
Simultaneous determination of iodide and creatinine in human urine by flow analysis with an on-line sample treatment column†‡
Jirayu
Sitanurak
ab,
Prawpan
Inpota
ab,
Thitirat
Mantim
ab,
Nuanlaor
Ratanawimarnwong
ac,
Prapin
Wilairat
ad and
Duangjai
Nacapricha
*ab
aFlow Innovation-Research for Science and Technology Laboratories (Firstlabs), Thailand. E-mail: duangjai.nac@mahidol.ac.th; dnacapricha@gmail.com; Fax: +66 2 201 5127; Tel: +66 2 201 5127
bDepartment of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Mahidol University, Bangkok 10400, Thailand
cDepartment of Chemistry, Faculty of Science, Srinakharinwirot University, Sukhumwit 23, Bangkok 10110, Thailand
dNational Doping Control Centre, Mahidol University, Rama VI Rd, Bangkok 10400, Thailand
First published on 13th October 2014
Abstract
This work presents the first flow system for direct analysis of iodide and creatinine suitable for screening of human urine samples. The system had a mini-column packed with strong anion exchange resin for on-line extraction of iodide. After injection of a sample on the column the unretained urine sample was analyzed for creatinine in one section of the flow system using the Jaffe's reaction with spectrometric detection at 520 nm. Iodide was eluted off with 1.42 mL 5 M NaNO3. A 150 μL fraction of the eluate was analyzed in another section of the same flow system for iodide using the kinetic–spectrometric Sandell–Kolthoff reaction. At the optimum condition, the sample throughput was 12 samples per h. The linear working range covered the normal levels of iodide and creatinine in human urine: 0–200 μg I L−1 and 50–1200 mg creatinine L−1, respectively. Recoveries tested in 10 samples were 87–104% for iodide and 89–104% for creatinine. Bland–Altman plots (n = 50) showed that the scatter of the differences between values obtained by this method and those of reference methods, for both iodide and creatinine, was within mean ± 2SD.
1. Introduction
Iodine Deficiency Disorder (IDD) is a term for various adverse health effects resulting from inadequate iodine intake leading to insufficient production of thyroid hormones.1 The consequence of iodine deficiency during pregnancy is well known for causing mental retardation in children.2 Iodine deficiency can occur at any age and it is estimated that more than 2 billion individuals worldwide have an insufficient intake of iodine.3 In order to ensure sufficient iodine intake many countries have implemented salt iodization and iodine supplementation.4 However, these strategies need efficient monitoring to avoid excessive intake of iodine as well as evaluation of the current IDD status of the population.Approximately 90% of the amount of ingested iodine is eventually excreted via urine.5 Therefore urinary iodine (UI) has been commonly used to monitor the intake of iodine and IDD status of the population. Since the iodide ion is the most predominant form of iodine in urine,6,7 quantitation of iodide provides information on total iodine excreted in urine. The measured level of iodide is used for assessing the ‘iodine intake’ and ‘IDD status’ according to WHO/UNICEF/ICCIDD guidelines. For school-aged children, the epidemiological criteria for iodine status are: <20 μg I L−1 (insufficient intake/severe IDD); 20–49 μg I L−1 (insufficient intake/moderate IDD); 50–99 μg I L−1 (insufficient intake/mild IDD); 100–199 μg I L−1 (adequate intake/no IDD); 200–299 μg I L−1 (more than adequate intake/risk of hyperthyroidism) and >300 μg I L−1 (excessive intake/risk of adverse health consequences).1,8
The most common method for determination of low levels of iodide in urine has been the kinetics-based Sandell–Kolthoff method.1 However there have also been other methods presented in the literature for measurement of iodide in human urine, such as electrospray ionization tandem mass spectrometry,9 ion-pairing reversed-phase high performance liquid chromatography with amperometric detection,10 single-drop microextraction coupled with gas chromatography11 and fluorescence enhancement, by iodide, of complex formation between carbon dots and mercury ion.12 Other methods have been recently reviewed by Shelor and Dasgupta.13
The classical kinetic method based on the Sandell–Kolthoff reaction14 is still a method of choice due to its high sensitivity and cost-effectiveness. The method utilizes the catalytic activity of iodide in the redox reaction of Ce(IV) and As(III) in acidic medium and requires only a common spectrometer. Analysis can be carried out in various formats, including batch,15,16 flow-based17–20 and microtitre plate.21–24 All these methods require sample treatment, such as ashing,15 digestion with chloric acid16,19–21,23 or persulfate17,18,21,22,24 to destroy organic interferences prior to the kinetic measurement. This step is labor intensive, especially when handling large number of samples. A previous method based on on-line dialysis was subject to positive error.25,26 Since then there have been various flow methods for on-line total digestion with subsequent spectrometric detection, such as UV-persulfate digestion27 or acid digestion with KMnO4–K2Cr2O7–H2SO4.28
We present a new flow method with an on-line anionic exchange treatment for determination of iodide in urine. The strong anionic exchange resin (SAX) was packed in a glass column incorporated into the flow system to extract iodide from other urine matrices. Iodide was then eluted with sodium nitrate for analysis in a section of the flow system, where the Sandell–Kolthoff reaction was employed. We also incorporated another flow section for simultaneous measurement of creatinine in the unretained urine sample from the SAX column. This employed the Jaffe's reaction.29 The system provided not only the iodide level but also the iodide/creatinine ratio (UI/UCr). This ratio of a spot urine sample is suitable for large scale screening of the iodine status of individuals, as compared to urine samples collected over a 24 h period.30
2. Experimental
2.1 Reagents and samples
All chemicals used in this work were of analytical reagent grade. Deionized distilled water, obtained from the Branstead EASYpure II (USA) unit, was employed for preparation of all aqueous solutions.Stock solution of iodide (1000 mg I L−1) was prepared by dissolving 0.1303 g of KI powder (Merck, Germany) in 100.0 mL of water. Creatinine stock solution (2000 mg creatinine L−1) was prepared by dissolving an accurate weight of approximately 0.2 g of anhydrous creatinine (Sigma, USA) in 100.0 mL of water. Working standard solutions for calibration were solutions of iodide and creatinine, serially diluted from the stock solutions.
