
Comparison of different calibration approaches for chloramphenicol quantification in chicken muscle by ultra-high pressure liquid chromatography tandem mass spectrometry
Xiao-Dong
Pan
*,
Wei
Jiang
and
Ping-Gu
Wu
Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou 310051, China. E-mail: zjupanxiaodong@hotmail.com
First published on 23rd October 2014
Abstract
Matrix-dependent signal suppression often occurs in quantitative analysis by ultra-high pressure liquid chromatography tandem mass spectrometry (UPLC-MS/MS). In this study, we investigated three calibration methods for compensation of signal suppression on chloramphenicol (CAP) quantification in chicken muscle. The data showed that the spiking recoveries by solvent standard calibration with a stable isotope labelled internal standard (SIL-IS) and matrix-matched standard calibration with a SIL-IS were significantly higher than by external matrix-matched standard calibration (P < 0.05). When the SIL-IS was used, standards prepared in the mobile phase solvent showed no significant difference as those prepared in the matrix (P > 0.05). The limit of detection (LOD) for external matrix matched standard calibration was 0.1 μg kg−1, and that for SIL-IS calibration (including matrix matched and solvent dissolved standard) was 0.03 μg kg−1.
Introduction
Chloramphenicol (CAP) with excellent antibacterial and pharmacokinetic properties has historical veterinary use in all major food-producing animals. Because of toxic side effects on the haemopoietic system and the emergence of drug resistant bacteria,1 their clinical applications are strictly controlled in many countries including China, USA and member states of European Union (EU).2–4 For example, CAP has been banned for use in food-producing animals in EU, and the minimum required performance limit (MRPL) or maximum residue limits (MRLs) have also been set for CAP (0.3 μg kg−1). Therefore, it is of great importance to develop sensitive and accurate methods for quantitative analysis of CAP in animal tissues.Much research in recent years has focused on antibiotic quantification by liquid chromatography tandem mass spectrometry (LC-MS/MS).5–7 LC-MS/MS has been found to be suitable for the analysis of food contaminants, because it provides a large amount of information on complicated mixtures.8,9 It also has higher selectivity and sensitivity than liquid chromatography. However, high selectivity of LC-MS/MS does not guarantee the effective elimination of interferences from residual matrix-dependant components. Quantitative analysis with electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) can be substantially affected by a matrix effect, which usually occurs with ion suppression.10 The matrix effect is variable and can be difficult to control or predict.
Some measures have been suggested to minimize the interferences of co-eluting matrix compounds.9,11–14 The common strategies are by sample preparation, chromatographic conditions and calibration methods.10 When it involves in the quantification, appropriate calibration techniques, for example matrix-matched standard and internal standard, are usually selected to compensate signal alterations.15 However, the studies on appropriate calibration techniques to compensate for signal interferences are seldom reported.11–13 Because of the complicated matrix components, it is reasonable to hypothesize that different calibration methods may result in variable performance in terms of selectivity, repeatability or accuracy of the analysis.
The aim of this study is to compare three calibration approaches for the compensation of signal suppression on chloramphenicol (CAP) quantification in chicken muscle by ultra-high pressure liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Three calibration methods are: (1) solvent standard calibration with a stable isotope labelled internal standard (SIL-IS); (2) external matrix-matched standard calibration; (3) matrix-matched standard calibration with a SIL-IS. Two extraction methods, liquid–liquid extraction (LLE) and solid-phase extraction (SPE), were adopted for the sample preparation and matrix effect investigation.
