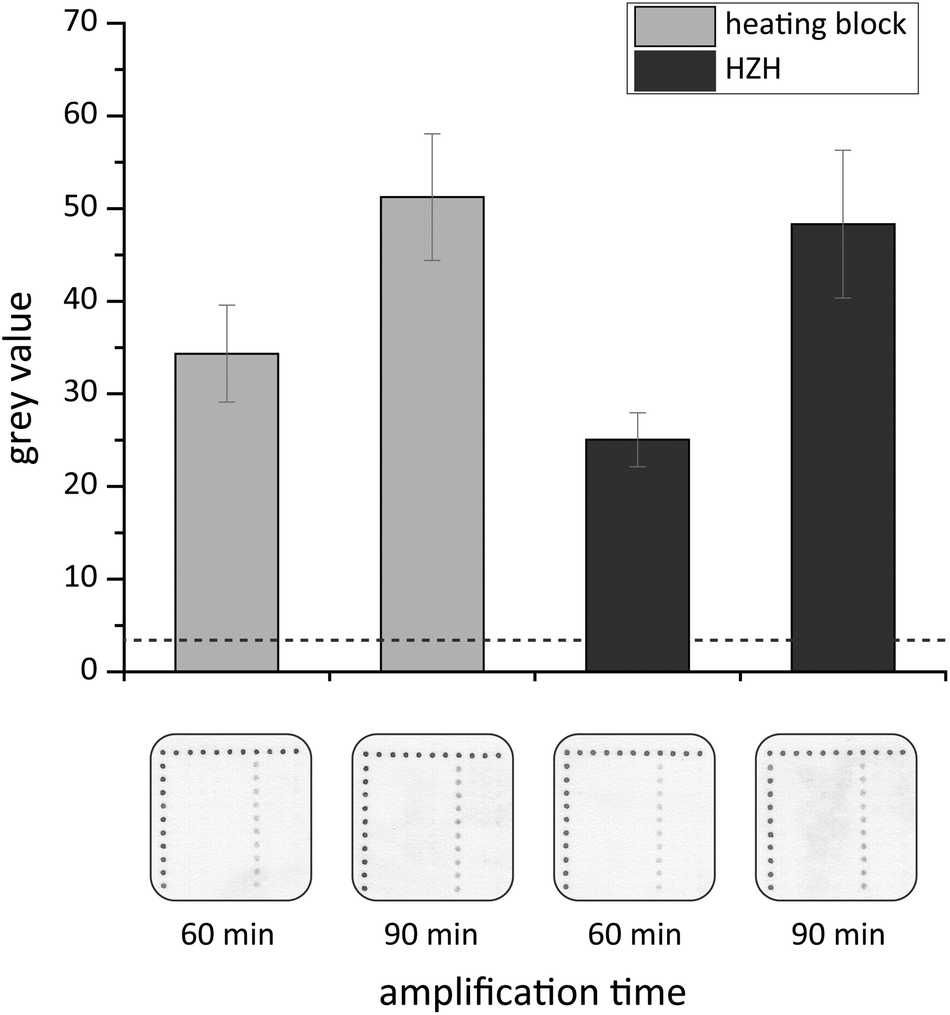 | ||
| Fig. 7 Chip-based detection of HDA-amplified P. kernoviae target DNA. The chip-based detection was realized by a short combined protocol at room temperature. HDA products were generated either by amplification in a heating block or the HZH. The indicated times represent the particular amplification times. Grey values were depicted as endpoint signals for successful on-chip hybridization. The dashed line indicates the calculated threshold (three times the standard deviation of negative controls). Black silver spot signals appear for P. kernoviae-specific capture probes (chip image, column 7, 10 replicates per column) when incubated with HDA products of P. kernoviae target DNA. Chip legend: black dot line on the left and upper side = process control, 1st column = negative control I, 2nd column = negative control II, 3rd column = P. lateralis, 4th column = P. ramorum, 5th column = P. pinifolia, 6th column = P. fragariae, 7th column = P. kernoviae, 8th column = P. austrocedrae, 9th column = P. cinnamomi, and 10th column = spotting buffer. | ||
In first experiments, the protocol for DNA hybridization, subsequent enzyme binding and visual detection was optimized. Since thermophilic HDA is essential to amplify short DNA fragments, we were able to perform the following hybridization step at room temperature. Applying a successive protocol, specific positive signals on the P. kernoviae capture probe positions (Fig. S1,† column 7) were detectable when using the 60 min or 90 min tHDA amplification products. These observations are in accordance with the appearance of the HDA products after 60 or 90 min amplification time (Fig. 5). Moreover, by conducting the tHDA reaction asymmetrically, sufficient amounts of single-stranded target DNA were generated to hybridize with the corresponding capture probes, even under room temperature conditions. We wish to point out that signal intensities on the chip using the 60 min HDA amplification products are sufficient and valid for a specific detection of P. kernoviae. Lastly, no false-positive signals were detectable on the chip (Fig. 7 and S1†).