Preparation of the reagents, used in the determination of iodide by the Sandell and Kolthoff reaction, was carried out similar to that reported in a previous work.19 The arsenious solution was prepared by first dissolving 10.0 g of As2O3 (Ajax, New Zealand) and 47.0 g of NaCl (Merck, Germany) in 500 mL of water. The mixture was heated until a clear solution was obtained. After cooling to room temperature, 27.8 mL of concentrated H2SO4 was added to the solution which was then made up with water to 1 L in a volumetric flask to give 0.10 M arsenious acid solution. The cerium(IV) solution (0.008 M) was prepared by dissolving 5.0 g of (NH4)4Ce(SO4)4·2H2O (Merck, Germany) in a 1 L volumetric flask with 1.75 M H2SO4. The solution was allowed to stand overnight before use.
The alkaline picrate solution (0.03 M), used for the Jaffe's method, was freshly prepared by dissolving approximately 1.7 g of picric acid powder (Merck, Germany) in 250 mL of 0.4 M NaOH. The 5 M NaNO3 eluting solution was prepared by dissolving 215 g of NaNO3 (Ajax, New Zealand) in 500 mL of water. This solution was filtered through a 0.45 μm cellulose acetate membrane (Sartorius, Germany) before use.
For the validation study, 50 samples of spot urine from healthy volunteers were used. The urine samples were stored at 4 °C until analyzed. Prior to analysis, 5.00 mL of the urine sample was diluted five-fold with water.
2.2 The flow manifold
The optimized flow system for simultaneous determination of iodide and creatinine is shown in Fig. 1. PTFE tubing (1.0 mm i.d., VICI, Switzerland) was employed in the assembly of the flow system. An autosampler (Perkin Elmer AS90, USA) was used for sample loading. The flow system consisted of three peristaltic pumps, P1, P2 and P3 (Ismatec/ISM827, Switzerland) and two 6-port injection valves (Upchurch, USA), IV1 and IV2, for loading and injection of the sample and the eluted zone of iodide, respectively. A 3-way switching valve, SV1 (Upchurch, USA), was used for selecting the flow of the sample or water into the system. A 4-way switching valve, SV2 (Upchurch, USA), was utilized for switching between the flow of the sample/water and the eluent, E, into the sample treatment column (SAX in Fig. 1). In ‘section b’ of the flow manifold, a relatively larger size tubing (3.17 mm i.d., 8 cm long), denoted ‘dilution tube’, was placed before the mixing coil MC to increase dilution of the urine sample in order to give a suitable signal amplitude in the analysis of creatinine. | ||
| Fig. 1 A schematic drawing of the optimized flow system with an on-line SAX column for simultaneous and direct analysis of urinary iodide and creatinine. The system is composed of three sections: ‘section a’ for sample treatment, ‘section b’ for creatinine analysis and ‘section c’ for iodide anlaysis. R1: 0.03 M alkaline pricrate; R2: 0.1 M As2O3 with 0.8 M NaCl in 0.5 M H2SO4; R3: 0.008 M (NH4)4Ce(SO4)4 in 1.75 M H2SO4; E: 5 M NaNO3; P1, P2 and P3: peristaltic pump 1, 2 and 3; IV1, IV2: injection valve 1 and 2; L1, L2: loop 1 (50 μL) and loop 2 (150 μL); SV1, SV2: switching valve 1 and 2; RC1, RC2: reaction coil 1 and 2 (1.0 mm i.d., 200 cm long); MC: mixing coil (1.0 mm i.d., 50 cm long); dilution tube: 3.17 mm i.d., 8 cm long; SAX minicolumn: glass tube (2.2 mm i.d., 25 mm long) packed with 30 mg of strong anion exchange resin; WB: water bath; D1 and D2: LED detector 1 (520 nm) and 2 (420 nm). | ||
As shown in Fig. 1, a water bath, WB (Fisher Scientific/Isotemp 205, UK), was used for maintaining the temperatures of reaction coils, RC1 and RC2, at 40 °C. A photometer, D1 (Bangkok High Lab Co. Ltd, Thailand), equipped with a SMB420/525/640-3100-I LED (Epitex, Japan) as the light source and a 10 mm flow-through cell (Helma, Germany), was used in the creatinine analysis (section b, Fig. 1). In the iodide determination, a second photometer D2 (Bangkok High Lab Co., Ltd, Thailand), equipped with a SMBD520-1100 LED (Epitex, Japan) as the light source and a 10 mm flow-through cell (Helma, Germany), was used for monitoring the absorbance of Ce(IV) (section c, Fig. 1). An in-house software written with Lab VIEW 8.2™ was used for recording the output from the photometers.