Materials and methods
Materials and reagents
Chloramphenicol (CAP, 99.0%) standard was brought from Sigma (St. Louis, MO, USA). Deuterated chloramphenicol-d5 (d5-CAP, 98.0%) as the internal standard was obtained from Dr Ehrenstorfer GmbH (Augsburg, German). Methanol, acetonitrile, ethyl acetate and hexane of HPLC grade were from DIMA Technology Inc. (Richmond, USA). The OASIS® HLB SPE cartridges (3 mL 60 mg) were from Waters (Milford, MA, USA). Water was purified by the Milli-Q reverse osmosis system (Millipore, Milford, MA, USA). Stock solutions at a concentration of 100 μg mL−1 were prepared by dissolving CAP in methanol. Working standard solutions at concentrations of 15, 50 and 250 μg L−1 for CAP, and a d5-CAP internal standard solution of 200 μg L−1 were prepared by dissolving the ampoule with 100 μg mL−1 in methanol. The stock solutions were stored at −20 °C and stable for 6 months. The standard solutions were stable for 1 month at 4 °C.Samples
The samples of chicken muscle were purchased from local supermarkets (TESCO, Hangzhou, China) and stored at −20 °C until analysis. The blank samples without residual of CAP were previously screened using an enzyme-linked immunosorbent assay (ELISA) kit (Beijing Wanger Biotechnology Co. Ltd., Beijing, China) and stored at −20 °C until analysis.UPLC-MS/MS system
A Waters Acquity UPLC instrument (Waters Corp., Milford, MA, USA) was used in the present study. Separation was carried out on an Acquity BEH C18 column (50 mm × 2.1 mm, 1.7 μm) maintained at 40 °C. The injection volume was 10 μL and the analysis was carried out with gradient elution using (A) methanol and (B) water as the mobile phase at a flow rate of 0.4 mL min−1. Chromatographic gradient conditions for the separation of CAP are shown in Table 1.| Time (min) | Flow (mL min−1) | (A) Methanol (%) | (B) Water (%) | Curve |
|---|---|---|---|---|
| 0 | 0.4 | 10 | 90 | — |
| 3.0 | 0.4 | 90 | 10 | 6 |
| 4.0 | 0.4 | 90 | 10 | 6 |
| 4.1 | 0.4 | 10 | 90 | 1 |
| 5.0 | 0.4 | 10 | 90 | 6 |
The Quattro LC triple-quadrupole mass spectrometer (Waters Corp., Milford, MA, USA) was connected to the LC system via an electrospray ionization (ESI) interface. The analysis of CAP was performed in negative ionization mode. The ESI source was operated with the capillary voltage set at 3.2 kV. The extractor and RF lens voltage was 40 V and 0.7 V, respectively. The temperature of the source and desolvation was set at 120 °C and 350 °C, respectively. The nitrogen flows were adjusted to 30 L per hour for the cone gas and 446 L per hour for the desolvation gas. For collision induced dissociation (CID), argon was used as the collision gas with a collision cell pressure of 2 × 10−3 mbar. For quantitative purposes, samples were analyzed by the mode of multiple reaction monitoring (MRM). The MRM parameters for the optimal yield of product ions were defined in individual time windows for each analyte and the internal standard as they eluted from the LC column (Table 2). All data were acquired by using MassLynx (Version 4.1) software.
| Analyte | Retention time (min) | Parent (m/z) | Daughter (m/z) | Cone voltage (V) | Collision energy (V) |
|---|---|---|---|---|---|
| CAP | 2.47 | 320.9 | 152 | 35 | 26 |
| 257 | 35 | 16 | |||
| d5-CAP | 2.46 | 326 | 157 | 35 | 26 |
Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v). The elution was evaporated to dryness under a nitrogen stream at 40 °C. The residue was reconstituted by 2 mL of 10% methanol solution and poured through a 0.20 μm filter.
:10, v/v). The elution was evaporated to dryness under a nitrogen stream at 40 °C. The residue was reconstituted by 2 mL of 10% methanol solution and poured through a 0.20 μm filter.
Matrix effect
To assay the matrix effect, CAP is spiked into the matrix before extraction, into the sample extract after extraction (matrix-matched standard) and into neat solution with a final level of 10 μg kg−1. As described by Matuszewski et al.,16 the ‘absolute’ matrix effect (ME) was calculated using the formula: ME (%) = X/Y × 100, where X is the peak area for analytes in solvent and Y is the peak area of standards spiked into the matched matrix after extraction. The recovery (RE) can be calculated using: RE (%) = Z/Y × 100, where Z is the peak area of standards spiked before the extraction. The overall process efficiency (PE) can then be calculated by: PE = Z/X × 100= (ME × RE)/100.Calibration
Three calibration methods used as described by Kang, Hick, & Price17 were: (1) solvent standard calibration with a SIL-IS; (2) external matrix-matched standard calibration; (3) matrix-matched standard calibration with a SIL-IS.The curves were all prepared by spiking CAP at the concentrations of 0.5, 1, 2, 5, 10, 20 and 50 μg L−1, and for IS of d5-CAP at 10 μg L−1. Calibration curves with internal standard were obtained by the ratio of the CAP area (m/z 152) to the CAP-d5 area (m/z 157). Calibration data of CAP are shown in Table 3. Peak area ratios of the analyte to the internal standard were computed by using MassLynx 4.1 software.