In order to significantly shorten the processing time, a combined protocol that encompasses DNA hybridization and enzyme binding within one step was examined. The results of the on-chip hybridization experiments are presented in Fig. 7. Specific signals for P. kernoviae occur on the white PP chips when incubating with target DNA that was amplified in the heating block or HZH for 60 or 90 min. For the heating block the grey values for P. kernoviae increased by one third when applying the 90 min amplification product. In the case of the HZH the grey values doubled for 90 min amplification time. Nevertheless, also the 60 min HDA product led to distinct hybridization signals. The obtained results implicate that the reduction of the hybridization assay time to 60 min (short protocol) is sufficient for a positive signal outcome. Thus, the HZH provides solid evidence to be comparable to electricity-driven lab equipment.
A further attempt was made to elucidate the sensitivity of the on-chip hybridization. For this purpose isolated gDNA was serially diluted to final DNA concentrations of 1 ng μl−1 to 1 pg μL−1, amplified by tHDA in the HZH and these amplification products were used for hybridization experiments. As illustrated in Fig. 8 dark spots of the generated silver nanoparticles could be observed for up to 10 pg μL−1. The highest grey values were detected for 1 ng μl−1 and they decreased gradually with falling gDNA amounts.
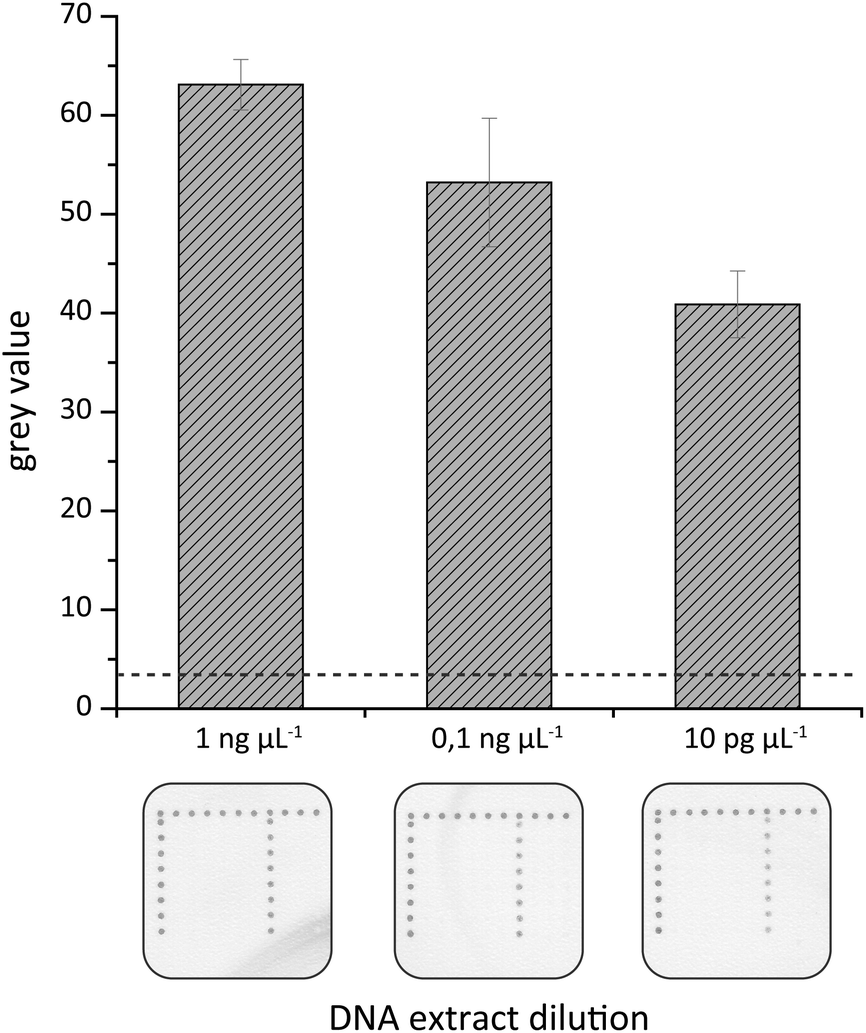 | ||
| Fig. 8 Sensitivity of the on-chip hybridization assay. Varying amounts of gDNA were used for tHDA amplification in the HZH. The generated amplification products were applied for on-chip hybridizations. The respective values were shown as the mean and standard deviation of eight spot replicates per species-specific P. kernoviae capture probe. The dashed line indicates the calculated threshold (three times the standard deviation of negative controls). Black silver spot signals only appear for P. kernoviae-specific capture probes (chip image, column 7, 8 replicates per column) when incubated with HDA products of P. kernoviae target DNA. Chip legend: black dot line on the left and upper side = process control, 1st column = negative control I, 2nd column = negative control II, 3rd column = P. lateralis, 4th column = P. ramorum, 5th column = P. pinifolia, 6th column = P. fragariae, 7th column = P. kernoviae, 8th column = P. austrocedrae, 9th column = P. cinnamomi, and 10th column = spotting buffer. | ||
Discussion
Within this study we highlight an entirely electricity-free solution for the DNA-based identification of plant pathogens exemplarily for P. kernoviae. Thus, three miniaturized real instrument-free tools for DNA isolation, DNA amplification as well as DNA detection are combined to guarantee easy handling and device portability. In the case of a suspected plant infection, the clarifying assay could be performed on-site without the transportation of expensive, heavy equipment or the necessity for an electricity infrastructure. Rhododendron leaves of P. kernoviae infected plants were chosen to demonstrate the functionality of the novel established assay.The collection and preparation of the samples is the first critical step in the detection of plant pathogenic oomycetes. Simply by homogenizing the leaves with a mortar and pestle symptomatic plant tissue disruption could be performed without the use of mills or centrifuges that require power supply. Afterwards, genomic DNA was easily extracted by a magnetic particle approach. Magnetic particles offer several benefits, e.g. convenient handling, short processing times and the absence of energy requirements.30–32
In order to prove a prospective field application, the isothermal amplification variant HDA was optimized to substitute PCR approaches, which commonly require cost-intensive and power-dependent thermocyclers. Amplification of P. kernoviae DNA was carried out in a miniaturized HDA-zeolite-heater. A fragment within the Ypt1 gene was chosen for amplification due to its high genetic variability that allows a clear discrimination between several Phytophthora species.25
In particular, tHDA enables true isothermal amplification without the need of prior heat denaturation or elaborate primer design, which is for instance mandatory for LAMP.33–35 We demonstrated that the minimal duration of HDA reactions is reduced to 60 min (plus 15 min warm-up time in the case of HZH), which resulted in a product yield that was comparable to the generally recommended 90 min reaction. The sample's presence in the device during the warm-up period had no influence on the amplification reaction. In contrast, a waiting time of more than 10 minutes before sample placement associated with device opening/closing steps is indispensable to ensure optimal LAMP assay sensitivity.21 Furthermore, the tHDA was carried out in an asymmetric manner to ensure the presence of single-stranded target DNA. Thus, a post-amplification treatment to generate single-stranded target DNA could be omitted, which favored its on-site operation purpose and additionally reduced the contamination risk.36–38 For application directly in the field, ice-packs are utilizable to cool the HDA components. Nevertheless, improvements regarding room temperature storable enzymes or even dry reagents, like that reported for LAMP,39 could eliminate the need for cold chain storage of kit components.
The major aim of this study was the realization of an electricity-free amplification protocol taking advantage of exothermal energy supply by zeolites40,41 and thermal coupling with PCM. Recent reports addressing the use of exothermic reactions for isothermal DNA amplification recommend calcium oxide powder, sodium acetate or magnesium iron alloy. However, there are several benefits of applying zeolites for exothermal reaction conditions. Firstly, the zeolite beads allow easy handling in terms of weighing, filling and emptying the HZH device without dust formation while filling or incrustation after the reaction, as is the case for calcium oxide powder.14,16,17,19 A heater cleaning as well as the final rinsing of the steel container to remove salt remnants21 after amplification is not necessary. Thus, the handling is incredibly user-friendly. Secondly, the ‘activated zeolite beads’ (dehydrated form) are storable and transportable in locked flasks and are immediately ready-to-use. A liquefaction by heating is not essential as is required for sodium acetate.20 Thirdly, they are cost-efficient and available in abundance, which is a major requirement when considering the design of a new bioanalytical device. One more important feature is their ability for reversible adsorption of water.40 The zeolites can be regenerated by an appropriate thermal treatment (200–300 °C) while maintaining their structural stability.42 This and the fact that zeolites are non-toxic mark them as environmentally-friendly. Moreover, the aluminum device is immediately usable after a cool down period without any further wax-refilling step. The thermal profile of a cheap paraffin wax as PCM meets the requirements for tHDA properly. The desired temperatures in the sample tubes, generated by the combined application of zeolites and PCM, persisted for a sufficient period of time. A batch-to-batch variation, as is reported for calcium oxide,21 was not observed for the zeolite beads when performing tHDA reactions. Furthermore, the system offers a small size that ensures its portability. One major advantage is the very precise heating profile, which implies the adaption of this heating method to other isothermal amplification variants like LAMP or thermophilic strand displacement amplification (SDA), which also demand a temperature range between 62–65 °C.34,43,44 The application of a NINA heater for isothermal amplification modes such as LAMP, EXPAR, NEAR and RPA was reported for the detection of Plasmodium falciparum,16,17Salmonella enterica,19Bacillus anthracis18 and human immunodeficiency virus.14,21 Additionally, Huang and colleagues introduced the use of HDA and a toe warmer material filled styrofoam insulator for the identification of Clostridium difficile.15 However, currently no report claims the performance of HDA in an electricity-free device for phytopathogen identification in the field. It can be stated that the developed HDA-zeolite-heater enabled a convenient isothermal amplification requiring only minimal equipment and its successful inclusion in an entire instrument-free analysis chain.
Subsequent on-chip hybridization experiments were conducted to prove the specificity and sensitivity of the assay and to exclude non-specific signals of the generated Ypt1-amplicons. Rating of the obtained endpoint signals was possible by the naked eye due to dark silver deposits on white polypropylene chips. The planar and inert chip material is characterised by its low price and its availability in large quantities. Furthermore, it is not necessary to chemically modify the surface prior to capture probe immobilization. In order to reduce the detection time ruled by labor-intensive multiple washing procedures, a combined protocol including both hybridization and enzyme immobilization was examined. Considering easy handling, the hybridization buffer was prestored in a vial, so the operator only had to supplement the amplified DNA and apply the solution to the respective chip. After 1 h of incubation with either the 60 min or 90 min HDA product, distinct silver spots could be observed for the capture probe of P. kernoviae. Due to the necessity of short fragments for the tHDA, hybridization could be performed at room temperature showing great efficiency and specificity. The target DNA bound selectively to the corresponding capture probe without showing any false-positive signals. The obtained results point out that 60 min asymmetric isothermal amplification and a subsequent 60 min chip-based detection are sufficient for a reliable and specific identification of the plant pathogen Phytophthora kernoviae. Considering the gDNA isolation procedure and various pipetting steps the whole analysis takes around 3 hours. Compared to the commonly applied microbiological identification, which needs several days the presented assay significantly minimized the sample-to-answer time. Furthermore, the described assay allowed the detection of 10 pg μL−1 target DNA isolated from infected plant tissue.
This is the first report on an entire non-instrumented detection scheme for a selected plant pathogen towards on-site operation. We highlight an electricity-free HDA-zeolite-heater based on an exothermic reaction of inexpensive starting material. The usage of this miniaturized, portable device generates results regarding amplicon yield that are comparable to thermal instruments. The functionality of this novel assay was exemplarily demonstrated for P. kernoviae, but could be easily adapted to other plant pathogens. The near-future implementation of the HDA-zeolite-heater for agriculture diagnostics directly in the field is highly desirable in order to prevent the further spreading of devastating plant pathogens. Besides, the simplicity of the assay steps indicates the realization of the full procedure by instructed personnel. This can significantly reduce the sample-to-answer times and enable a faster intervention in the case of an in-field detected plant pathogen or also a human pathogen in POCT applications.
Acknowledgements
We thank Stephan König, Stephan Wagner and Sabine Werres from the Julius Kühn-Institute for the capture probe design, for the preparation of artificially inoculated Rhododendron leaves and for the extraction and reprocessing of the Phytophthora DNA. The Federal Ministry of Food and Agriculture (BMEL) finances the project “PhytoChip-Validierung” (28-1-54.081-10) based on a decision of the Parliament of the Federal Republic of Germany via the Federal Office for Agriculture and Food (BLE) under the innovation support programme. The projects “JBCI 2.0” (03IPT513Y, InnoProfile-Transfer, Unternehmen Region) and InfectoGnostics Forschungscampus (13GW0096F) supported by the Federal Ministry of Education and Research (BMBF), Germany are gratefully acknowledged.References
- P. A. O'Brien, N. Williams and G. E. S. Hardy, Crit. Rev. Microbiol., 2009, 35, 169–181 CrossRef PubMed.
- O. K. Ribeiro and K. Lamour, Phytophthora: A Global Perspective, 2013, vol. 2, p. 1 Search PubMed.
- D. C. Erwin and O. K. Ribeiro, Phytophthora diseases worldwide, APS Press, St. Paul, Minn., 1996 Search PubMed.
- C. Brasier and J. Webber, Nature, 2010, 466, 824–825 CrossRef CAS PubMed.
- C. M. Brasier, P. A. Beales, S. A. Kirk, S. Denman and J. Rose, Mycol. Res., 2005, 109, 853–859 CrossRef PubMed.
- F. N. Martin, Phytophthora: A Global Perspective, 2013, vol. 2, p. 19 Search PubMed.
- F. N. Martin, Z. G. Abed, Y. Baldi and K. Ivors, Plant Dis., 2012, 96, 1080–1103 CrossRef.
- G. J. Bilodeau, C. A. Levesque, A. W. A. M. de Cock, C. Duchaine, S. Briere, P. Uribe, F. N. Martin and R. C. Hamelin, Phytopathology, 2007, 97, 632–642 CrossRef CAS PubMed.
- L. Schena and D. E. L. Cooke, J. Microbiol. Methods, 2006, 67, 70–85 CrossRef CAS PubMed.
- P. J. Asiello and A. J. Baeumner, Lab Chip, 2011, 11, 1420–1430 RSC.
- P. Craw and W. Balachandran, Lab Chip, 2012, 12, 2469–2486 RSC.
- P. Gill and A. Ghaemi, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 224–243 Search PubMed.
- L. Schwenkbier, S. Pollok, S. Koenig, M. Urban, S. Werres, D. Cialla-May, K. Weber and J. Popp, Anal. Methods, 2015, 7, 211–217 RSC.
- K. A. Curtis, D. L. Rudolph, I. Nejad, J. Singleton, A. Beddoe, B. Weigl, P. LaBarre and S. M. Owen, PLoS One, 2012, 7 Search PubMed.
- S. Huang, J. Do, M. Mahalanabis, A. Fan, L. Zhao, L. Jepeal, S. K. Singh and C. M. Klapperich, PLoS One, 2013, 8 Search PubMed.
- P. Labarre, J. Gerlach, J. Wilmoth, A. Beddoe, J. Singleton and B. Weigl, Conference proceedings: Annual International Conference of the IEEE Engineering in Medicine and Biology Society, IEEE Engineering in Medicine and Biology Society, Annual Conference, 2010, 2010, 1097–1099.
- P. LaBarre, K. R. Hawkins, J. Gerlach, J. Wilmoth, A. Beddoe, J. Singleton, D. Boyle and B. Weigl, PLoS One, 2011, 6 CrossRef CAS PubMed.
- B. Hatano, T. Maki, T. Obara, H. Fukumoto, K. Hagisawa, Y. Matsushita, A. Okutani, B. Bazartseren, S. Inoue, T. Sata and H. Katano, Jpn. J. Infect. Dis., 2010, 63, 36–40 CAS.
- R. Kubota, P. Labarre, B. H. Weigl, Y. Li, P. Haydock and D. M. Jenkins, Chin. Sci. Bull., 2013, 58, 1162–1168 CrossRef CAS.
- L. Lillis, D. Lehman, M. C. Singhal, J. Cantera, J. Singleton, P. Labarre, A. Toyama, O. Piepenburg, M. Parker, R. Wood, J. Overbaugh and D. S. Boyle, PLoS One, 2014, 9, e108189–e108189 CrossRef PubMed.