2.3 SAX column
The flow system also comprised a glass column (2.2 mm i.d., 25 mm long), packed with strong anion exchange resin (75–150 μm SAX, Alltech, USA), which had a maximum capacity of 900 μmol anion equivalent/600 mg resin. Slurry of the SAX-resin, made by mixing 30 mg of the resin with water, was loaded into the glass column which was stoppered at each end with cotton wool. Before use, the resin was conditioned by flowing a solution of 5 M NaNO3 for 15 min, followed by washing with water for 5 min.2.4 Flow procedure
The optimized operating sequence is shown in Fig. 2. The system operated under continuous-flow mode throughout the analysis. The steps in Fig. 2 describe the operations of the four electronic components SV1, SV2, IV1 and IV2. Fig. 2 shows the steps for triplicate analysis of a sample together with the signal profiles for iodide and creatinine. According to the procedure in Fig. 2, a 5.0 mL urine sample (after 5-fold dilution) or a standard mixture was selected by switching valves SV1 and SV2 to flow into the SAX column. Iodide ions were retained by the quaternary amine groups of the resin. Unretained components in the urine, including creatinine, flowed through the loop L2 (IV2 at the ‘load’ position) and loop L1 (IV1 at the ‘load’ position) to waste line W2. In the middle of the period of trapping of iodide by the resin, the urine in loop L1 was injected (IV1 is at the ‘inject’ position) into ‘section b’ (Fig. 1) for measurement of creatinine. The urine sample from L1 was mixed with a stream of water at the first confluence point before entering the dilution tube and then into the mixing coil MC. This diluted plug of urine then merged with the stream of the picrate reagent R1, being mixed inside the reaction coil RC1, before flowing into the detector, D1. The signal profile for the first measurement of creatinine is shown in Fig. 2 with a signal height C1. At the start of detection of creatinine (t = 2.5 min) injection valve IV1 was set back to the ‘load’ position and switching valve SV1 was switched, allowing water to rinse the SAX column for 30 seconds. The next procedure was the elution of iodide from the SAX column into sample loop L2. For this step, SV2 was switched to allow 5 M NaNO3 eluent (E in Fig. 1) to flow into the SAX column at a flow rate of 1 mL min−1 for 1.42 min. The eluate, containing iodide, flowed into the sample loop L2. At 3.42 min, the valve IV2 was switched to the ‘inject’ position to inject the eluate in loop L2 into ‘section c’ for analysis of iodide by the Sandell–Kolthoff reaction. This sample zone merged and mixed with arsenious acid R2 and ceric reagent R3, respectively. The reaction zone passed through the heated reaction coil RC2 for acceleration of the kinetics before flowing into the detector D2. In the Sandell–Kolthoff reaction the iodide ion catalyzes the reduction of Ce(IV) and thus there is a decrease in absorbance when the sample zone is monitored at D2. Thus for an injection of a sample there would be 2 flow profiles with the peaks for creatinine and iodide appearing almost simultaneously (see Fig. 2). Fig. 2 shows the flow profiles for triple injections of a sample. | ||
| Fig. 2 A graphical representation of the timing sequence of the valves and the corresponding absorbance signals for three replicate injections of a sample for the developed flow system (refer to Fig. 1). Section (a) gives the steps for the sample treatment, section (b) for the creatinine analysis and section (c) for the iodide analysis. At the start of the first injection, the switching valves SV1 and SV2 are set to allow the sample to flow through the SAX column to trap the iodide ion. Injection valves IV1 and IV2 are in the “load” positions. Unretained urine flows through sample loops L2 and L1, respectively, to waste line W2. At 1.0 min injection valve IV1 is set to the “inject” position to introduce the urine in the sample loop L1 into section (b) of the system for analysis of creatinine by reacting with a flow of picric acid (R1). The profile of the absorbance of the product of the Jaffe's reaction is shown as the signal (D1). At 2.5 min, the time at which creatinine begins to be observed, switching valve SV1 is set to allow H2O to flow through the SAX column for 0.5 min. Also at 2.5 min injection valve IV1 is set back to the load position. Then at 3.0 min the switching valve SV2 is set to allow the eluent E (5 M NaNO3) to elute the trapped iodide off the SAX column for 1.42 min and then set back. The eluted zone flows into the loop L2. At 3.42 min the injection valve IV2 is set to the “inject” position to introduce the iodide eluate in the sample loop L2 into section (c) of the system for analysis of the iodide by the Sandell–Kolthoff kinetic method. The resulting absorbance signal as recorded at detector D2 is shown as Signal (D2). At 4.42 min injection valve IV2 is set back to the “load” position. At 5.0 min switching valve SV1 is set to allow the sample solution to flow into the SAX column, thus starting the second cycle of the analysis. | ||
3. Results and discussion
3.1 System design and operation
 | ||
| Fig. 3 Comparison of the two flow configurations for sample pretreatment (section a) and iodide analysis (section c). The recording of the absorbances at detector D2 are for 3 replicate injections of blank sample and a standard solution of 20 μg I L−1. (a) Configuration I: the SAX column is fitted to the injection valve IV2, replacing the sample loop. (b) Configuration II: a second selection valve SV2 is added in section a and the SAX column placed between SV2 and injection valve IV2, which now has a sample loop L2 installed. This is the configuration selected for the flow system employed for analysis of urine samples. Using Configuration II the solution in sample loop L2, when injected into section c (for iodide analysis) always has 5 M NaNO3 (eluent E) as the solvent. Thus the blank signal is due to the effect of 5 M NaNO3 on the kinetics of the Sandell–Kolthoff reaction (see Section 3.4). | ||
| Step operation | Configuration I | Configuration II | ||
|---|---|---|---|---|
| Device connection | Duration | Device connection | Duration | |