| Calibration | Matrix | Linear equation | Correlation coefficient (r) |
|---|---|---|---|
| a 1, Solvent standard calibration with a SIL-IS; 2, External matrix-matched standard calibration; 3, Matrix-matched standard calibration with a SIL-IS. | |||
| 1 | Mobile phase solvent | y = 0.2425x − 0.0648 | 0.9998 |
| 2 | LLE | y = 32.793x + 27.109 | 0.9995 |
| SPE | y = 37.521x + 26.957 | 0.9996 | |
| 3 | LLE | y = 0.2511x − 0.1327 | 0.9997 |
| SPE | y = 0.2232x − 0.0968 | 0.9996 | |
Results and discussion
Assessment of the matrix effect
Matrix effects, originally discussed by Kebarle and Tang18 in the early 1990s, can be described as the difference between the mass spectrometric response for an analyte in standard solution and the response for the same analyte in a biological matrix. Matrix constituents may alter the ionization efficiency of the target analyte thereby compromising the efficiency and reproducibility of the quantitative method. As a consequence this will affect the precision and sensitivity of the LC-MS method.19 In this study, we observed the ion suppression of CAP (m/z, 320.9 > 152) in the matrix of liquid–liquid extraction where the area of the CAP response in a matrix was decreased comparing with the standard in solvent (Fig. 1). The area of the CAP response in matrix spiked post-extraction was larger than spiked before extraction.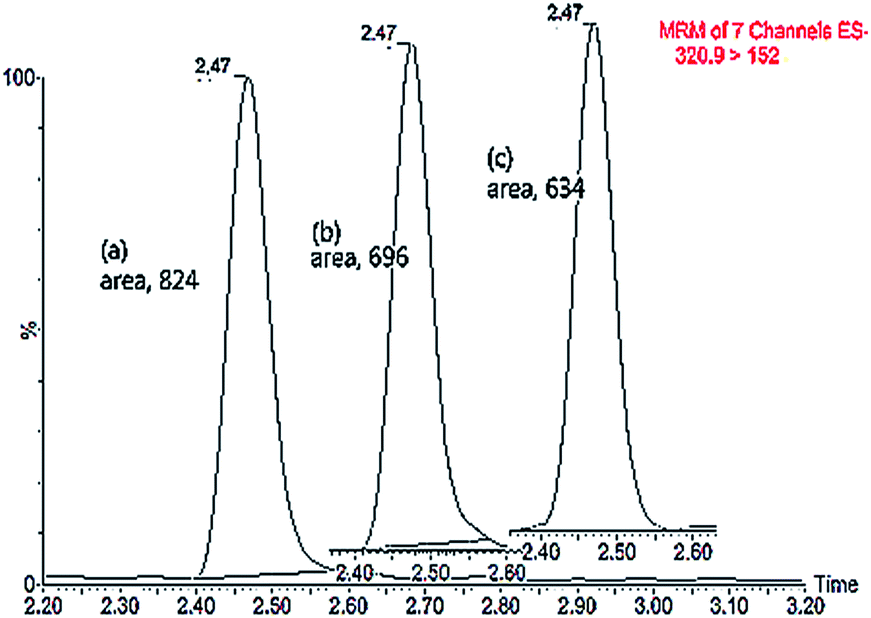 | ||
| Fig. 1 Chromatographs after liquid–liquid extraction showing differences in response between the standard in solvent (a), the standard in matrix spiked post-extraction (b), and the standard in matrix spiked before extraction (c). | ||
For minimizing the matrix effect, we adopted two extraction methods, solid phase extraction (SPE) and liquid–liquid extraction (LLE). SPE and LLE are the most commonly used techniques to circumvent the matrix effect.11 As shown in Table 4, we observed the matrix effect with ion suppression both in LLE and SPE (where ME > 1). The averages of ME (110.6%) and RE (86.0%) in SPE were all less than those in LLE (ME, 118.4%; RE, 90.1%). However, there was no statistically difference between the two extraction methods in terms of ME and RE (P > 0.05). Both two extraction methods cannot completely eliminate the matrix effect for CAP quantification in chicken muscle. Furthermore, average PEs of LLE and SPE were 76.9% and 77.8%, respectively. PE represents the combination between the ME and the recovery of the sample extraction method. Low PE can be deleterious for method precision and sensitivity.