- J. Singleton, J. L. Osborn, L. Lillis, K. Hawkins, D. Guelig, W. Price, R. Johns, K. Ebels, D. Boyle, B. Weigl and P. LaBarre, PLoS One, 2014, 9, e113693–e113693 CrossRef PubMed.
- J. Singleton, C. Zentner, J. Buser, P. Yager, P. LaBarre and B. H. Weigl, SPIE MOEMS-MEMS, 2013 Search PubMed.
- J. A. Tomlinson, M. J. Dickinson and N. Boonham, Phytopathology, 2010, 100, 143–149 CrossRef CAS PubMed.
- S. Werres, R. Marwitz, W. Veld, A. De Cock, P. J. M. Bonants, M. De Weerdt, K. Themann, E. Ilieva and R. P. Baayen, Mycol. Res., 2001, 105, 1155–1165 CrossRef CAS.
- S. König, L. Schwenkbier, S. Pollok, M. Riedel, S. Wagner, J. Popp, K. Weber and S. Werres, Plant Pathol., 2015 DOI:10.1111/ppa.12357.
- S. Julich, M. Riedel, M. Kielpinski, M. Urban, R. Kretschmer, S. Wagner, W. Fritzsche, T. Henkel, R. Moeller and S. Werres, Biosens. Bioelectron., 2011, 26, 4070–4075 CrossRef CAS PubMed.
- L. Schena, J. M. Duncan and D. E. L. Cooke, Plant Pathol., 2008, 57, 64–75 CAS.
- L. Schwenkbier, S. Koenig, S. Wagner, S. Pollok, J. Weber, M. Hentschel, J. Popp, S. Werres and K. Weber, Microchim. Acta, 2014, 181, 1669–1679 CrossRef CAS.
- L. X. An, W. Tang, T. A. Ranalli, H. J. Kim, J. Wytiaz and H. M. Kong, J. Biol. Chem., 2005, 280, 28952–28958 CrossRef CAS PubMed.
- R. Gong and S. Li, Int. J. Nanomed., 2014, 9, 3781 CrossRef CAS PubMed.
- J. H. Min, M.-K. Woo, H. Y. Yoon, J. W. Jang, J. H. Wu, C.-S. Lim and Y. K. Kim, Anal. Biochem., 2014, 447, 114–118 CrossRef CAS PubMed.
- M. Schwarz, S. Pahlow, T. Bocklitz, C. Steinbruecker, D. Cialla, K. Weber and J. Popp, Analyst, 2013, 138, 5866–5870 RSC.
- Y. Mori, M. Kitao, N. Tomita and T. Notomi, J. Biochem. Biophys. Methods, 2004, 59, 145–157 CrossRef CAS PubMed.
- Y. Mori, K. Nagamine, N. Tomita and T. Notomi, Biochem. Biophys. Res. Commun., 2001, 289, 150–154 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28 Search PubMed.
- K. Boissinot, A. Huletsky, R. Peytavi, S. Turcotte, V. Veillette, M. Boissinot, F. J. Picard, E. A. Martel and M. G. Bergeron, Clin. Chem., 2007, 53, 2020–2023 CrossRef CAS.
- A. Brinker, H. Schulze, T. Bachmann and R. Moeller, Biosens. Bioelectron., 2010, 26, 898–902 CrossRef CAS PubMed.
- R. Wilson, Nucleic Acid Ther., 2011, 21, 437–440 CrossRef CAS PubMed.
- Z. K. Njiru, PLoS Neglected Trop. Dis., 2012, 6 Search PubMed.
- P. Maier-Laxhuber, R. Schmidt and C. Grupp, Air Ventilated Heating and Cooling Based on Zeolite Technology, DTIC Document, 2002.
- A. Primo and H. Garcia, Chem. Soc. Rev., 2014, 43, 7548–7561 RSC.
- M. Ramos, R. L. Espinoza, M. J. Horn and A. P. F. Leite, ISES Solar World Congress, 2003 Search PubMed.
- S. Ehses, J. Ackermann and J. S. McCaskill, J. Biochem. Biophys. Methods, 2005, 63, 170–186 CrossRef CAS PubMed.
- N. Tomita, Y. Mori, H. Kanda and T. Notomi, Nat. Protoc., 2008, 3, 877–882 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an00855g |
| This journal is © The Royal Society of Chemistry 2015 |