| 1. SAX loading with urine | SV1(std)-IV2(SAX)-W2 | 2 min and 30 s | SV1(std)-SV2(SAX)-IV2-W2 | 2 min and 30 s |
| 2. SAX cleanup using water | SV1(H2O)-IV2(SAX)-W2 | 30 s | SV1(H2O)-SV2(SAX)-IV2-W2 | 30 s |
| 3. SAX elution using 5 M NaNO3 and iodide analysis in ‘section c’ | ‘Section c’ (E)-IV2(SAX)-W3 | 3 min and 15 s | (3.1) ‘Section a’ (E)-SV2(SAX)-IV2-‘section c’(H2O) | 25 s |
| (3.2) ‘Section a’ (E)-SV2(SAX)-IV2-W2 | 1 min | |||
| 4. SAX washing with water | SV1(H2O)-IV2(SAX)-W2 | 30 s | SV1(H2O)-SV2(SAX)-IV2-W2 | 30 s |
| Total operation time/cycle | 6 min and 45 s | 4 min and 55 s | ||
The design of ‘configuration I’ used ‘section a’ for introducing urine into the SAX column to retain iodide (step 1 in Table 1), followed by rinsing with water (step 2 in Table 1). Iodide was eluted from the column with 5 M NaNO3 (carrier E) by the six-port injection valve IV2 (step 3 in Table 1). The eluate directly merged and mixed with the reagents R2 and R3 in ‘section c’. It was observed that using the configuration there was not only the signal of iodide (i.e. I1, I2 or I3), but also a large positive peak in front of the negative signal for iodide. This positive peak was also obtained for a blank sample. Water from the rinsing step that remained in the SAX column and in the connecting tubes was the cause of the positive peak. Since the carrier solution in ‘section c’ is 5 M NaNO3 this water plug reduced the ionic strength of the carrier solution leading to a lower reaction rate and an increase in the background absorbance (vide infra). Thus ‘configuration I’ was not selected.
In ‘configuration II’ (Fig. 3b) a switching valve SV2 was installed in ‘section a’ and the SAX column connected between this valve and the six-port injection valve IV2. A sample loop L2 (150 μL) was installed on the injection valve IV2. The carrier line of ‘section c’ was changed from NaNO3 (eluent E) to water (Fig. 3b). The NaNO3 line now formed a part of ‘section a’ and its flow line was connected to the valve SV2. The elution of iodide from SAX now constituted ‘section a’.
‘Configuration II’ (Fig. 3b) was operated according to steps 1 to 4 of Table 1. SV1 was used in conjunction with SV2 to flow urine, water or eluent (E) through the SAX column. In step 3.1 of Table 1, the volume of the total eluate zone that was in the sample loop L2 was delivered to ‘section c’ on switching injection valve IV2. The rest of the eluate was vented to waste line W2 in step 3.2 of Table 1. The signal from the catalytic effect of iodide was now observed (Fig. 3b). The signal of the blank sample resulted from the increase of the uncatalyzed reaction rate due to the NaNO3 in the eluent (E). This was shown by a separate experiment using only ‘section c’. The detector D2 showed a baseline absorbance of 0.9 au, when water was used as the carrier. The baseline absorbance decreased to about 0.45 au when 5 M NaNO3 was used as the carrier. This showed that increase in ionic strength led to an increase in the rate of the Sandell–Kolthoff reaction.
In the final system the waste line W2 of ‘section c’ was connected to the six-port injection valve IV1, which had a 50 μL sample loop L1 (Fig. 1). Urine that passed through the resin column flowed into this sample loop L1 (50 μL). The urine in L1 was then injected into the carrier stream of water for further dilution and reaction with alkaline picrate (R1) in ‘section b’.
3.2 Selection of the iodide eluate zone for kinetic analysis in ‘section c’
In the final manifold in Fig. 1, only the NaNO3 eluate (containing iodide ions) in the 150 μL sample loop of injection valve IV2 was sent to ‘section c’ for kinetic analysis. We therefore need to find the suitable time at which to inject the 150 μL eluate so that the eluate in the sample loop contained the highest concentration of iodide. Fig. 4 shows the signals for a blank (water) sample and iodide standard (100 μg I L−1) in the NaNO3 eluate at various injection times. The signals for the blank sample increased, reaching a constant value for injection times >30 s. As discussed previously, NaNO3 increases the kinetics of the uncatalyzed reaction due to the increase in ionic strength. The head zone of the eluate will be diluted by the water remaining in the column, but the later eluting solution (eluting after 30 s) will be 5 M NaNO3. The signals for the iodide sample also steadily increased with injection times, but reaching a maximum between 25 and 30 s. For longer injection times the signal gradually decreased to the level found for the blank sample (injection times >85 s). This indicated that there was no iodide remaining in the eluate, only 5 M NaNO3. Thus injection of the eluate in the injection loop at 25 s was the most suitable injection time. The results also showed that a volume of 1420 μL of 5 M NaNO3 was sufficient to elute all the sample iodide trapped on the column. | ||
| Fig. 4 Signals obtained for a blank (water) sample and 100 μg I L−1 iodide (analyte) eluted from the on-line ion-exchange column with 5 M NaNO3 (eluent). The signals were obtained from solution in the 150 μL sample loop (of injection valve IV2) injected into ‘section c’ at various injection times. | ||
3.3 Volume and flow rate of the samples
In this work urine samples were diluted 5-fold with water prior to analysis. Therefore a standard iodide solution of 20 μg I L−1 was chosen as representative of the urine at the median level of iodine (100 μg I L−1, the cut off level between mild IDD and normal). The amount of resin packed in the SAX column was capable of trapping 45 μmol of iodide ion. Volume, from 1 to 5 mL, of this solution was loaded on the SAX resin at a flow rate of 1 mL min−1 (1.6 × 10−4 to 7.9 × 10−4 μmol I−). As expected, the iodide signal recorded at detector D2 increased linearly with the loading volume (r2 = 0.994, data not shown) indicating that the system had high efficiency in both trapping and elution of iodide ion. In this work, 5 mL was selected as the sample volume.Loading flow rates of 0.5, 1.0, 1.5, 2.0 and 2.5 mL min−1 were tested with no difference in the observed signals. However, it was found that the flow rate of 2.5 mL min−1 led to back pressure after long use. A flow rate of 2.0 mL min−1 was selected for loading the sample.
3.4 Investigation of the kinetics
For determination of iodide using the kinetics of Ce(IV)–As(III) reaction, it was proposed that the reaction should exhibit pseudo-first order kinetics in Ce(IV) concentration.20,32 Then the relationship between Ce(IV) concentration and time can be expressed by [Ce(IV)]t = [Ce(IV)]0 e−kt: where [Ce(IV)]t is the concentration of Ce(IV) at time t; [Ce(IV)]0 is the initial concentration of Ce(IV); k is the observed pseudo-first order rate constant. This rate constant is equal to (kuncatalyzed + kcatalyzed[I−]), where the two rate constants are for the uncatalyzed and catalyzed processes, respectively. In flow-based analysis the absorbance of Ce(IV) is monitored at a fixed time t after mixing of the reagent(s) and sample. It can be shown that −ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) At and [I−] are linearly related, as shown in eqn (1).
At and [I−] are linearly related, as shown in eqn (1).| −lnAt = −lnAo + tkuncatalyzed + tkcatalyzed[I−] | (1) |
The flow manifold of ‘section c’ (Fig. 1) was constructed similar to the manifold presented for the determination of total inorganic iodine in drinking water.32 However, the current manifold has a 40 °C water bath to accelerate the kinetics and improve the sensitivity. For this new flow system, it was necessary to adjust the system to ensure that the Sandell–Kolthoff reaction followed first-order kinetics. A stopped-flow mode20,32 was employed in the flow system ‘section c’ to investigate the kinetics for five concentrations of iodide (20 to 100 μg I L−1), prepared in 5 M NaNO3 (to simulate the iodide in the eluate). An exponential decrease in absorbance with time (Fig. S1‡) was observed for all the iodide solutions. Using Sigma plot software33 it was shown that the exponential fittings had less than 1% fitting errors, comparable to a batch mixing.
3.5 Length of the dilution tube for creatinine analysis
In this work, creatinine was measured using the flow manifold ‘section b’ (Fig. 1). It had been recommended that for the Jaffe's method the urine sample should be diluted approximately 50-fold before reacting with the picrate reagent.34 Thus a large diameter tube (3.17 mm i.d.), the dilution tube, was added to further dilute the urine sample and also to improve the mixing efficiency. Table 2 shows the analysis times and percent recoveries obtained when using various lengths of dilution tube. The experiment was carried out using pooled urine from 5 subjects. It was observed that the recovery decreased to almost 100% as the length of the dilution tube increased, i.e. with increasing dilution. 8 cm tubing was selected for a faster analysis time.| Tube length (cm) | Calculated value | Analysis time (minutes per injection) | %Recovery (n = 3) | |
|---|---|---|---|---|
| Tube volume (μL) | Dilution factora (fold) | |||
| a Dilution factor = (volume of dilution tube)/50 μL of L1. | ||||
| 2 | 160 | 3.2 | 3.00 | 118 ± 2.5 |
| 4 | 320 | 6.4 | 3.50 | 102 ± 4.7 |
| 6 | 470 | 9.4 | 4.00 | 100 ± 6.5 |
| 8 | 630 | 12.6 | 4.50 | 100 ± 0.7 |
| 10 | 790 | 15.8 | 5.00 | 101 ± 0.4 |
3.6 Analytical performance and validation
Using the optimal conditions discussed previously, the flow system in Fig. 1 gave simultaneous measurements of iodide and creatinine in urine. The linear range for iodide was 0–200 μg I L−1 (−lnAt = (0.0109 ± 0.0001) μg I L−1 + (0.2485 ± 0.0045): r2 = 0.999) and for creatinine the linear range was 50–1200 mg creatinine L−1 (ΔA = (0.0007 ± 0.0001) mg creatinine L−1 + (0.0015 ± 0.0045): r2 = 0.999) (Fig. S2‡). The limits of detection (LOD, 3σ) were 1 μg I L−1 and 12 mg creatinine L−1 for iodide and creatinine, respectively. The method had a precision of 1.5% RSD (n = 10) (for 20 μg I L−1) and 6.1% RSD (for 50 mg creatinine L−1). Recoveries for 10 urine samples were 87–104% for iodide and 90–104% for creatinine, respectively. The method had a throughput of 12 samples per hour. The SAX column was robust and self-regenerated to the nitrate form during the elution of iodide with the sodium nitrate eluent.
The developed method was applied to the determination of iodide and creatinine in 50 urine samples collected from volunteers with no known history of IDD. These values were compared with values obtained using the reference methods by means of the Bland–Altman plot35 (see Fig. 5a and b for iodide and creatinine, respectively). The plots showed that all data lay within ±2SD of the mean of their differences, showing that this method was equivalent to the reference methods. Pearson's correlations (Fig. S3‡)35 also confirmed that our method gave results that did not differ significantly from values using the reference methods (r = 0.988 for iodide, r = 0.992 for creatinine).
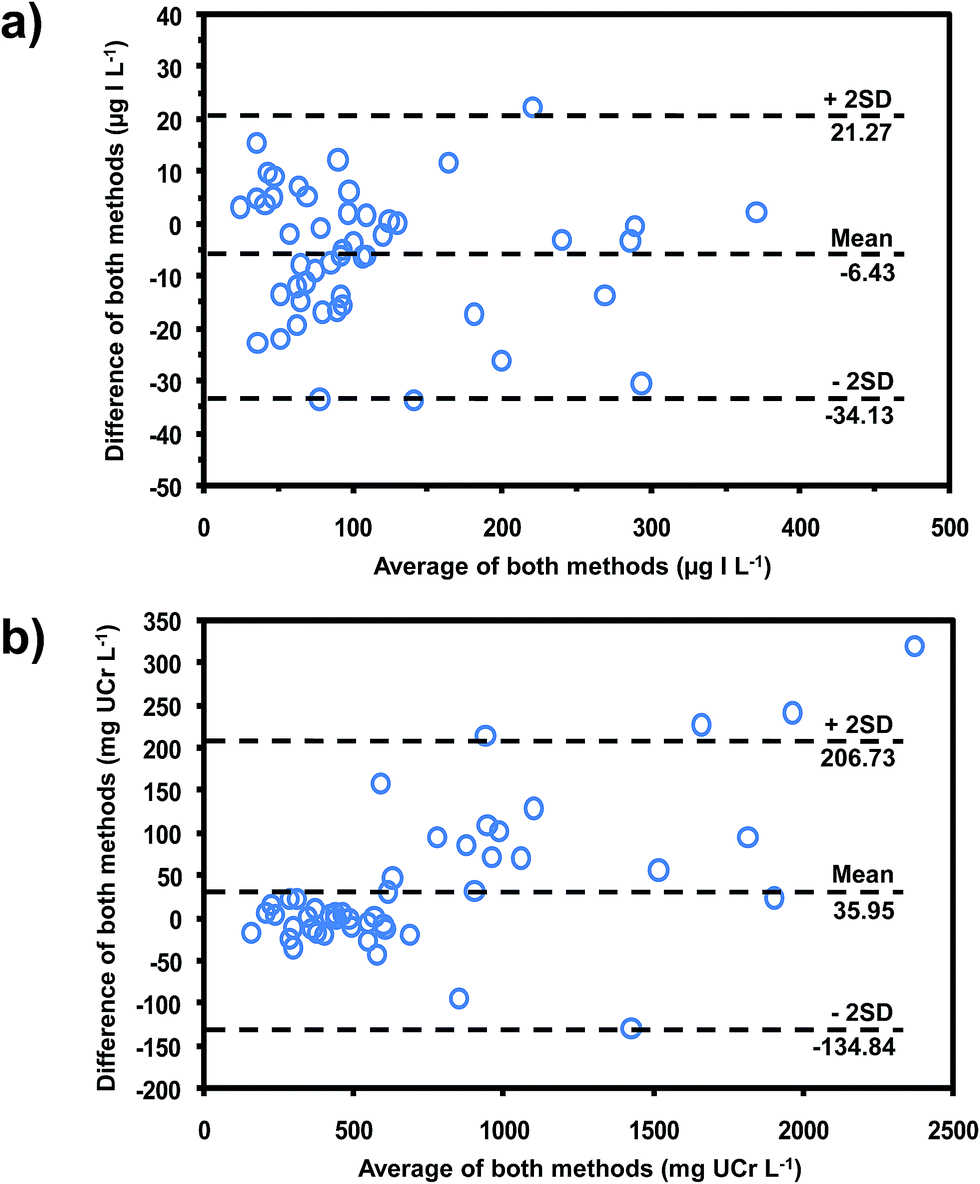 | ||
| Fig. 5 Bland–Altman plots for comparing the data obtained from our method for (a) iodide, compared with chloric acid digestion method19 and (b) creatinine, compared with the standard Jaffe's method.29 | ||
As reported in the literature,36,37 the levels of urinary iodine and creatinine showed diurnal fluctuation over a 24 hour period (Fig. 6). However, the iodide to creatinine ratios (UI/UCr) almost eliminated this diurnal fluctuation (see Fig. 6a and b). Therefore, this method allows the evaluation of the UI/UCr ratio of a spot urine sample which can replace a 24 h urine sample for evaluating the IDD status of a large number of subjects. A recent paper38 also measured the UI/UCr ratio in urine dried on filter paper strips. The extracted sample was oxidized with persulphate prior to analysis by the Sandell–Kolthoff reaction. A separate Jaffe's method was employed for creatinine determination.
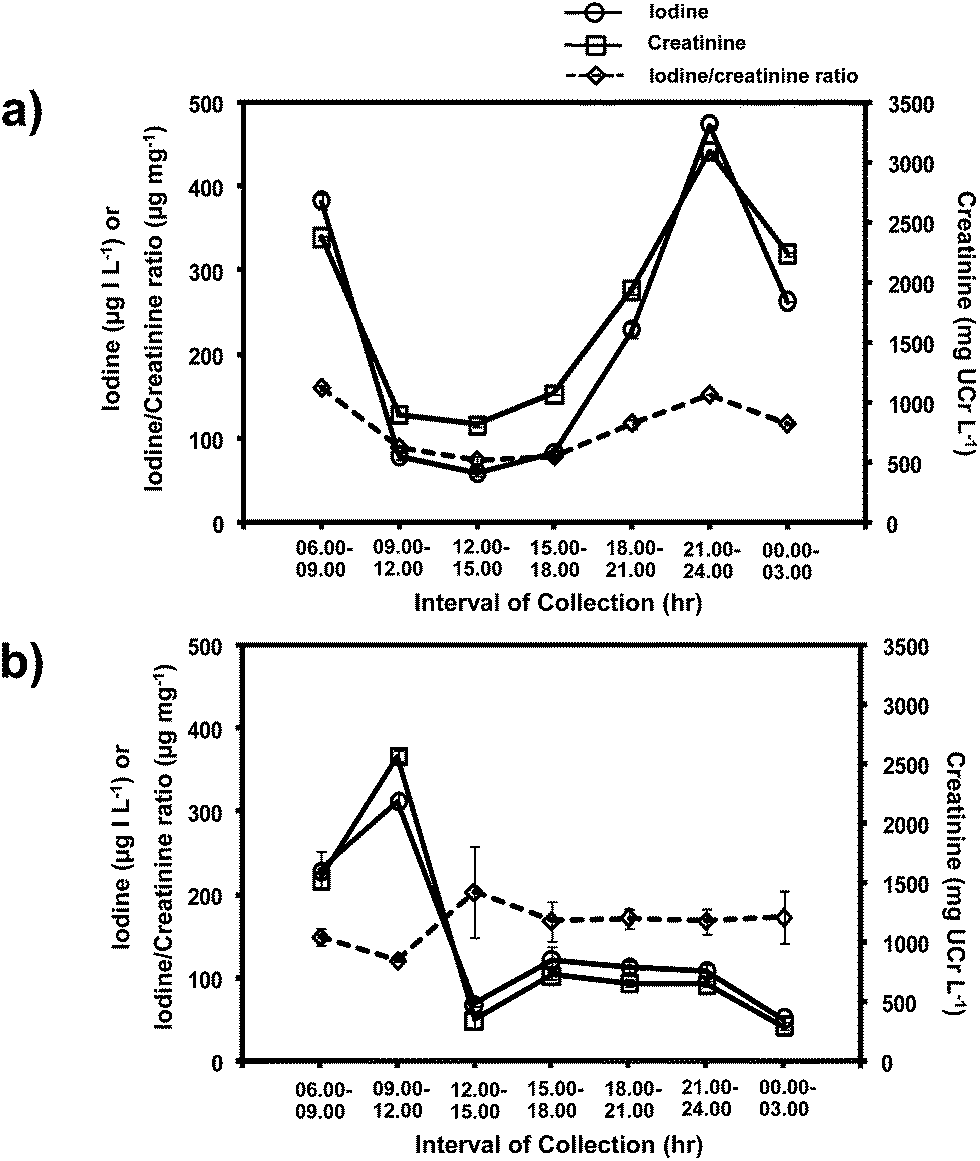 | ||
| Fig. 6 Levels of iodide, creatinine and iodide/creatinine ratio of spot urine samples collected over 24 hours from two subjects, (a) and (b). | ||
4. Conclusion
To our knowledge, this is the first system that provides simultaneous determination of iodide and creatinine in urine, and hence the UI/UCr ratio. Thus survey of the iodine status of a population can be conveniently carried out using spot urine. The urine sample is directly injected into the flow manifold without any sample pre-treatment, except a 5-fold dilution with water. An in-line SAX column in the nitrate form extracts iodide from the urine matrix while the non-retained urine is analyzed for creatinine content using alkaline picrate. A selected fraction of the eluate from the SAX column is analyzed for iodide using the Sandell–Kolthoff reaction. A 5 M NaNO3 solution is used for eluting iodide from the SAX column as well as for regenerating the SAX resin for the next sample. The SAX column has been employed for more than 200 injections. We believe that this system is a step towards resolving the outstanding problem of a facile sample preparation as commented in the review of Shelor and Dasgupta.13Acknowledgements
The work was financially supported by the research grant (B04/2554) of Mahidol University (DN), S&T Research Grant of the Thailand Toray Science Foundation, 2010 (DN) and the Office of Higher Education Commission and Mahidol University under the National Research Universities Initiative, chaired by Prof. Manat Pohmakotr. We would like to thank the Thailand Research Fund (TRF) for providing the student scholarship of the Royal Golden Jubilee Ph.D. program to JS. We are grateful to the postdoc scholarship of Mahidol University (TM). The authors would also like to acknowledge the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education for the support of equipment and partial scholarships for students (JS and PI). The authors wish to thank all the volunteers for providing us with urine samples.References
- World Health Organization, United Nations Children’s Fund, International Council for the Control of Iodine Deficiency Disorders, in Assessment of iodine deficiency disorders and monitoring their elimination – A guide for programme managers, WHO, Geneva, 3rd edn, 2007 Search PubMed.
- A. Melse-Boonstra and N. Jaiswal, Best Pract. Res., Clin. Endocrinol. Metab., 2010, 24, 29–38 CrossRef CAS PubMed.
- M. B. Zimmermann, Endocr. Rev., 2009, 30, 376–408 CrossRef CAS PubMed.
- M. Andersson, B. D. Benoist and L. Rogers, Best Pract. Res., Clin. Endocrinol. Metab., 2010, 24, 1–11 CrossRef CAS PubMed.
- M. B. Zimmermann, P. L. Josste and C. S. Pandav, Lancet, 2008, 372, 1251–1262 CrossRef CAS.
- B. Michalke and P. Schramel, Electrophoresis, 1999, 20, 2547–2553 CrossRef CAS.
- B. Michalke, P. Schramel and H. Witte, Biol. Trace Elem. Res., 2000, 78, 67–79 CrossRef CAS.
- P. L. Jooste and E. Strydom, Best Pract. Res., Clin. Endocrinol. Metab., 2010, 24, 77–88 CrossRef CAS PubMed.
- K. Minakata, I. Yamagishi, S. Kanno, H. Nozawa, M. Suzuki and O. Suzuki, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 1683–1686 CrossRef CAS PubMed.
- V. T. P. Nguyen, V. Piersoel and T. E. Mahi, Talanta, 2012, 99, 532–537 CrossRef CAS PubMed.
- M. Hu, C. Chen, Y. Jiang and H. Zhu, Chem. Pap., 2013, 67, 1255–1261 CrossRef CAS.
- F. Du, F. Zeng, Y. Ming and S. Wu, Microchim. Acta, 2013, 180, 453–460 CrossRef CAS.
- C. P. Shelor and P. K. Dasgupta, Anal. Chim. Acta, 2011, 702, 16–36 CrossRef CAS PubMed.
- E. B. Sandell and I. M. Kolthoff, J. Am. Chem. Soc., 1934, 56, 1426 CrossRef CAS.
- G. Aumont and J. C. Tressol, Analyst, 1986, 111, 841–843 RSC.
- J. T. Dunn, H. E. Crutchfield, R. Gutekunst and A. D. Dunn, Thyroid, 1993, 3, 119–123 CrossRef CAS.
- S. Pino, S. L. Fang and L. E. Braverman, Clin. Chem., 1996, 42, 239–243 CAS.
- S. Pino, S. L. Fang and L. E. Braverman, Exp. Clin. Endocrinol. Diabetes, 1998, 106, S22–S27 CrossRef CAS PubMed.
- D. Nacapricha, S. Muangkaew, N. Ratanawimarnwong, J. Shiowatana and K. Grudpan, Analyst, 2001, 126, 121–126 RSC.
- D. Nacapricha, N. Ratanawimarnwong, J. Suwannachoat, P. Wilairat, J. Shiowatana and K. Grudpan, Anal. Sci., 2001, 17, i33–i36 Search PubMed.
- T. Ohashi, M. Yamaki, C. S. Pandav, M. G. Karmarkar and M. Irie, Clin. Chem., 2000, 46, 529–536 CAS.
- H. Hussain and W. N. W. Mohamud, Trop. Biomed., 2006, 23, 109–115 Search PubMed.
- M. Hedayati, M. Khazan, P. Yaghmaee, M. Z. Yeghaneh, L. Behdadfar and M. S. Daneshpour, Clin. Chem. Lab. Med., 2011, 49, 281–284 CrossRef CAS PubMed.
- A. Mina, E. J. Favaloro and J. Koutts, J. Trace Elem. Med. Biol., 2011, 25, 213–217 CAS.
- W. May, D. Wu, C. Eastman, P. Bourdoux and G. Maberly, Clin. Chem., 1990, 36, 865–869 CAS.
- H. C. Ford and L. A. Johnson, Clin. Chem., 1991, 37, 759 CAS.
- K. Tsuda, H. Namba, T. Nomura, N. Yokoyama, S. Yamashita, M. Izumi and S. Nagataki, Clin. Chem., 1995, 41, 581–585 CAS.
- Z. Yaping, Y. Dongxing, C. Jixiang, L. Tianshiu and C. Huiqin, Clin. Chem., 1996, 42, 2021–2027 CAS.
- R. W. Bonsnes and H. H. Taussky, J. Biol. Chem., 1945, 158, 581–591 CAS.
- P. Vejbjerg, N. Knudsen, H. Perrild, P. Laurberg, S. Andersen, L. B. Rasmussen, L. Ovesen and T. Jørgensen, Thyroid, 2009, 19, 1281–1286 CrossRef CAS PubMed.
- G. E. Abraham, R. C. Handal and J. C. Hakala, The Original Internist, 2006, 13, 125–135 Search PubMed.
- N. Choengchan, K. Lukkanakul, N. Ratanawimarnwong, W. Waiyawat, P. Wilairat and D. Nacapricha, Anal. Chim. Acta, 2003, 499, 115–122 CrossRef CAS PubMed.
- Sigma plot®, Programming Guide, SPSS Inc., Chicago, 2001 Search PubMed.
- T. Songjaroen, T. Maturos, A. Sappat, A. Tuantranont and W. Laiwattanapaisal, Anal. Chim. Acta, 2009, 647, 78–83 CrossRef CAS PubMed.
- S. A. Glantz, in Primer of Biostatistics, The McGRAW-HILL, New York, 3rd edn, 2005, ch. 8, pp. 305–310 Search PubMed.
- L. B. Rasmussen, L. Ovesen and E. Christiansen, Eur. J. Clin. Nutr., 1999, 53, 401–407 CAS.
- F. KÖnig, M. Andersson, K. Hotz, I. Aeberli and M. B. Zimmermann, J. Nutr., 2011, 14, 2049–2054 CrossRef PubMed.
- T. T. Zava, S. Kapur and D. T. Zava, Anal. Chim. Acta, 2013, 764, 64–69 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated to the 60th Birthday of Prof. Dr Kate Grudpan. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an01224k |
| This journal is © The Royal Society of Chemistry 2015 |