| Extraction | ME (%) | RE (%) | PE (%) |
|---|---|---|---|
| a LLE, liquid–liquid extraction; SPE, solid phase extraction; mean ± SD, n = 6. | |||
| LLE | 118.4 ± 4.1 | 90.1 ± 3.5 | 76.9 ± 4.9 |
| SPE | 110.6 ± 5.3 | 86.0 ± 4.2 | 77.8 ± 3.8 |
Comparison of different calibration methods
Besides the selection of sample preparation, calibration is another strategy to overcome the matrix effect. The standard-addition method probably represents the most effective way to compensate adverse influence of the matrix on method performances.20 However, this approach is laborious and time-consuming because the spiking step must be prepared for each sample. Internal standard calibration is the most widely used method to compensate for matrix effects. The external matrix-matched standard is also proposed in the literature.17 As shown in Fig. 2, both for LLE and SPE extraction, the spiking recoveries by mobile phase solvent standard calibration with a SIL-IS and matrix-matched standard calibration with a SIL-IS were significantly higher than by external matrix-matched standard calibration (P < 0.05). In terms of the reduction of the matrix effect, the usage of internal standard calibration is better than matrix matched calibration.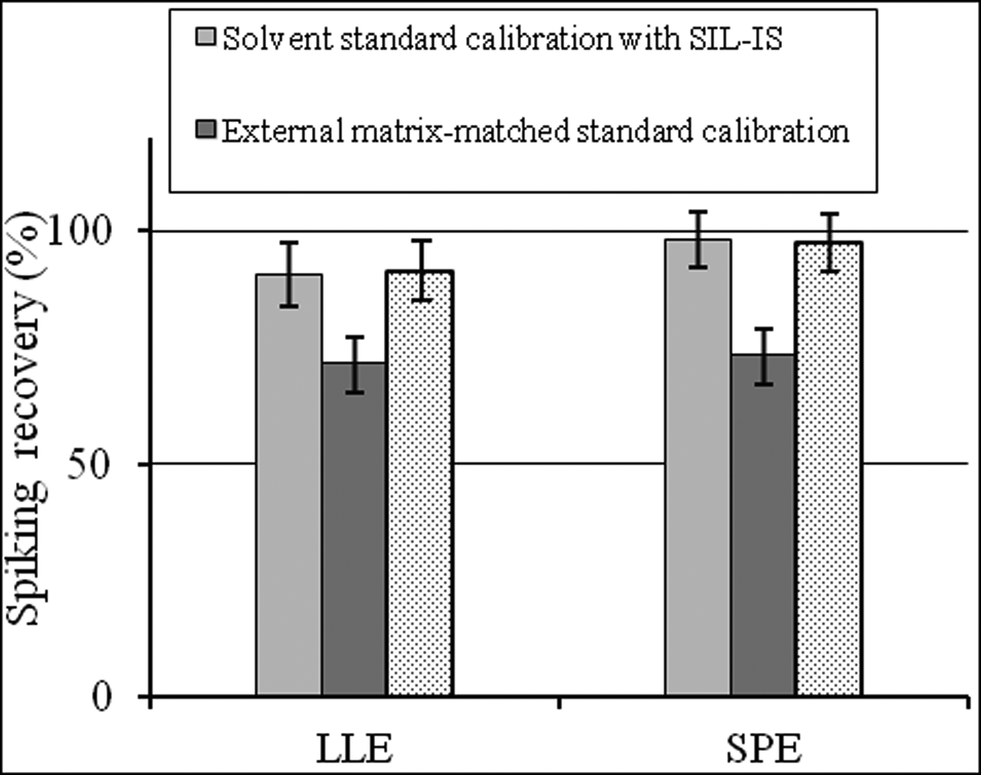 | ||
| Fig. 2 The spiking recovery by the three standard calibration methods to the analysis of CAP in chicken muscle (mean ± SD, n = 6). | ||
Noticeably, there was no significant difference between solvent standard calibration with a SIL-IS and matrix-matched standard calibration with a SIL-IS (P > 0.05). It indicates that once the appropriate internal standard is adopted, there is no difference in the results for standards prepared in the matrix and mobile phase solvent. Hewavitharana21 also pointed out that mass spectrometry presents accurate quantitation without the need for matrix matching, as long as the internal standard co-elutes with the analyte of interest. If the analyte and internal standard co-elute, the slope of the calibration curve analyte response/internal standard vs. analyte concentration is independent of the matrix composition.
Application to real samples
Sixty-four samples of chicken were analysed for CAP by LLE. Three calibration methods were all used for quantification. The limits of detection (LODs), defined as the spiking level in chicken muscle that produce a signal-to-noise ratio of 3:1 were 0.1 μg kg−1 for external matrix matched standard calibration, and 0.03 μg kg−1 for SIL-IS calibration (including matrix matched and solvent dissolved standard).
One of 64 samples was detected for containing CAP more than 0.03 μg kg−1. The calibrated values were 1.06 ± 0.03 μg kg−1 for external matrix matched calibration, 1.67 ± 0.04 μg kg−1 for solvent dissolved SIL-IS calibration, and 1.65 ± 0.05 μg kg−1 for matrix matched SIL-IS calibration (n = 6).
Conclusion
Sample preparation, such as LLE and SPE, is the common method to minimize interferences of the matrix. If the matrix effect cannot be reduced by that, then an appropriate calibration technique should be used to compensate for signal alterations. In this study, LLE and SPE cannot completely overcome the ion suppression. Three calibration methods were selected for the compensation of the matrix effect. The usage of SIL-IS calibration can more significantly reduce the matrix effect than external matrix matched standard calibration. And no statistical difference was observed between solvent standard calibration with a SIL-IS and matrix-matched standard calibration with a SIL-IS.Acknowledgements
This research work was financially supported by Health Bureau of Zhejiang Province (2013RCA008), and the National High Technology Research and Development Program of China (863 Program, no. 2012AA101603), the program For Zhejiang Leading Team of Science and Technology Innovation (2011R50021)Notes and references
- S. Zhang, Z. Liu, X. Guo, L. Cheng, Z. Wang and J. Shen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 875, 339–404 Search PubMed.
- R. L. Epstein, C. Henry, K. P. Holland and J. Dreas, J. AOAC Int., 1994, 77, 570–576 CAS.
- 235th Bulletin, Agricultural Department of the People's Republic of China, 2002.
- Council Regulation (EEC) 37/2010 of 22 December 2009, Off. J. Eur. Commun., 2010, vol. L15, pp. 1–76 Search PubMed.
- E. Alechaga, E. Moyano and M. T. Galceran, Analyst, 2012, 137, 2486–2494 RSC.
- V. Matamoros, D. Calderón-Preciado, C. Domínguez and J. M. Bayona, Anal. Chim. Acta, 2012, 722, 8–20 CrossRef CAS PubMed.
- X. Q. Li, Z. Yang, Q. H. Zhang and H. M. Li, Anal. Chim. Acta, 2014, 807, 75–83 CrossRef CAS PubMed.
- R. P. Lopes, É. E. de Freitas Passos, J. F. de Alkimim Filho, E. A. Vargas, D. V. Augusti and R. Augusti, Food Control, 2012, 28, 192–198 CrossRef CAS PubMed.
- H. Trufelli, P. Palma, G. Famiglini and A. Cappiello, Mass Spectrom. Rev., 2011, 30, 491–509 CrossRef CAS PubMed.
- C. Ghosh, C. P. Shinde and B. S. Chakraborty, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 893–894, 193–200 CrossRef CAS PubMed.
- M. Fiori, C. Civitareale, S. Mirante, E. Magaro and G. Brambilla, Anal. Chim. Acta, 2005, 529, 207–210 CrossRef CAS PubMed.
- F. Moragues and C. Igualada, Anal. Chim. Acta, 2009, 637, 193–195 CrossRef CAS PubMed.
- D. L. Buhrman, P. I. Price and P. J. Rudewicz, J. Am. Soc. Mass Spectrom., 1996, 7, 1099–1105 CrossRef CAS.
- F. Gosetti, E. Mazzucco, D. Zampieri and M. C. Gennaro, J. Chromatogr. A, 2010, 1217, 3929–3937 CrossRef CAS PubMed.
- A. Furey, M. Moriarty, V. Bane, B. Kinsella and M. Lehane, Talanta, 2013, 115, 104–122 CrossRef CAS PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS.
- J. Kang, L. A. Hick and W. E. Price, Rapid Commun. Mass Spectrom., 2007, 21, 4065–4072 CrossRef CAS PubMed.
- P. Kebarle and L. Tang, Anal. Chem., 1993, 65, 972–986 Search PubMed.
- H. Li, H. Sun, J. Zhang and K. Pang, Food Control, 2013, 31, 359–365 CrossRef CAS PubMed.
- M. Stuber and T. Reemtsma, Anal. Bioanal. Chem., 2004, 378, 910–916 CrossRef PubMed.
- A. K. Hewavitharana, J. Chromatogr. A, 2011, 1218, 359–361 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |