
Lawsone in organic synthesis
Alessandro K. Jordão
ab,
Maria D. Vargas
a,
Angelo C. Pinto
c,
Fernando de C. da Silva
*a and
Vitor F. Ferreira
*a
aUniversidade Federal Fluminense, Institute of Chemistry, Campus do Valonguinho, 24020-141, Niterói, RJ, Brazil. E-mail: gqofernando@vm.uff.br; cegvito@vm.uff.br
bUnidade Universitária de Farmácia, Fundação Centro Universitário Estadual da Zona Oeste, 23070-200, Rio de Janeiro, RJ, Brazil
cInstituto de Química, Universidade Federal do Rio de Janeiro, CT, Cidade Universitária, Ilha do Fundão, 21945-970, Rio de Janeiro, RJ, Brazil
First published on 3rd August 2015
Abstract
Lawsone (1) is a special naphthoquinone that is useful for many applications in various scientific and technological fields. For over 100 years, it has been used as the starting material for the synthesis of a variety of biologically active compounds and materials with interesting properties. In organic synthesis, it has been used in many reactions. This review aims to address the various aspects of its use in organic synthesis.
 Alessandro K. Jordão | Alessandro K. Jordão received his Industrial Pharmacist degree from the Fluminense Federal University, Brazil in 2005. In 2007 he obtained his MSc and in 2010, his DSc in Organic Chemistry under the supervision of Vitor F. Ferreira and Anna C. Cunha at the same university. Currently he is a lecturer at Fundação Centro Universitário Estadual da Zona Oeste (UEZO) in Rio de Janeiro. His research interests focus on the synthesis of heterocyles, quinones and nitroxide radicals. |
 Maria D. Vargas | Maria D. Vargas is a full professor in Inorganic Chemistry at the Fluminense Federal University. After her PhD at the University of Cambridge and a Sydney Sussex College research fellowship, she took a position at the State University of Campinas (UNICAMP) in Brazil, in 1988. In 2003 she moved to her present position. Her recent research focuses on the synthesis of biologically active naphthoquinone metal complexes and nanodevices for drug delivery. |
 Angelo C. Pinto | Angelo C. Pinto is a full professor in Organic Chemistry at the Rio de Janeiro Federal University. His current research interests include natural product chemistry, organic synthesis and medicinal chemistry. |
 Fernando de C. da Silva | Fernando de C. da Silva received his Industrial Chemistry degree in 2002 and his PhD in 2007, both from the Fluminense Federal University (UFF). He then completed a two-year post-doctoral fellowship at Universidade de Aveiro, Portugal, with J. A. S. Cavaleiro, working on porphyrin chemistry. He has been a professor at UFF since 2009. The synthesis of 1,2,3-triazoles, quinones, carbohydrates, diazocompounds, β-enaminones and porphyrins are his present research interests. |
 Vitor F. Ferreira | Vitor Francisco Ferreira is a full professor of Organic Chemistry at the Fluminense Federal University. He obtained his BS in chemistry in 1976 and MSc in the chemistry of natural products in 1980 under the guidance of Antonio Ventura Pinto at the Rio de Janeiro Federal University. He is a Fellow of the RSC and former President of the Brazilian Chemical Society. His research interests include organic syntheses using readily available carbohydrates, synthetic methods using diazocompounds and the synthesis of bioactive compounds against neglected diseases. |
1. Introduction
Quinones are widely distributed in nature, and several of their synthetic and natural products are very important in many diverse areas of chemistry and biochemistry.1 They play a fundamental role in several living cells as electron carriers in the respiratory chain, as well as in blood coagulation and carboxylation of glutamates. Due to the intimate relationship between quinones and the biochemical processes of cells, these compounds have been extensively explored in the synthesis2 of several bioactive compounds with antitumor,3–5 molluscicidal,6,7 antiparasitic,8–10 leishmanicidal,11 antiinflammatory,12 antifungal,13 antimicrobial14 and trypanocidal15–17 activities. Various reports note that the biological activities of these molecules stem from their ortho or para-quinonoid group that accepts one and/or two electrons (redox cycling) to form the corresponding radical anion or dianion species in situ.18,19 Thus, the semi-quinone radicals accelerate the creation of intracellular hypoxic conditions by producing superoxide anions.20,21 Through this mechanism, quinones may present cytotoxicity in mammalian cells, possibly by affecting enzymes such as topoisomerases, a group of enzymes that are critical for DNA replication in cells.22The quinone class includes several important synthetic and natural compounds bearing a hydroxy group on the quinone ring. Specifically, the lawsone (1) motif and that of its isomeric form 4-hydroxy-1,2-naphthoquinone are common subunits of compounds with a number of valuable biological activities,23 examples of which are illustrated in Fig. 1: lapachol (2), atovaquone (3), parvaquone (4), NQ1 (5),24 β-lapachone (6) and α-lapachone (7).
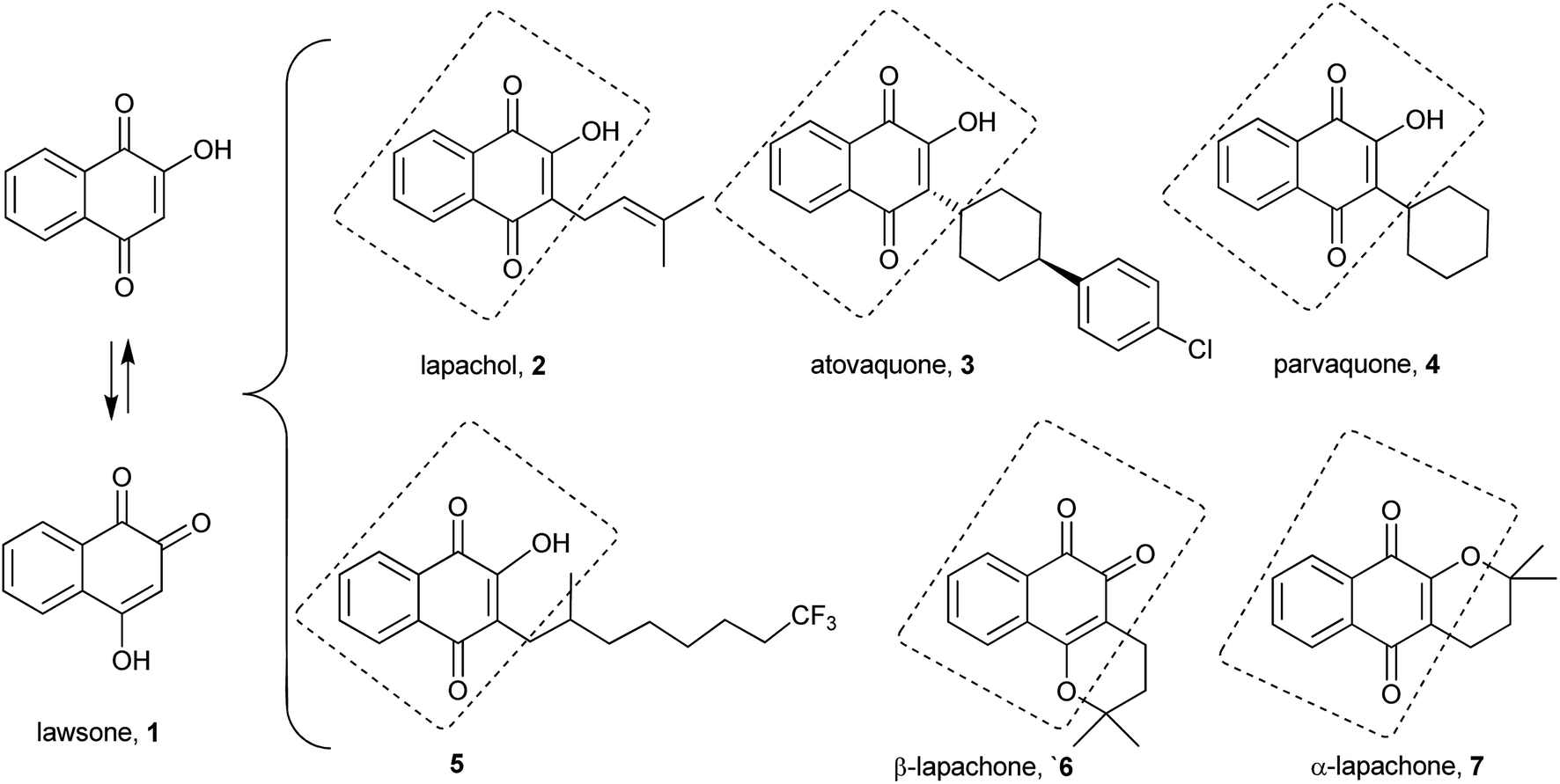 | ||
| Fig. 1 Natural and synthetic bioactive naphthoquinones containing the lawsone fragment. | ||
2-Hydroxy-1,4-naphthoquinone (1), or lawsone, or hennotannic acid, is one of the simplest naturally occurring naphthoquinones. This red-orange pigment, classified as Natural Orange 6 (C.I. 75480), can be obtained from the extract of dried powdered leaves of henna (Lawsonia spp., family Lythraceae), a branched shrub or small tree with grayish-brown bark. The most common species are Lawsonia alba, Lawsonia spinosa and Lawsonia inermis Linn. (mignonette tree)25,26 that contain approximately 0.5–1.5% of 1.27,28 This compound is also found in the jewelweed Impatiens balsamina29 and in Juglans regia (or walnut).30 An important isomer of 1 is 5-hydroxy-1,4-naphthoquinone or juglone, which is also a natural 1,4-naphthoquinone found in several species of the Juglandaceae family, such as Juglans nigra, J. cinerea, and J. regia. The position of the hydroxyl group in these compounds is crucial to their biological activities as it affects their redox potentials and prooxidant activities.31–34
Lawsone (1) is responsible for the red dye properties of henna, which is traditionally used for coloring hair and dying nails and skin,35–37 and served as an inspiration for the development of new bioactive compounds.38 This dye has been used since the most ancient times by the Egyptians and Indians. Presently, henna extracts are still commercialized for skin,39 wool40 and cotton41 dyeing purposes in several countries from Asia and Africa, with the Pali district of Rajasthan in India being the area with the highest production of henna extract, mainly from L. inermis Linn.42,43
Extracts of henna are widely used in folk medicine, especially in developing countries,44,45 to treat burn wounds infected by microorganisms46 such as Mycobacterium tuberculosis,47 Staphylococcus aureus, Pseudomonas aeruginosa, Fusarium oxosporum, Aspergillus niger and Candida albicans, due to its antibacterial properties,48 as well as headaches, lumbago, bronchitis, ophthalmia, syphilitis, sores and amenorrhoea.43 In several countries, henna is a sacred plant that is widely used in religious ceremonies, and it symbolizes prosperity, fertility and happiness. Despite its long history of use, the toxicity of henna extracts has only been investigated in recent decades.49 According to these studies, extracts of henna and 1 are highly toxic, causing hemolytic anemia and renal tubular necrosis when administered orally to rats,50 as well as hypersensitivity with urticaria, rhinitis and bronchial asthma.51 Recently, this compound was evaluated by the Scientific Committee on Cosmetic Products and Non-Food Products Intended for Consumers of the European Community,52 which concluded that it is moderately toxic on acute ingestion, with mortalities occurring at 300 mg kg−1 and above. In addition, two 13 week studies have shown clear signs of toxicity in the haemopoietic system,53 kidneys, forestomach and liver, but when compound 1 was used on the skin, most of it remained there with only low absorption levels by the fluids.54 McMillan et al. found that henna ingestion can induce severe hemolytic anemia and that administration of 1 to rats induces a hemolytic response that is associated with oxidative damage to erythrocytes.55 Several deaths of children to whom henna was administered during joyous, celebratory events have been reported.56,57
Because 1 is easily obtained from natural sources and is currently commercially available in bulk from Yucca Enterprises (India) and Sigma-Aldrich, it is not surprising that its activity against various microorganisms including fungi, bacteria and viruses has been investigated.58–60
Marzin and Kirkland found that 1, obtained from the natural dye of henna, is non-genotoxic in the mouse bone marrow micronucleus test and does not produce oxidative DNA damage in Chinese hamster ovary cells, and they therefore concluded that 1 poses no or negligible genotoxic risk.61 Recently, Klotz and co-workers studied the oxidative damage and stress signaling pathway in HaCaT human keratinocytes induced by several 1,4-naphthoquinones and stated that neither lawsone (1) nor-lapachol (2) (up to 100 μM) were active in any of these assays, but they discovered that these compounds induce phosphorylation of the epidermal growth factor receptor (EGFR) and the related ErbB2 receptor tyrosine kinases.62 In a report published in 2005, the Norwegian Scientific Committee for Food Safety was of the opinion that “if 1 is considered as an in vivo mutagen and using an estimated systemic exposure dose based on a worst case use situation, the possible cancer risk associated with the use of Henna products for hair-dyeing containing 1 would be negligible”.63
Besides its cosmetic applications, lawsone (1) has several other uses. A literature search reveals a long list of applications, including skin protection from ultraviolet radiation,64,65 cloth dyeing, antiaging additive to vulcanized natural rubber,66 corrosion inhibition for steel,67,68 and oxidation of chlorinated compounds.69,70 Furthermore, it reacts with the amino acids of latent fingermark deposits on paper surfaces to yield a clean purple-brown detailed impression that is also photoluminescent.71 Recently, it was demonstrated that 1 is a sensitive colorimetric and electrochemical sensor for anions, such as cyanide, acetate, fluoride and dihydrogen phosphate (DHP) in acetonitrile. These anions cause a color change of the solution of 1 from yellow to orange-red, which is not observed in the presence of other anions, such as chloride, bromide, iodide or perchlorate.72
The complexation reactions of 1 have been investigated for various purposes, and a number of metal compounds exhibiting a variety of structures have been described in the literature, examples of which are illustrated in Fig. 2.73–76 Potentially, 1 and analogous compounds can bind to metal ions in three different oxidation states (Fig. 2), quinone, semiquinone (the one-electron reduced form of quinone)77 and catechol (the two-electron reduced form), and this binding ability is, in part, believed to be responsible for the important role of quinones in biological systems.78 Hence, iron complexes of 1 have been studied in an attempt to mimic the iron–quinone couple found in bacterial reaction centers,79–82 whereas copper complexes of 1 have been investigated mainly to understand metal–quinone interactions and to mimic the structural and functional models of copper-containing enzymes.83 Metals can also play an important role in modifying the pharmacological properties of known drugs. In this way, metal complexes of 1 (Fig. 2) were also evaluated for their potential cytotoxicity against cancer cells, and the copper(II) complex 9 was found to induce apoptosis in HepG2 human cancer cells by a mechanism that involves activation of caspases and modulation of apoptotic related proteins.84 Last but not least, the ruthenium(III) lawsonate complex 10a has been shown to oxidize primary and secondary alcohols to aldehydes and ketones, respectively, in the presence of N-methylmorpholine-N-oxide as co-oxidant.85 The catalytic cycle of the oxidation is activated by the metal center that undergoes oxidation and is capable of stabilizing the coordinated semiquinone(II) and catechol(III) forms of the naphthoquinone.
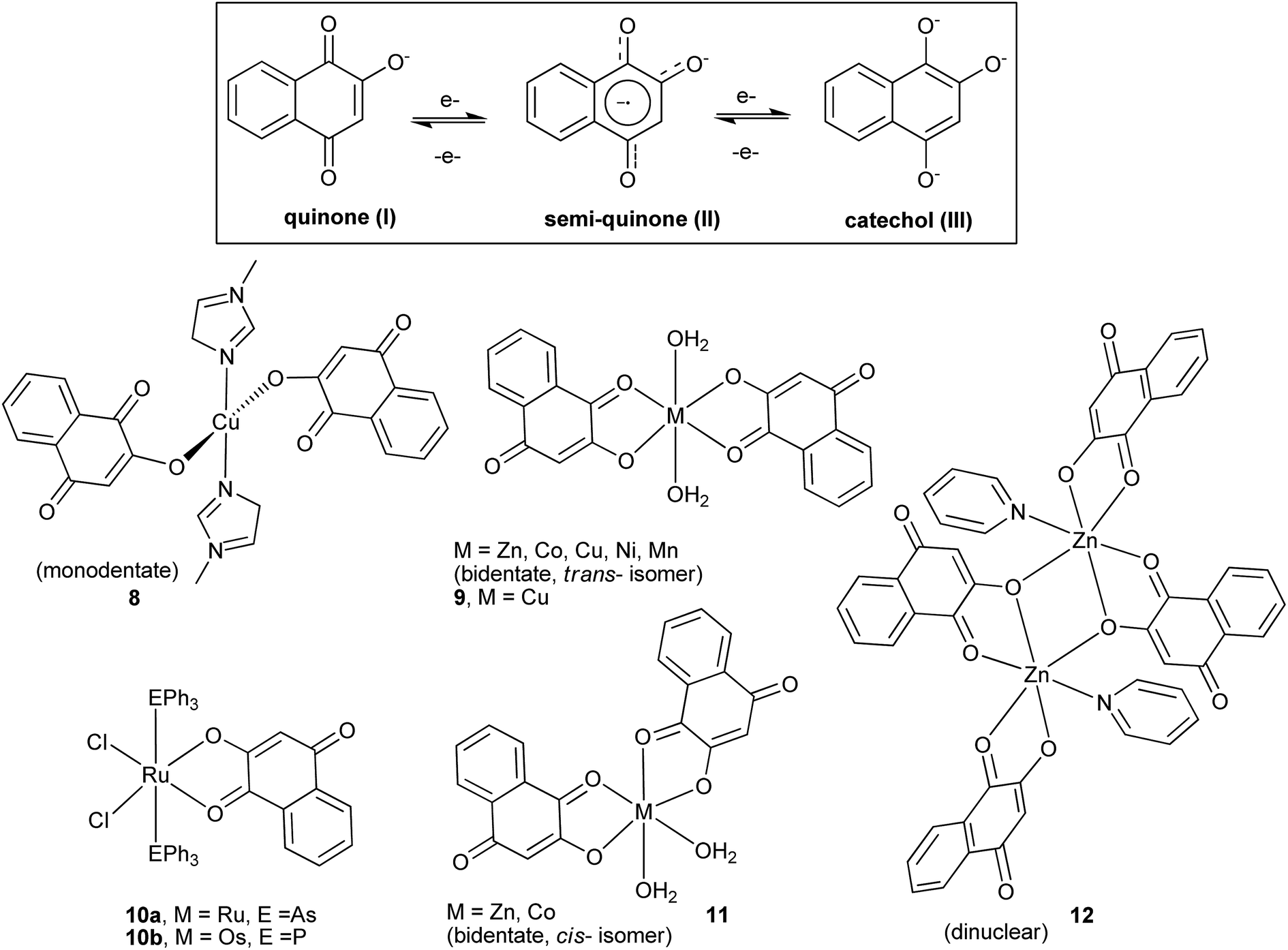 | ||
| Fig. 2 Redox forms of the lawsone anion and schematic structure of some of its metal(II) complexes. | ||
If controversy exists over the use of 1 as a hair and skin dye because of its toxicological and biological effects,86–89 the same does not exist for its use as a starting material in organic synthesis. Hydroxyquinone moieties90 are present in a number of natural products with interesting biological activities, but this review will only focus on the use of 2-hydroxy-1,4-naphthoquinone (1) in organic synthesis.
2. Methods for the preparation of lawsone (1)
Several methods have been reported in the literature for the synthesis of 1, which are based on either the de novo construction of the naphthoquinone system or the use of naphthalene derivatives as starting materials. In some cases, the two methods are complementary.2.1 Preparation of lawsone (1) by ring constructing methods
Hooker described the first synthesis of 1 in 1892 via construction of the naphthalene core from 3-methyl-1H-indane 13. This compound was oxidized and cleaved with sodium dichromate to phenylacetic acid 14, which upon cyclization and oxidation produced 1 (Scheme 1).91,92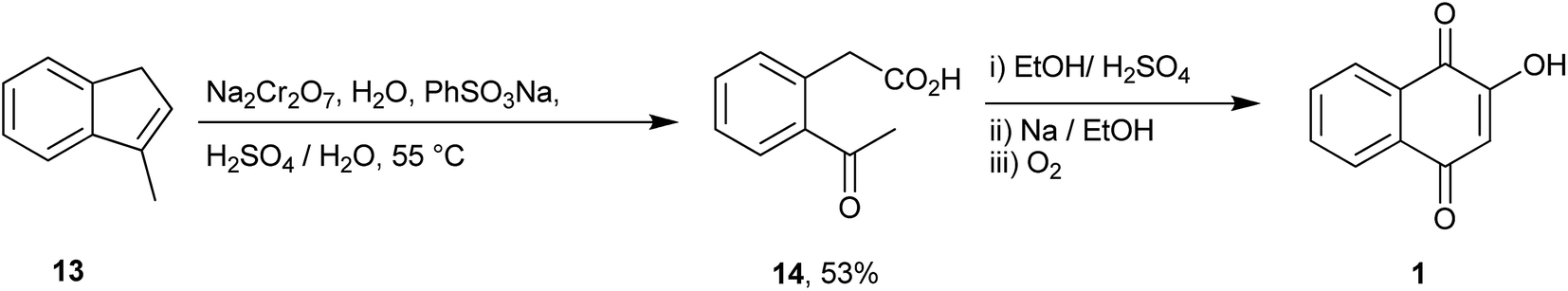 | ||
| Scheme 1 | ||
A short route to 1 and other similar naphthoquinones was described by Heerding and Moore from 4-alkynyl-3-tert-butoxy-4-hydroxycyclobutenones 16 as illustrated in Scheme 2. Addition of the alkynyllithium reagent to 3-tert-butoxycyclobutene-l,2-dione 15 gave the cyclobutenones in excellent (91–96%) yields. Thermolysis to promote ring expansion, performed in refluxing acetonitrile, provided naphthoquinones 17a and 17b in 63 and 48% yields, respectively. Deprotection of the tert-butoxy quinones led to lawsone (1) and quinone 18 in quantitative yields (Scheme 2).93 The same research group used a variation of this ring expansion method to produce hydroxynaphthoquinone 22 from 3-t-butoxy-4-aryl-4-hydroxycyclobutenone 20. Hooker oxidation of 22 yields 23, which can be used to prepare the natural naphthoquinone lomandrone (24).94 This synthetic sequence was employed to prepare two analogues of parvaquone (4) in good yields, starting from diisopropyl squarate, by addition of the appropriate aryl lithium compounds and ring expansion-cyclization induced by microwave irradiation.95
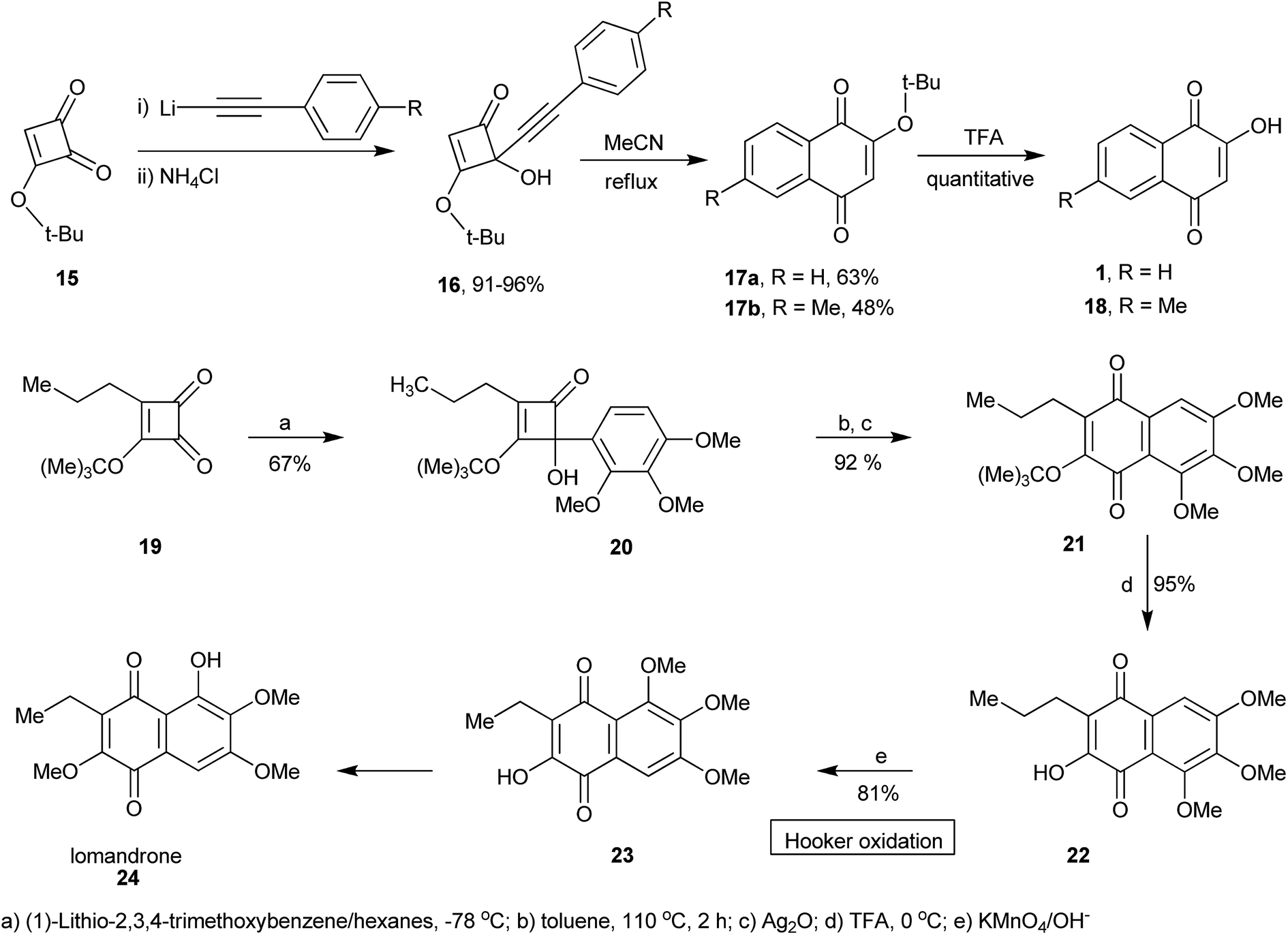 | ||
| Scheme 2 | ||
2.2 Preparation of lawsone (1) from naphthalene derivatives
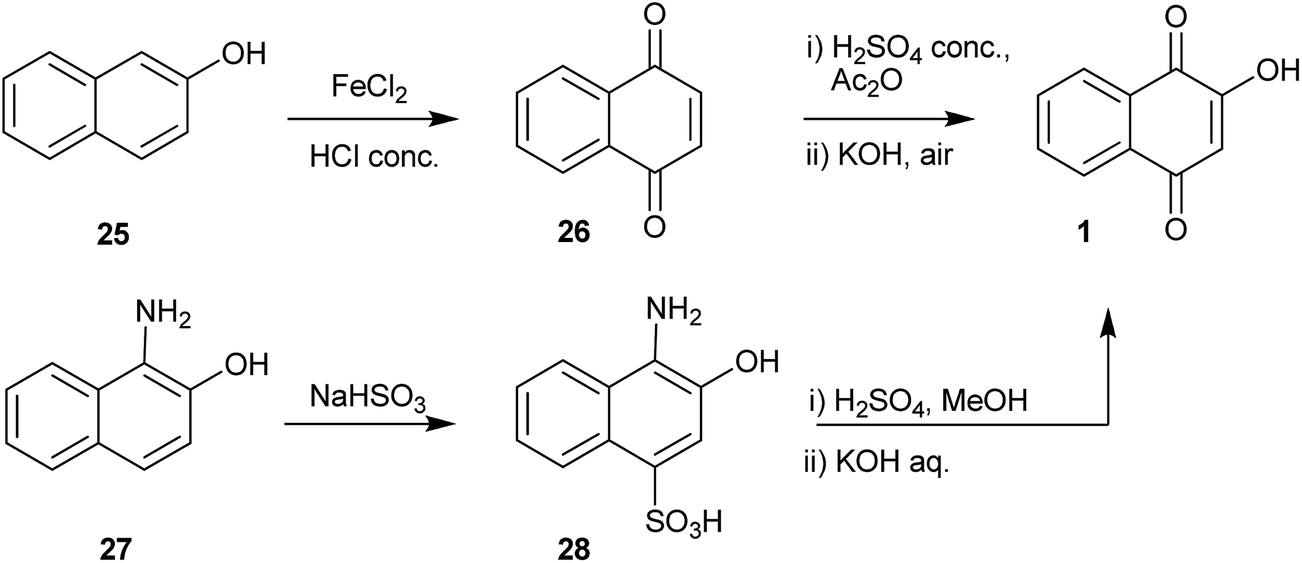
Because several naphthol derivatives are easily obtained from naphthalene, these are the most convenient starting materials for the preparation of 1. Indeed, the most efficient methods used to prepare 1 involve the oxidation of naphthoquinones, 1- or 2-naphthols, dihydroxy naphthalenes, aminonaphthols and tetralones. The variety of existing methods for the preparation of 1 (ref. 96) is a good indication of its importance as a starting material in organic synthesis.2-Naphthol 25 can be converted into 1,4-naphthoquinone 26 by reaction with a solution of ferric chloride hexahydrate in concentrated hydrochloric acid.97 Compound 26 reacts with acetic anhydride-sulfuric acid to give 1,2,4-trihydroxynaphthalene triacetate, which is hydrolyzed and oxidized to 1.98 1-Amino-2-naphthol (27)99 has been used an alternative starting material for the synthesis of 1. Its reaction with sodium bisulfite in aqueous 6 N sodium hydroxide100 produces 1-amino-2-naphthol-4-sulfonic acid (28), which upon hydrolysis with concentrated sulfuric acid in methanol leads to 2-methoxy-lawsone. Hydrolysis of the methoxy group in aqueous sodium hydroxide solution gives 1.101 By using this sequence starting from β-naphthol, 1 can be obtained in 82–84% overall yields (Scheme 3).
 | ||
| Scheme 3 | ||
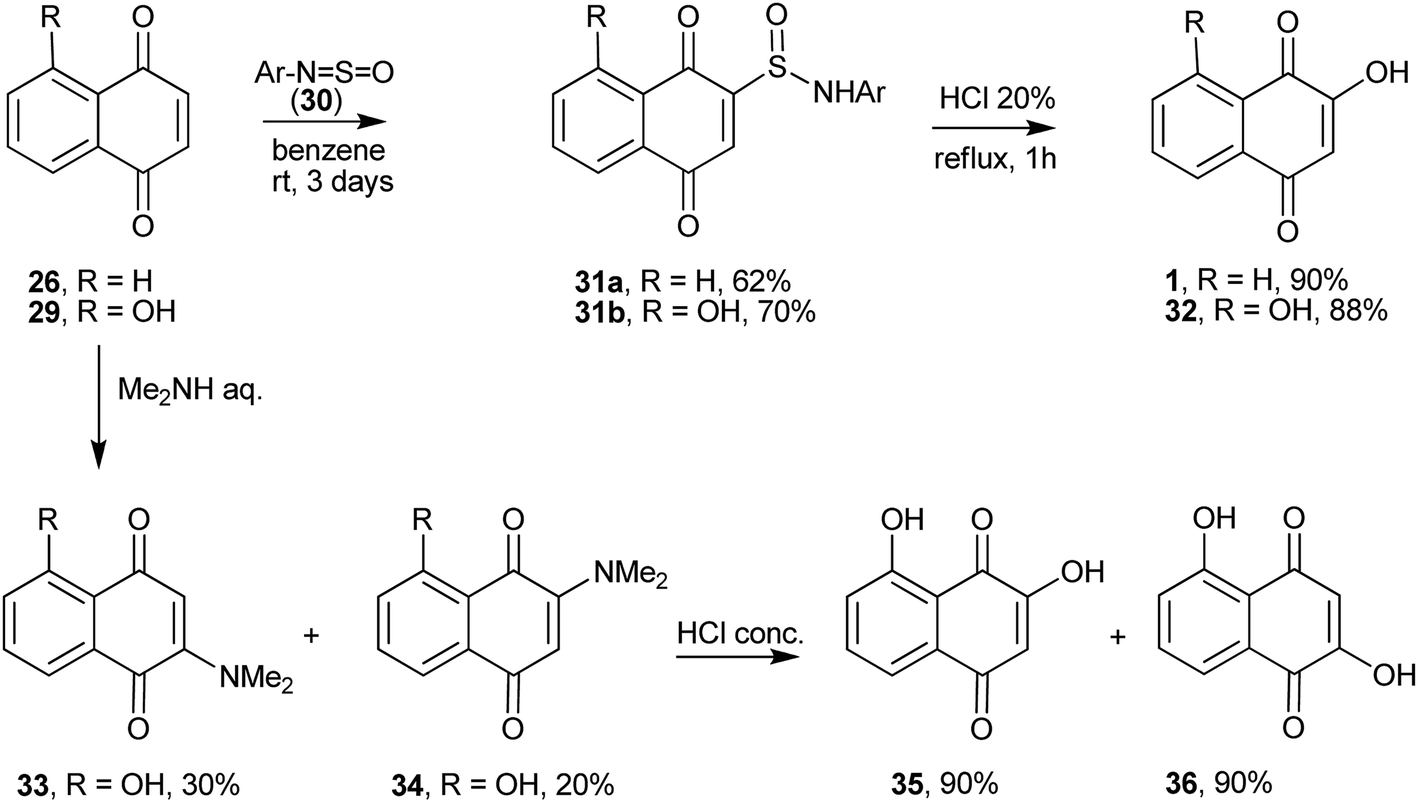
The hydroxyl group can be introduced in position 2 of 1,4-naphthoquinones by several procedures. For instance, 2 + 2 cycloaddition of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond of N-sulfinylarylamines (30) with 1,4-naphthoquinone, followed by a 1,3-H sigmatropic shift, produces 2-(N-arylsulfinamoyl)-1,4-naphthoquinones 31a–b. Hydrolysis of the latter compounds in aqueous HCl gives 1 (Scheme 4).102 By adding amines into the 1,4-unsaturated system 29 (R = OH), Oliveira et al. obtained the necessary intermediate for the preparation of the natural product α-caryopterone.103
S bond of N-sulfinylarylamines (30) with 1,4-naphthoquinone, followed by a 1,3-H sigmatropic shift, produces 2-(N-arylsulfinamoyl)-1,4-naphthoquinones 31a–b. Hydrolysis of the latter compounds in aqueous HCl gives 1 (Scheme 4).102 By adding amines into the 1,4-unsaturated system 29 (R = OH), Oliveira et al. obtained the necessary intermediate for the preparation of the natural product α-caryopterone.103
 | ||
| Scheme 4 | ||
With the emergence of green chemistry as a response to the need to minimize the environmental impact of the classic chemical processes, many alternative, more sustainable and profitable methods have been developed. Thus, Yan et al. selectively converted 2-naphthol into 1 in high yield (62%) and selectivity (100%) by reaction with molecular oxygen under air pressure in an alkaline solution over iron porphyrin catalysts, such as Fe-porphyrin (TMOPPFeCl),104 5,10,15,20-tetra (4-methoxylphenyl) porphyrin iron chloride,105 or by cobalt porphyrins immobilized on polymer microspheres (80%).106 These methods are relatively cheap and use an environmentally benign oxidant. Naphthalene diols are also competent substrates for the oxidation to 1. The performance of the oxidation depends on the relative position of the hydroxy groups on the naphthalene. Maurette and co-workers107,108 developed a method to produce 1 involving the oxidation of dihydroxynaphthalenes 37 or 38 with potassium superoxide (KO2) in a heterogeneous aprotic medium (Scheme 5). Other options for the oxidation of naphthalene diols to 1 include tetraethylammonium superoxide for the oxidation of 1,2-naphthalene diols (56% yield)109 and the use of supported metallated phthalocyanine on montmorillonite K10 or zirconium phosphate (ZrPO4) as the oxidation catalysts (80% yield).110
 | ||
| Scheme 5 | ||
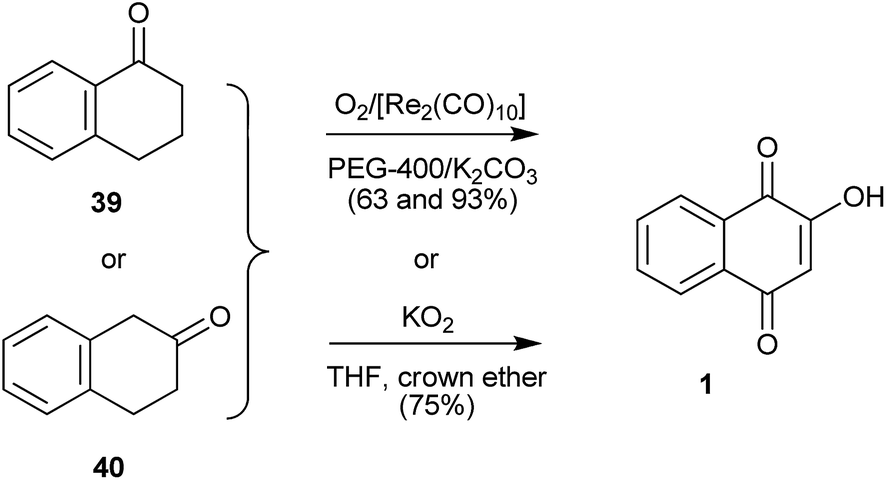
Tetralones 39 or 40 also can be oxidized to 1 by oxygen, potassium hydroxide, potassium carbonate, and catalytic amounts of rhenium carbonyl, [Re2(CO)l0], and polyethylene glycol (PEG-400)111 with potassium superoxide in THF with crown ether in good yields,112 or by Thomson's method of autoxidation with O2/t-BuOK/t-BuOH.113,114 Ullas115 prepared 14C-labelled 1 ([phenyl-14C(U)] 2-hydroxy-1,4-naphthoquinone) from [14C(U)]benzene. The last step of the synthesis involved oxidation of [phenyl-14C(U)]α-tetralone to [phenyl-14C(U)] 1 in 40% by Thomson's method. Likewise, Ashwell et al.116 used Thomson's oxidation method to transform substituted α-tetralones into hydroxy-naphthoquinones in the total synthesis of β-lapachones substituted on the aromatic ring (Scheme 6).
 | ||
| Scheme 6 | ||
3. Lawsone (1) as a starting reagent in organic synthesis
3.1 C-Alkylation and O-alkylation of lawsone
Lawsone (1) can be considered as the stable enol form of a β-diketone and can therefore be alkylated either at the oxygen (O-alkylation) or at the carbon (C-alkylation). The first attempt to alkylate 1 was described in 1927 by Fieser, who reported a preparation for lapachol (2) by reacting the silver salt of 1 with isoprenyl bromide, as shown in Scheme 7. However, the yields of both C-alkylated 2 (5%)117–119 and O-alkylated 42 (3%) products were very low. Interestingly, upon heating, the O-alkylated product 42 underwent a Claisen rearrangement, resulting in the C-alkylated natural product 43 in quantitative yield,120 whose carbon connectivity is different from that of the product obtained from direct C-alkylation, 2. This latter strategy is another alternative for the C-alkylation of 1, which was subsequently rearranged and used in the synthesis of the dunnione pigment.121,122 Recently, Frydman and co-workers developed an improved method for the alkylation of 1 by reacting its lithium salt in DMSO with dimethylallyl bromide to produce lapachol (2) in 30% yield.123 Direct C-alkylation of 1 was employed for the synthesis of an important intermediate used for the preparation of compounds, which were found to be active against various cancer cell lines. The reactions of 1 with α-bromoacetate ethyl ester or allyl halide in DMF catalyzed by carbonate salts resulted in C-alkylated products 44 and 45a–b,124 respectively, in moderate yields. A mild procedure for the O-alkylation of 2-hydroxynaphthoquinones using phase-transfer catalysts was developed recently. In spite of being selective for the O-alkylation of 1, this procedure gave only 20% of the desired product.125 The examples presented above indicate that, in general, direct C-alkylation of 1 is not a good route to C-alkylated products. However, when the reactions of 1 with ferrocenemethanol derivatives are carried out in water, in the absence of a catalyst and at moderate temperatures, the alkylated products 46a–h are obtained in moderate to high yields. These naphthoquinones have shown in vitro antiplasmodial activity against chloroquine-sensitive and resistant Plasmodium falciparum strains.126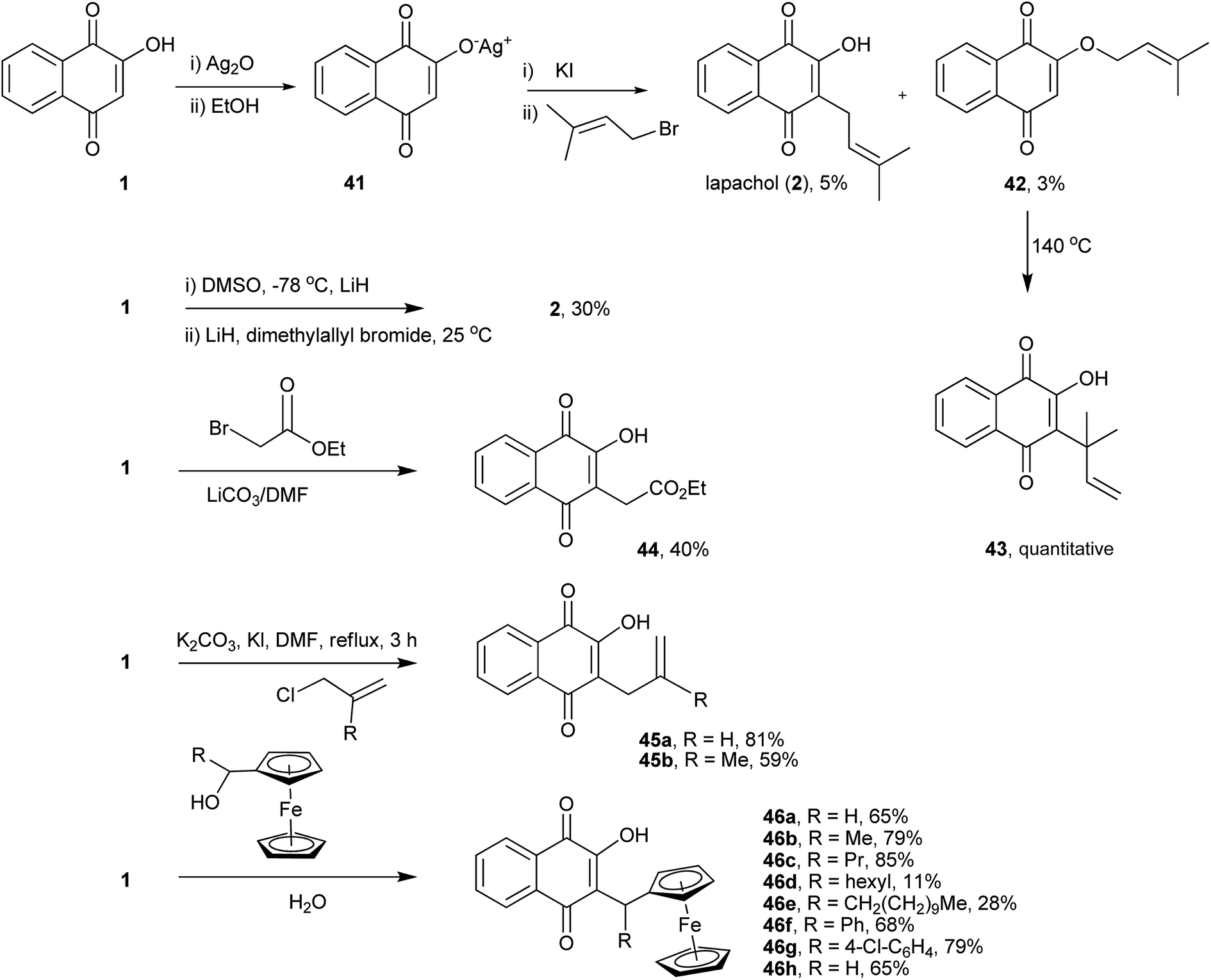 | ||
| Scheme 7 | ||
Other similar 1,4-naphthoquinones undergo C-alkylation with higher degrees of regioselectivity than 1. For example, recently Padrón and coworkers developed a synthetic route to 5-hydroxylapachol (47) through alkylation of 2-hydroxyjuglone (32) with 1-bromo-3-methyl-2-butene. The desired product 47 was formed in moderate yields with side products 48 and 49. Thus, the C-alkylation of (32) gave better results than the analogous reaction of 1 (Scheme 8).127
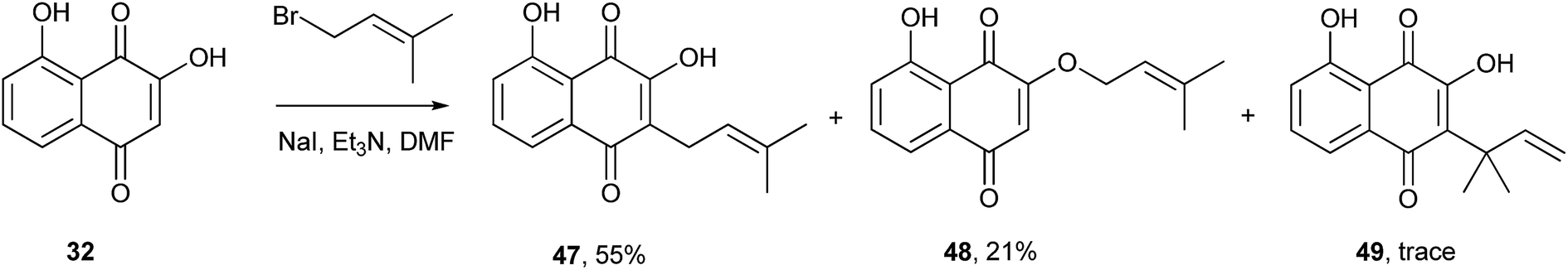 | ||
| Scheme 8 | ||
A very interesting copper-catalyzed regioselective C-alkylation of 1 was carried out with 3-chloro-3-methyl-1-butyne (Scheme 9), and this method was used for the preparation of the related natural isoprenylated naphthoquinone pigments streptocarpone (51) and (±)-α-dunnione (52). The major product of this one-pot formal [3 + 2] cyclization is the furanonaphthoquinone 50, which, upon acid treatment, is converted into streptocarpone (51) or, upon hydrogenation, into (±)-α-dunnione (52).128
 | ||
| Scheme 9 | ||
Bieber and co-workers129 developed a direct regioselective alkylation of 1 using trialkylboranes. The initially formed product, a reduced boranequinone complex, was oxidatively hydrolyzed to 3-alkyl-lawsones 53a and 53b, which are the only two reported examples (Scheme 10).
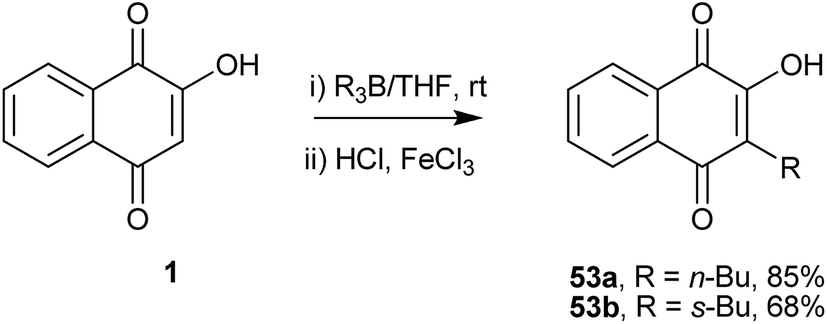 | ||
| Scheme 10 | ||
Ferreira and co-workers have developed a method for the C-alkylation of 1 by reacting it with various aldehydes in ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), forming in situ o-quinone methide (o-QM) intermediates that are immediately reduced by formic acid. Using this protocol, they prepared several 3-alkyl derivatives of 2-hydroxy-1,4-naphthoquinone 54a–e, including lapachol (2), in moderate to excellent yields. This one-pot reaction was used to prepare 54a, a commercial product known as phthiocol that possesses molluscicidal activity (Scheme 11).130 The same research group developed a new three-component process for the C-alkylation of 1 with simultaneous thiol addition. With this method, a series of new C-alkyl or C-arylsulfanylmethyl-[1,4]-naphthoquinones 55a–i were synthesized in good to high yields (Scheme 11). The reaction involved in situ generation of o-quinone methide intermediates (o-QM), which are formed via Knoevenagel condensation and react with thiols through 1,4-addition reactions. The compounds synthesized by this method showed moderate to good activity against the human malaria parasite Plasmodium falciparum (3D7)131 and Leishmania (L.) infantum.132 Very recently, Dmitrenok et al. used the same protocol to produce a series of N-acetyl-S-[(3-hydroxy-1,4-dioxo-1,4-dihydronaphthalen-2-yl)methyl]-L-cysteine conjugates in good yields. Based on the products of this reaction, fibrostatins B, C, and D were synthesized.133
:1), forming in situ o-quinone methide (o-QM) intermediates that are immediately reduced by formic acid. Using this protocol, they prepared several 3-alkyl derivatives of 2-hydroxy-1,4-naphthoquinone 54a–e, including lapachol (2), in moderate to excellent yields. This one-pot reaction was used to prepare 54a, a commercial product known as phthiocol that possesses molluscicidal activity (Scheme 11).130 The same research group developed a new three-component process for the C-alkylation of 1 with simultaneous thiol addition. With this method, a series of new C-alkyl or C-arylsulfanylmethyl-[1,4]-naphthoquinones 55a–i were synthesized in good to high yields (Scheme 11). The reaction involved in situ generation of o-quinone methide intermediates (o-QM), which are formed via Knoevenagel condensation and react with thiols through 1,4-addition reactions. The compounds synthesized by this method showed moderate to good activity against the human malaria parasite Plasmodium falciparum (3D7)131 and Leishmania (L.) infantum.132 Very recently, Dmitrenok et al. used the same protocol to produce a series of N-acetyl-S-[(3-hydroxy-1,4-dioxo-1,4-dihydronaphthalen-2-yl)methyl]-L-cysteine conjugates in good yields. Based on the products of this reaction, fibrostatins B, C, and D were synthesized.133
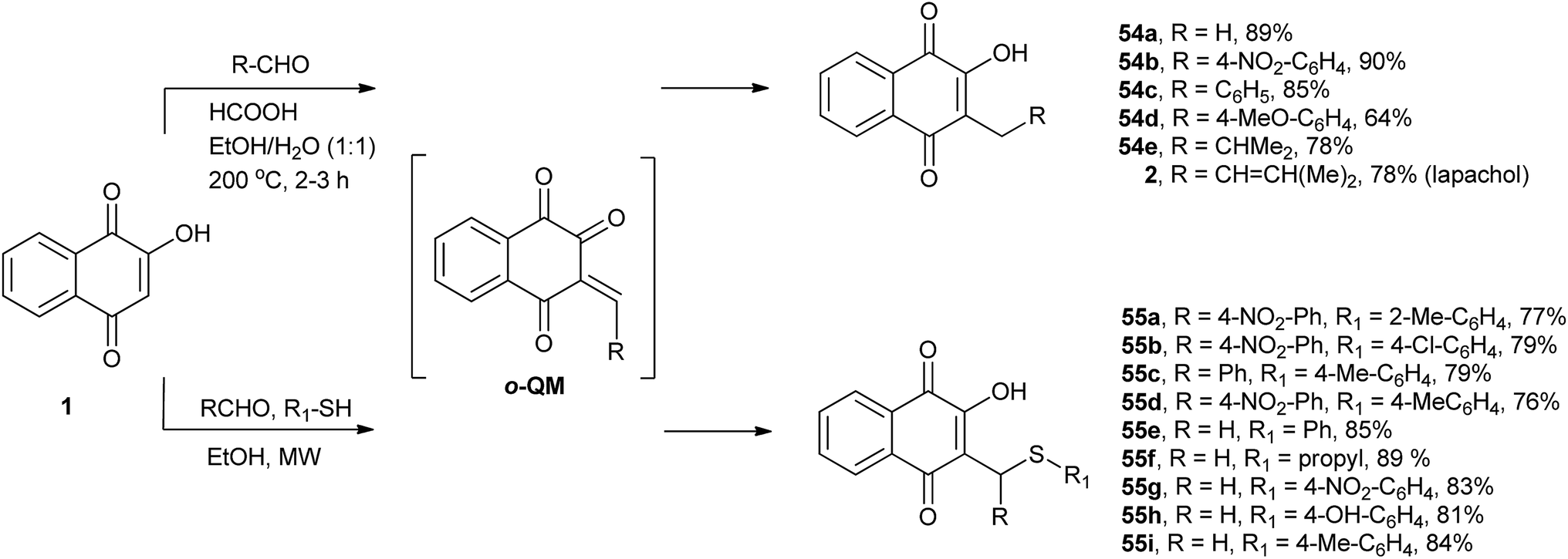 | ||
| Scheme 11 | ||
Alexandre-Moreira and co-workers134 have studied the synthesis of a series of 3,3′-(R-methylene)bis-2-hydroxy-1,4-naphthoquinone derivatives 57a–h that were prepared in high yields through the condensation of 1 with aromatic and aliphatic aldehydes 56a–h under mild acidic conditions (β-alanine/AcOH) (Scheme 12). The compounds were evaluated in vitro against Leishmania amazonensis and Leishmania braziliensis promastigotes. Some compounds showed good activity without significant toxic effects, and the compound with the highest activity (57a) was used for an in vivo assay with Leishmania amazonensis.
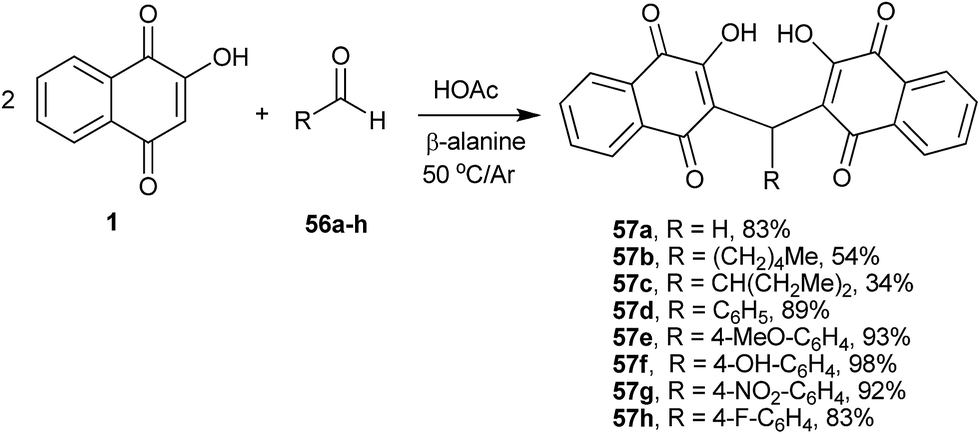 | ||
| Scheme 12 | ||
The Mannich reaction is a very useful one-pot C-aminoalkylation adjacent to a carbonyl functional group. The final product is a β-amino-carbonyl compound that is also known as a Mannich base. In this reaction, nucleophilic addition of an amine to a carbonyl group, followed by dehydration, forms a Schiff base that reacts in a second step with nucleophiles, such as enol or enone derivatives, to form an amino C-alkylated adduct (Scheme 13). This easy-to-perform three-component reaction of 1 (an enone) with aldehydes and amines produces the amino C-alkylated adduct in high yields and high atom economy. This reaction is therefore ideal for the preparation of a library of compounds for biological screening.
 | ||
| Scheme 13 | ||
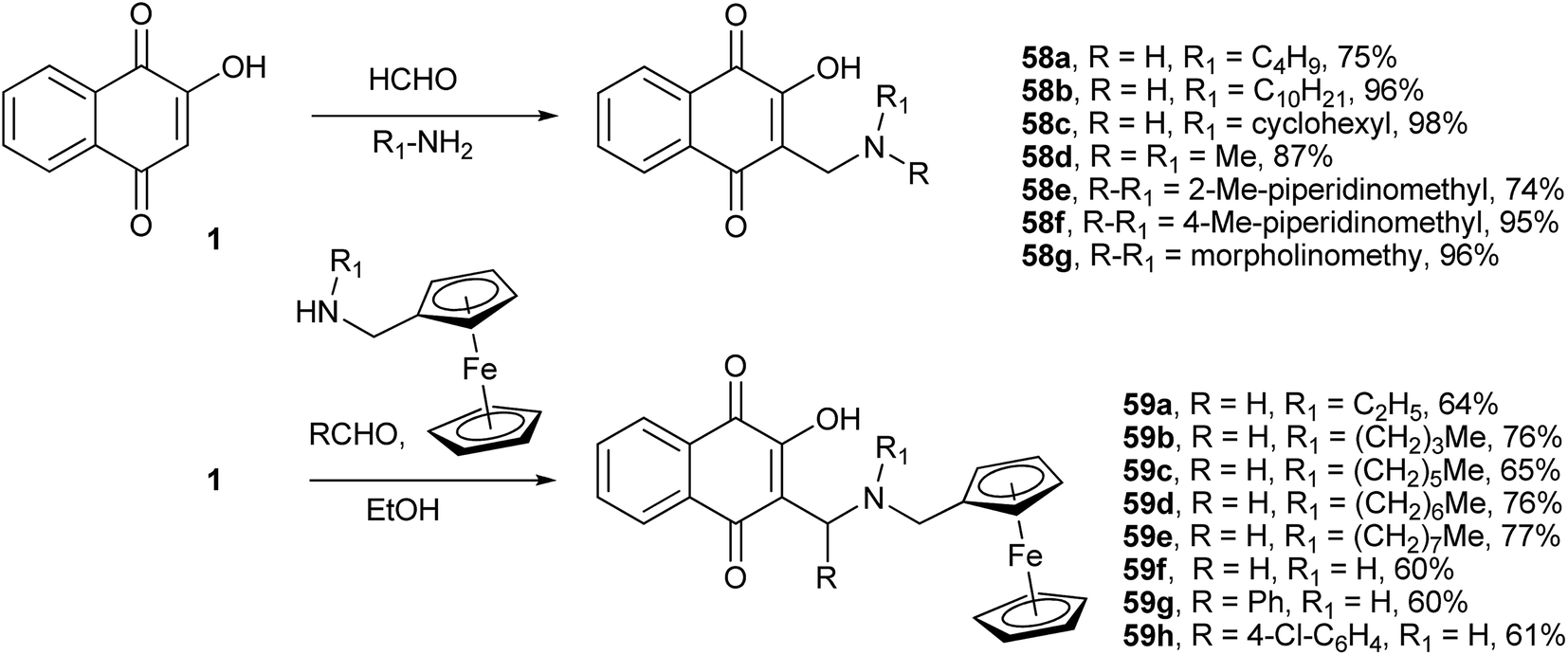
During their search for 3-alkyl-1,4-naphthoquinones with antimalarial activity, Leffler and Hathaway were the first to investigate this reaction.135 By using 1, formaldehyde and several amines, they produced a series of compounds 58a–g containing an aminomethyl side chain in high yields (Scheme 13 for R1 = H). Afterward, Dalgliesh reinvestigated this reaction and produced other Mannich bases derived from 1.136 He also proposed a mechanism to explain the formation of 3,3′-(R-methylene)bis-2-hydroxy-1,4-naphthoquinones (Scheme 13) from the reactions with bulky secondary amines instead of the Mannich bases that are obtained in excellent yields from the reactions with primary amines. Various 2-hydroxy-3-alkyl-1,4-naphthoquinones thus synthesized showed significant molluscicidal activity against the Biomphalaria glabrata snail, which is the intermediate host of Schistosoma mansoni.137 Following the same methodology, Brocard and co-workers synthesized a number of Mannich bases of 1 from ferrocenylmethylamines. The ferrocenyl aminohydroxynaphthoquinones 59a–g containing aliphatic chains with 6–8 carbon atoms were found to be significantly active against the atovaquone (3)-resistant strain of Toxoplasma gondii.138
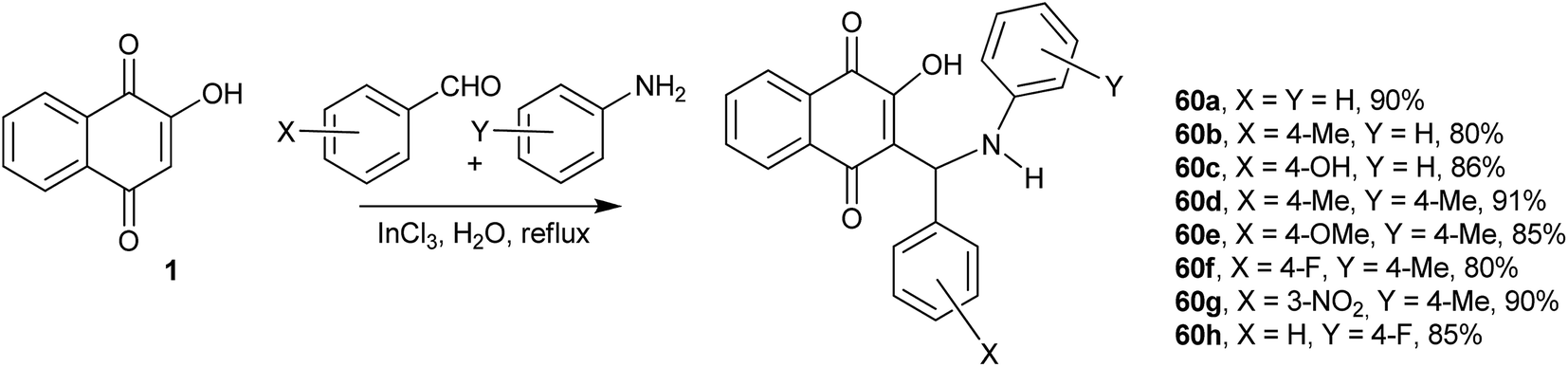
Recently, Greco and co-workers showed that the one-pot, three-component reactions of 1 with aromatic aldehydes and the secondary amine pyrrrolidine or 4-nitroaniline occur in the presence of catalytic amounts of p-TsOH at room temperature to give the respective Mannich bases in good yields.139 Bazgir and coworkers investigated the synthesis of Mannich bases derived from 1, aromatic aldehydes and heterocyclic or carbocyclic amines to form fluorescent hydroxyl naphthalene-1,4-dione derivatives 60a–h. They used catalytic amounts of InCl3 in refluxing water, which makes this process very simple, green and highly atom-economic. The authors speculated that the Lewis acid interacts with the nitrogen of the Schiff base intermediate, increasing its electrophilicity. The reaction works very well for aromatic aldehydes containing both electron-withdrawing and electron-donating substituents as well as with heterocyclic amines. The products were fluorescent in solution and emitted in the green part of the spectrum (546–560 nm) (Scheme 14).140 The use of environmentally friendly ionic liquids as solvents in the condensation reactions of 1 with aromatic aldehydes and aromatic amines was also investigated, and similar Mannich bases were obtained in high yields.141
 | ||
| Scheme 14 | ||
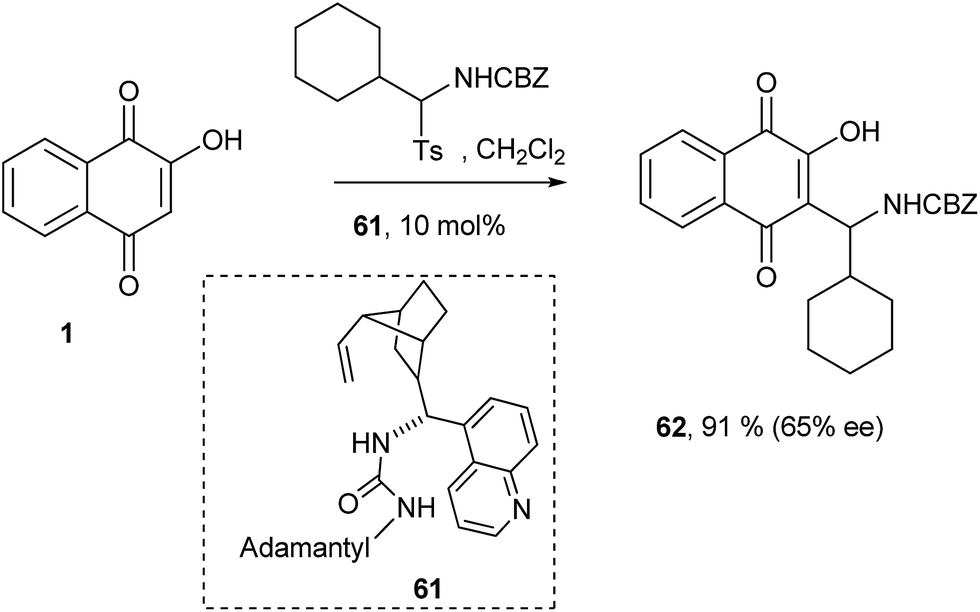
Ayaz and Westermann also have attempted the nucleophilic addition of 1 to imines under organocatalytic conditions. In this particular case, several commonly used organocatalysts were screened to give Mannich bases in excellent yields but with moderate enantioselectivities. The use of catalyst 61, an analogue of Takemoto's catalyst, gave the best enantioselectivity for compound 62 (Scheme 15).142
 | ||
| Scheme 15 | ||
The 1,4-naphthoquinone Mannich bases derived from 1 can coordinate to several metal ions, thus potentially generating new molecules with improved biological activities.143 Within this context, Neves and coworkers were the first research group to report metal complexes of Mannich bases derived from 1, benzaldehydes or pyridinecarboxaldehyde, and a variety of amines. The copper(II) complexes 63, for example,144 (Scheme 16) were easily prepared but showed less antimicrobial activity than the free Mannich bases, which was attributed to their low solubility. The same group demonstrated that the coordinating ability of the Mannich bases can be controlled by judicious choice of the amine and the aldehyde.145 Thus, from the bases derived from aminomethyl pyridine, and by slight variation of the complexation reaction conditions, different dinuclear copper(II) complexes were synthesized.146 Furthermore, they also synthesized a series of platinum(IV)147 and platinum(II)148 complexes of the Mannich bases derived from pyridinecarboxaldehyde, the latter of which was structurally related to picoplatin, and evaluated their cytotoxicity against various cancer cell lines149 and studied their mechanism of action.150 Complex 66, for instance, is responsible for the formation of mono-adducts with 5′-GMP, suggesting that it can interact similarly with DNA (Fig. 3).
 | ||
| Scheme 16 | ||
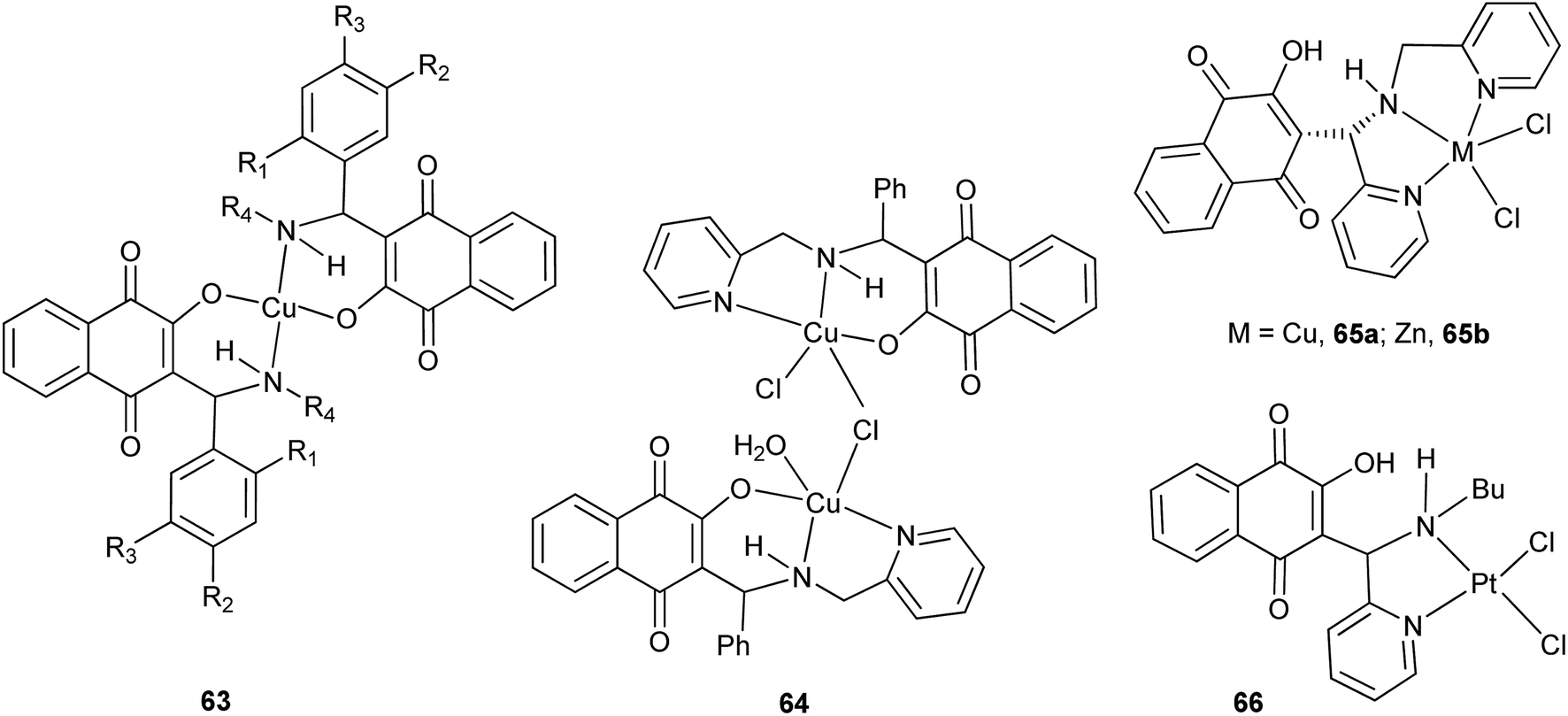 | ||
| Fig. 3 Examples of metal complexes formed from Mannich bases derived from 1. | ||
Pinto and co-authors151 evaluated the antiviral potential of several of the Mannich bases described above against bovine herpesvirus type 5 (BoHV-5), the etiologic agent of meningoencephalitis in cattle. The most potent compound of the 67 series (Fig. 4) is a selective inhibitor of this virus and exhibits better CC50 and EC50 values than acyclovir.
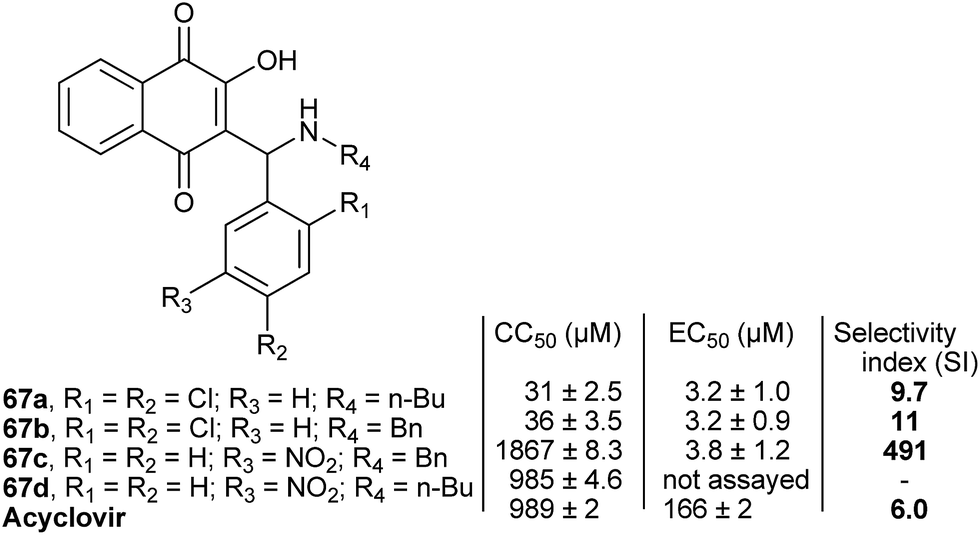 | ||
| Fig. 4 Antiviral evaluation of selected aminomethylnaphthoquinones against BoHV-5. | ||
3.2 C-alkylation via the Michael addition reaction
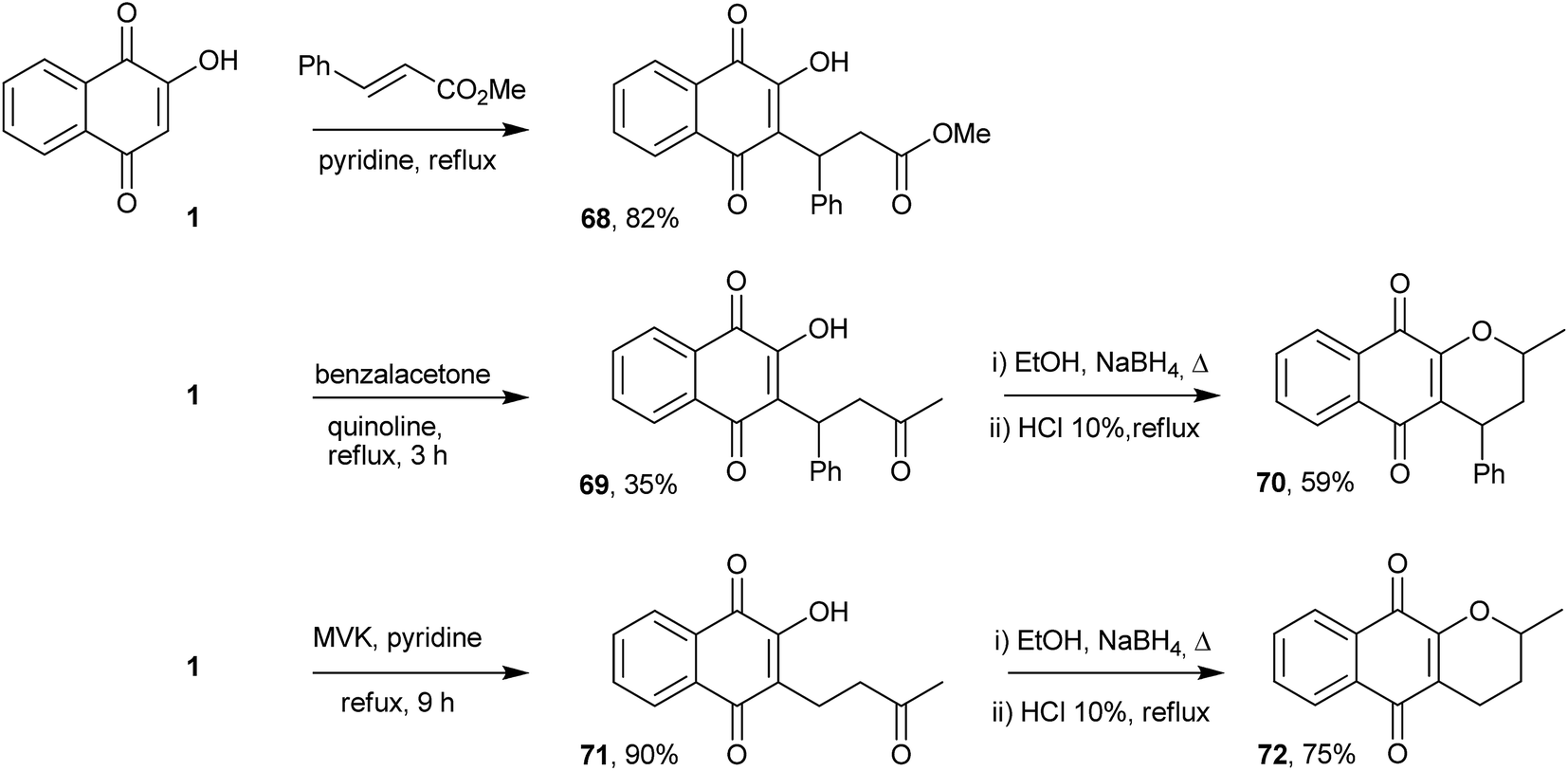
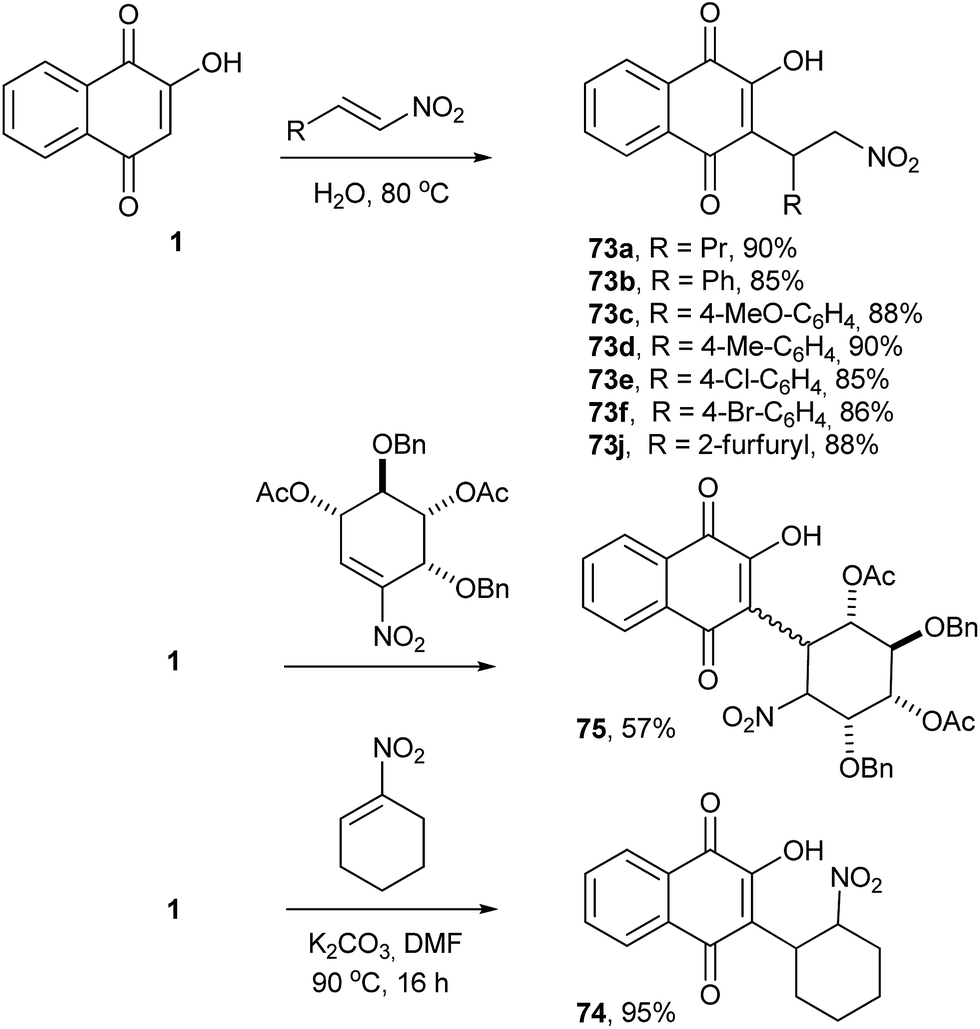
The Michael addition reaction represents one of the most important and general methods for the construction of a carbon–carbon bonds in organic synthesis. Thus, direct alkylation of 1 by Michael addition to α,β-unsaturated carbonyl compounds occurs selectively at position 3 and was initially explored by Zaugg for the production of pyranonaphthoquinones (see compound 50 in Scheme 9).152 Tapia and co-workers used this strategy to perform the C-alkylation of 1 and transform the Michael adducts into pyranonaphthoquinones with potential biological activities. The Michael adducts 69 and 71 were reduced with sodium borohydride and cyclized under acid conditions to form the pyranaphthoquinones 70 and 72 in 64–75% yields (Scheme 16).153,154Nitroolefins are very versatile building blocks in organic synthesis due to their ready availability and ease of transformation into a wide variety of functionalities.155,156 C-Alkylations through Michael additions of 1 to various nitroolefins were easily performed in good yields under aqueous conditions.157 The mechanism of these reactions can be explained on the basis of dual activation of the nitroalkene and 1 via hydrogen bonding. C-Alkylation of 2-hydroxy-1,4-naphthoquinone (1) was controlled by electronic factors. This reaction has attractive features, such as operational simplicity, high product yields (85–90%), high atom economy, versatility with respect to the β-nitrostyrenes and being environmentally benign (Scheme 17). Recently, Otero and coworkers reported the Michael addition of 1 to nitrocyclohexene, producing 74 in 95% yield, and to a chiral nitrocyclohexene, producing a diastereoisomeric mixture of 75 in moderate yields (Scheme 17).158
 | ||
| Scheme 17 | ||
Organocatalysis has been applied to various asymmetric reactions. This methodology is ideal for the C-alkylation of 1 (described above in Schemes 16 and 17) because it generates a chiral center. Thus, enantioselective Michael addition of 1 to nitroolefins was first studied by Du and co-workers who reported good yields of the products 77 with excellent enantioselectivities (up to >99% ee) and high atom economy. This first report of an organocatalytic asymmetric Michael addition of 1 showed that it is possible to produce these optically active compounds and that the presence of electron-rich substituents on the 7-position of the 2-hydroxy-1,4-naphthoquinone does not affect the efficiency of the reaction (Scheme 18).159
 | ||
| Scheme 18 | ||
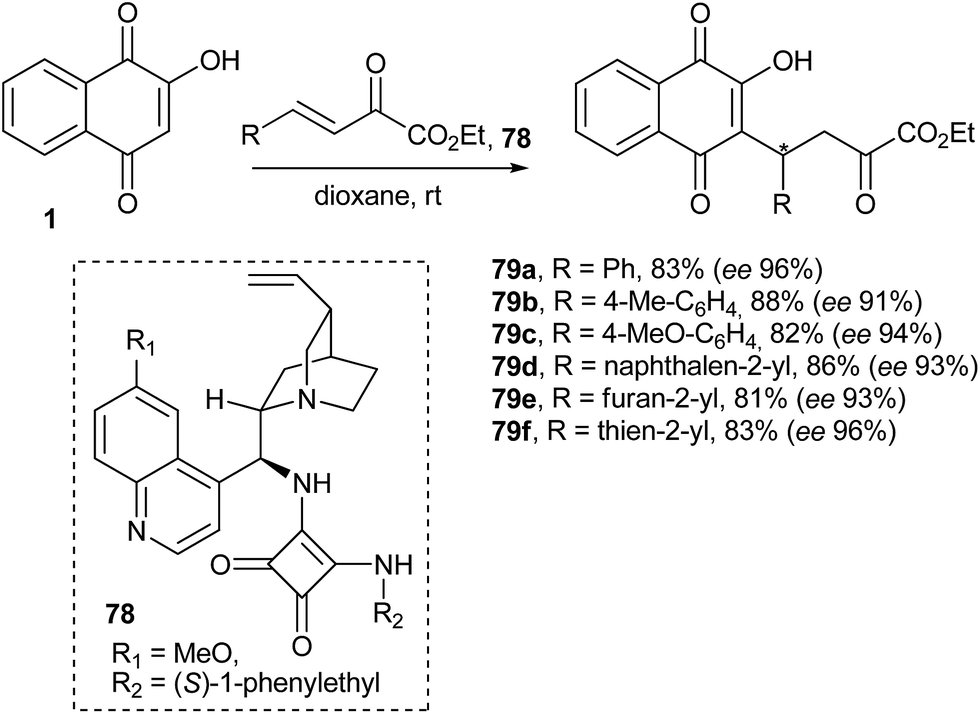
Wang and co-workers160 have studied the analogous reaction with β,γ-unsaturated α-oxo esters and bifunctional chiral amine-derived squaramides (78) as catalysts that function through a hydrogen-bond mediated activation mechanism, and they obtained the chiral adducts in high yields and excellent enantioselectivities under mild conditions. This organocatalytic asymmetric Michael addition provides an efficient route to optically active functionalized naphthoquinones 79a–f (Scheme 19).
 | ||
| Scheme 19 | ||
Tang and co-workers developed an organocatalytic Michael addition process to synthesize some novel optically active β,γ-unsaturated α-derivatives of 1 by employing a cinchonine-based thiourea catalyst (Scheme 20). This process produced the desired ketophosphonate adducts of 1 with high enantioselectivity. The chiral ketophosphonates were then transformed into the corresponding β-substituted carboxylates 81a–j in excellent yields and with high levels of enantioselectivities upon treatment with DBU in MeOH.161
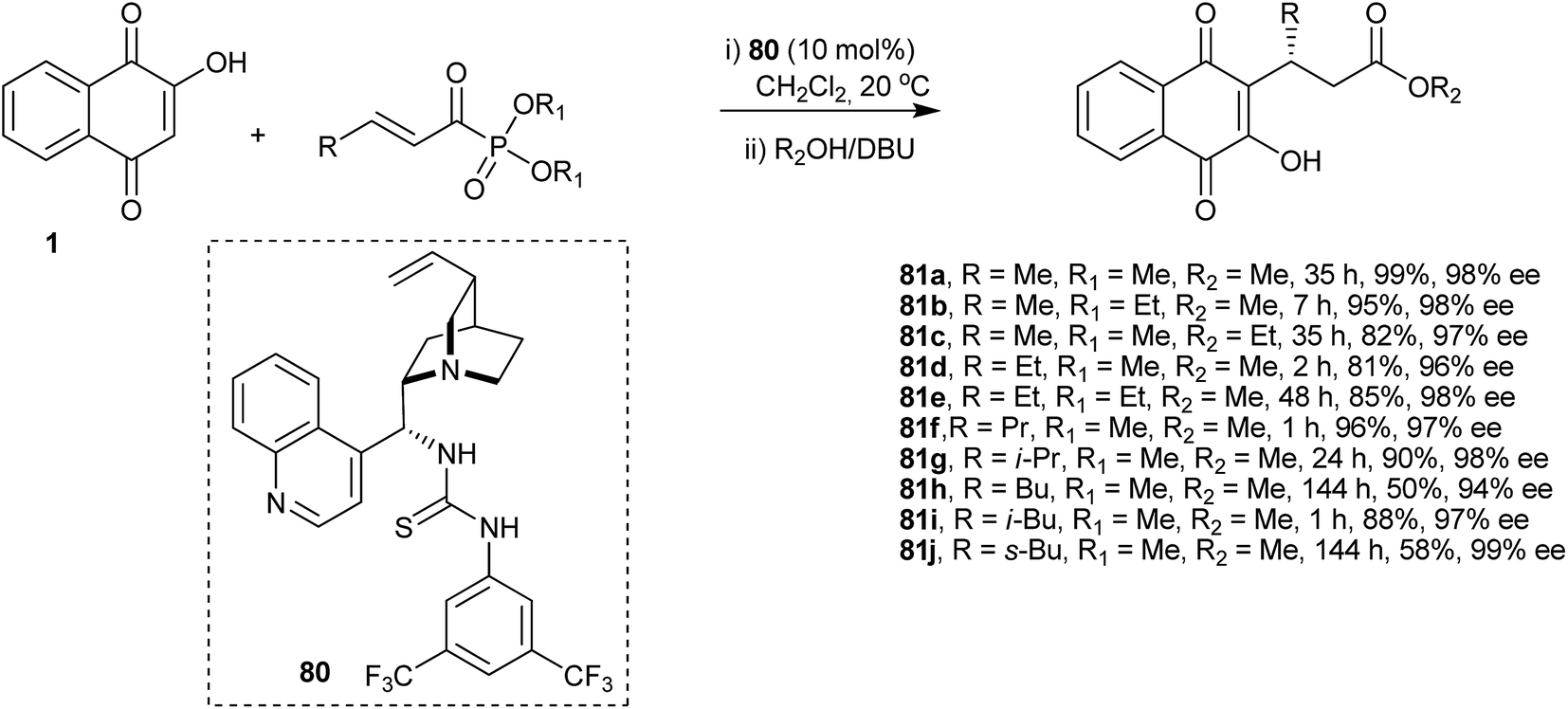 | ||
| Scheme 20 | ||
3.3 Dimerization of lawsone (1)
In the 1890s, Hooker and co-workers162 noted that aqueous solutions of 1 deposited the dimeric compound 2,2′-bis(3-hydroxy-1,4-naphthoquinone) 82 when left exposed for long periods of time. Later, they showed that this compound could be prepared in yields over 40% by exposing a hot aqueous solution of 1 to ultraviolet light (Scheme 21). This reaction was reinvestigated by Chandrasenan and Thomson,163 who speculated that the dimerization process occurred via a radical coupling reaction and, therefore, carried out the persulfate oxidation of 1, producing 82 in ca. 60% yield. In this case, only the coupling at C-3 was observed. This reaction is also promoted by ammonium metavanadate in the presence of diluted perchloric acid164 as well as by FeCl3; however, the principal products in the latter case are the iron complexes of the dimer.165 Recently Lanznaster and coworkers reported the dimerization of 1 upon reaction with Co(BF4)2·6H2O and N,N′-bis(pyridin-2-ylmethyl)ethylenediamine (py2en) in the presence of Et3N. The product from this reaction is the metal complex 83 that contains the deprotonated form of 84, (bhnq),166 and dissociation of this dianion was promoted by reduction with ascorbic acid. Coordination of two lawsonate molecules to Co(II) was shown to be a necessary step prior to dimerization.167 The anticancer168 and antimycobacterial169 activities among others of 82 have been investigated, but the main interest lies in the complexing capability of its dianion, a flexible hingelike ligand that has been used for the construction of metal organic frameworks (Scheme 21).170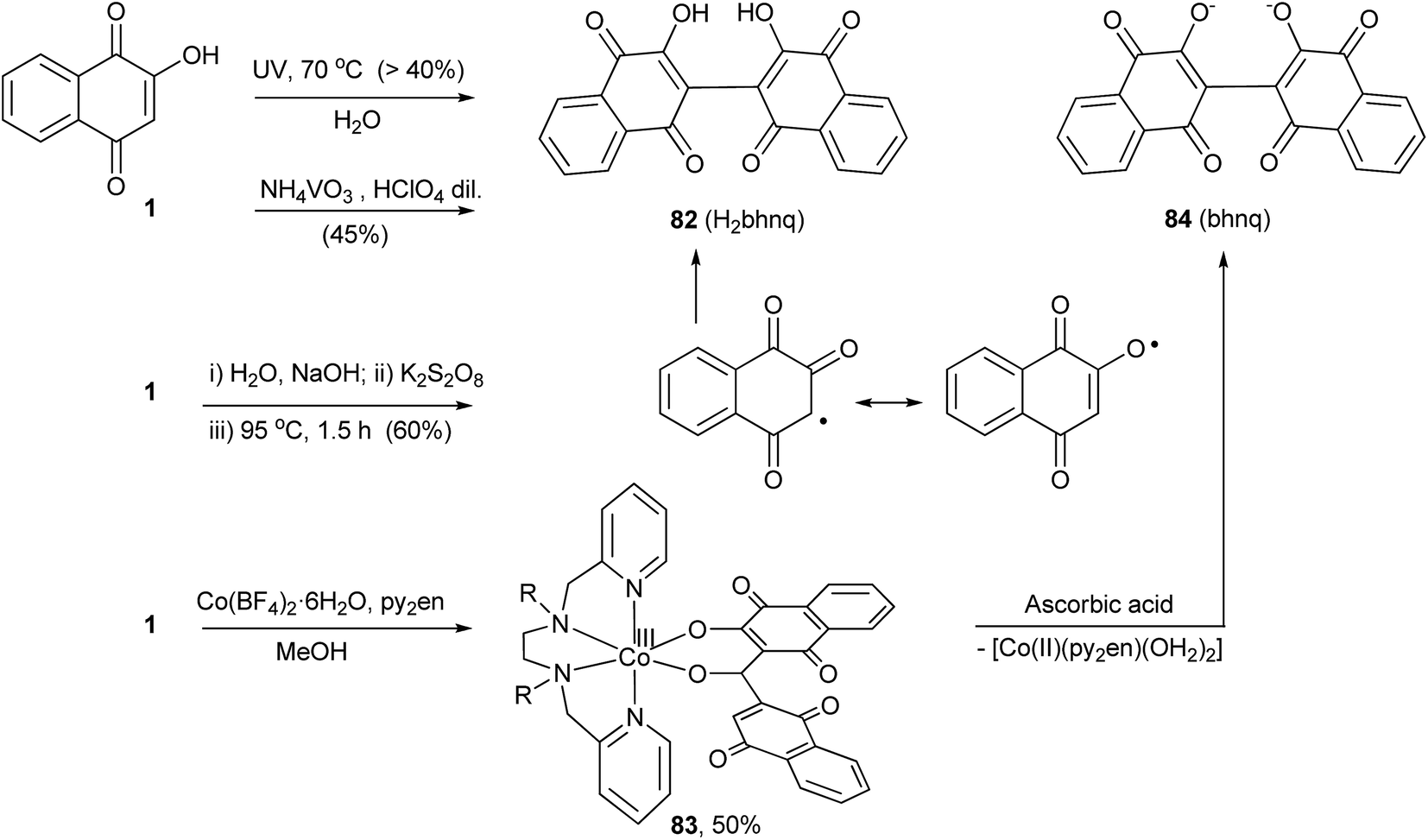 | ||
| Scheme 21 | ||
3.4 Free radical alkylation of lawsone (1)
Free radical reactions have proved to be especially useful for numerous cascade or domino sequences because they offer many advantages when compared with ionic or organometallic reactions.171,172 The reaction of 1 with radical species was one of the first radical methods employed to obtain C-alkylated lawsone derivatives. The method, developed by Fieser and co-workers in 1945, involved the alkylation of hydroxynaphthoquinones by heating them with diacyl peroxide (Scheme 22). This general one-step alkylation process gives moderate to good yields and pure products that can be readily isolated from the reaction mixture by acid–base extraction. The diacyl peroxides used in this reaction can be obtained from the reaction of a carboxylic acid with an acyl chloride. The yields of the alkylation process are generally in the range of 30–60%, with some sharp decreases in yield with high-molecular-weight compounds. One limitation of this peroxide alkylation reaction is poor yields (1–20%) when using peroxides obtained from α-branched acids and cycloalkane carboxylic acids (Scheme 22).173,174 Jacobsen and Torssell improved this reaction by replacing diacyl peroxide for carboxylic acids in a radical process generated by decarboxylation with ammonium peroxodisulfate and catalytic amounts of silver nitrate.175,176 The reaction is widely applicable to the synthesis of 2-C-alkylated-2-hydroxynaphthoquinones and 1,4-quinones (Scheme 22).177,178 In this regard, Liu and co-workers synthesized several plastoquinone derivatives with ω-substituted alkyl side chains,179 and Tauraité synthesized lipophilic 1,4-naphthoquinone derivatives with redox properties.180 Boehm and co-workers have also used this procedure to prepare a series of naphthoquinones that were evaluated in vitro against the causative agent of the cattle disease East Coast Fever or Theileria parva infection.181 Recently, the same methodology was used for the synthesis of novel naphthoquinones from lipids, which were assayed against the infective bloodstream form of Tripanosoma cruzi (Scheme 22). Among the compounds, 91e exhibited excellent ability to inhibit the growth of the microorganism with an IC50 of 7.8 μM.182 It is worth noting that the use of acylated lawsone gives better yields of C-alkylated products by peroxysulfate-mediated radical decarboxylation.121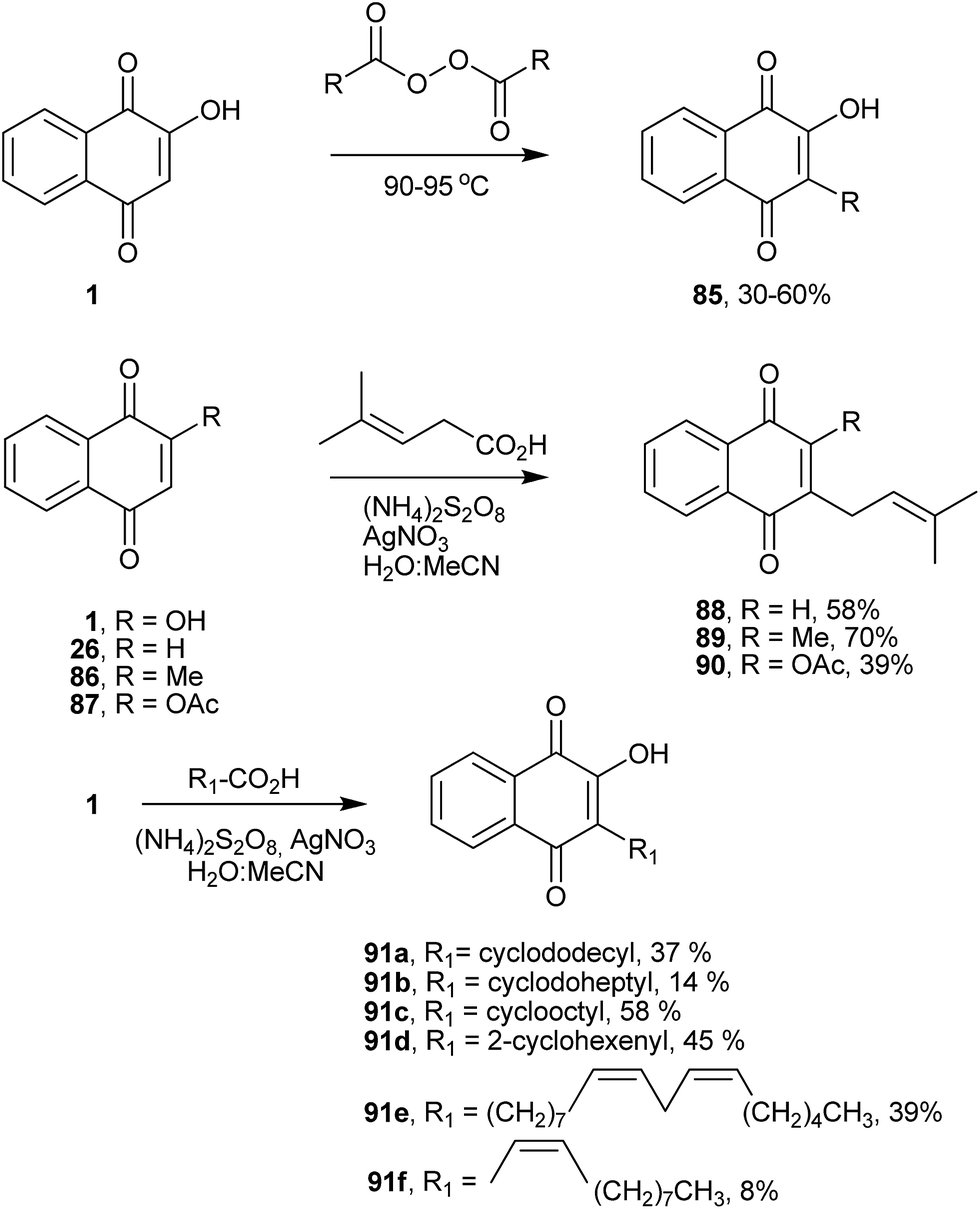 | ||
| Scheme 22 | ||
3.5 C-Alkylated derivatives of 1 by the Hooker oxidation method
Lapachol (2) stands out among the natural naphthoquinones and may be considered one of the main representatives of the group of natural quinones. First isolated from Tabebuia avellanedae (Bignoniaceae) by E. Paternò, in 1882,183 it was later isolated also from other plants, e.g., of the Verbenaceae and Proteaceae families. Paternò also proposed a structure for it, but with the double bond of the side chain in the wrong position.184,185 Hooker assigned its structure correctly and also made several derivatives of this molecule between 1889–1896.186,187 When studying the degradation reactions of lapachol (2) with potassium permanganate in alkaline medium, he found a remarkable degradation reaction, which was of general scope and could transform 3-alkyl-2-hydroxy-1,4-naphthoquinones into other 3-alkyl-2-hydroxy-1,4-naphthoquinones with one less CH2 in the side chain.185,188 This reaction has been used for the synthesis of a number of new naphthoquinones.189–192 Fieser wrote the following in his memoir published in 1964: “A second reaction which proved to be extremely useful is a remarkable one discovered by Dr Hooker in retirement and known as the Hooker oxidation. On reaction of M-1926 (R = (CH2)9CH3 in Scheme 23) with alkaline potassium permanganate in the cold, a CH2 unit mysteriously disappear from the side chain”.193 Some examples of this methodology are illustrated in Scheme 23. Hooker's proposed mechanism for this reaction was that oxidation would occur by cleavage of the 3-alkyl-2-hydroxy-naphthoquinone 92 ring by the permanganate ion, followed by cyclization with release of one CO2 molecule, and subsequent tautomeric equilibrium, resulting in the lower homologue of the 3-alkyl-2-hydroxy-1,4-naphthoquinone 93.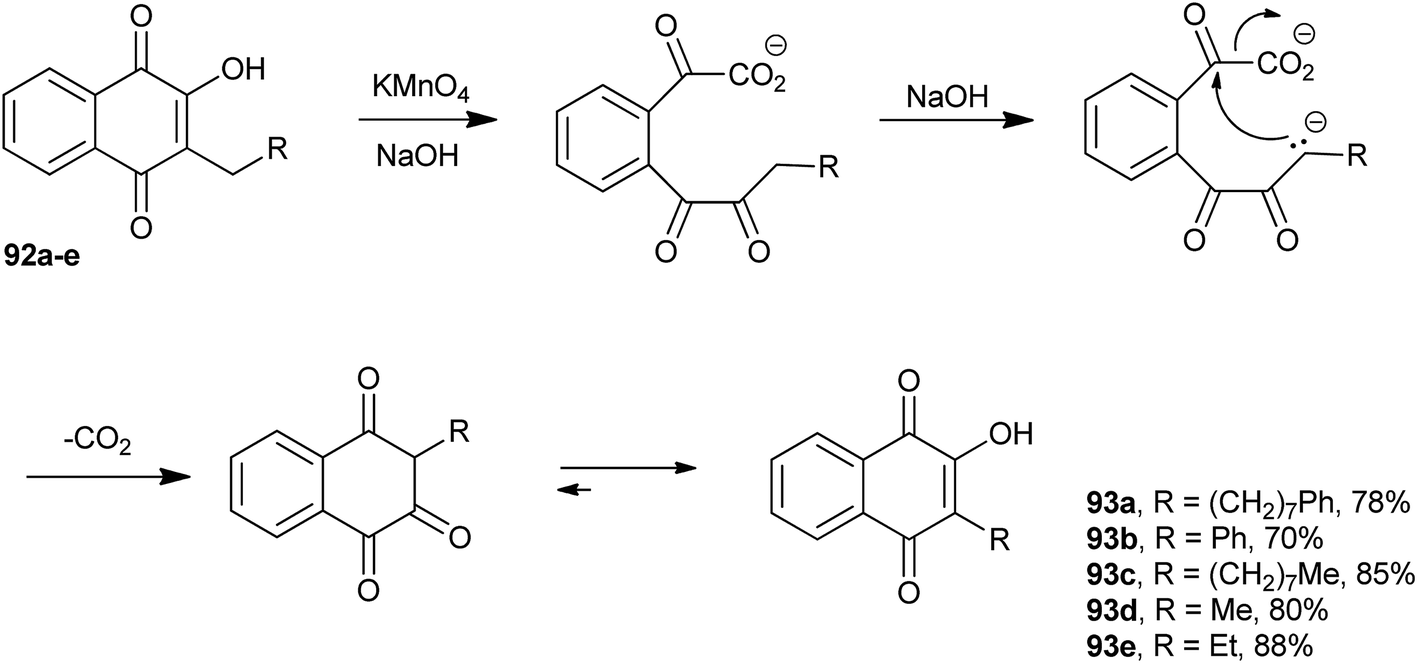 | ||
| Scheme 23 | ||
The Hooker oxidation works very well in the 1–2 g scale, but reaction with larger quantities results in the formation of undesirable overoxidation products. Fieser et al. solved this problem by developing a two-step process that used peroxide and copper sulfate and gave the desired products in 85–90% yields. By treating the appropriate 2-hydroxy-3-alkyl-1,4-naphthoquinone 94 with hydrogen peroxide in a dioxane-soda solution, he obtained the colorless acid known as Hooker's intermediate 95 (ref. 194) that was converted by the action of copper sulfate and alkali into the corresponding 2-hydroxyl-3-nor-alkylnaphthoquinone 97 in very high overall yields.195 By using this modified Hooker oxidation methodology, Cram synthesized a series of lipophilic naphthoquinones in high yields.196 Fieser proposed a structure for the Hooker's intermediate obtained from lapachol (2) (1,2-dihydroxy-2-(3-methylbut-2-enyl)-3-oxo-2,3-dihydro-1H-indene-1-carboxylic acid, 95), which was later confirmed by 13C-labeling studies197 and, recently, by an X-ray diffraction analysis (Scheme 24).198 In a recent study, however, evidence was presented for the formation of another intermediate (96) before the classical Hooker's intermediate (Scheme 24).199
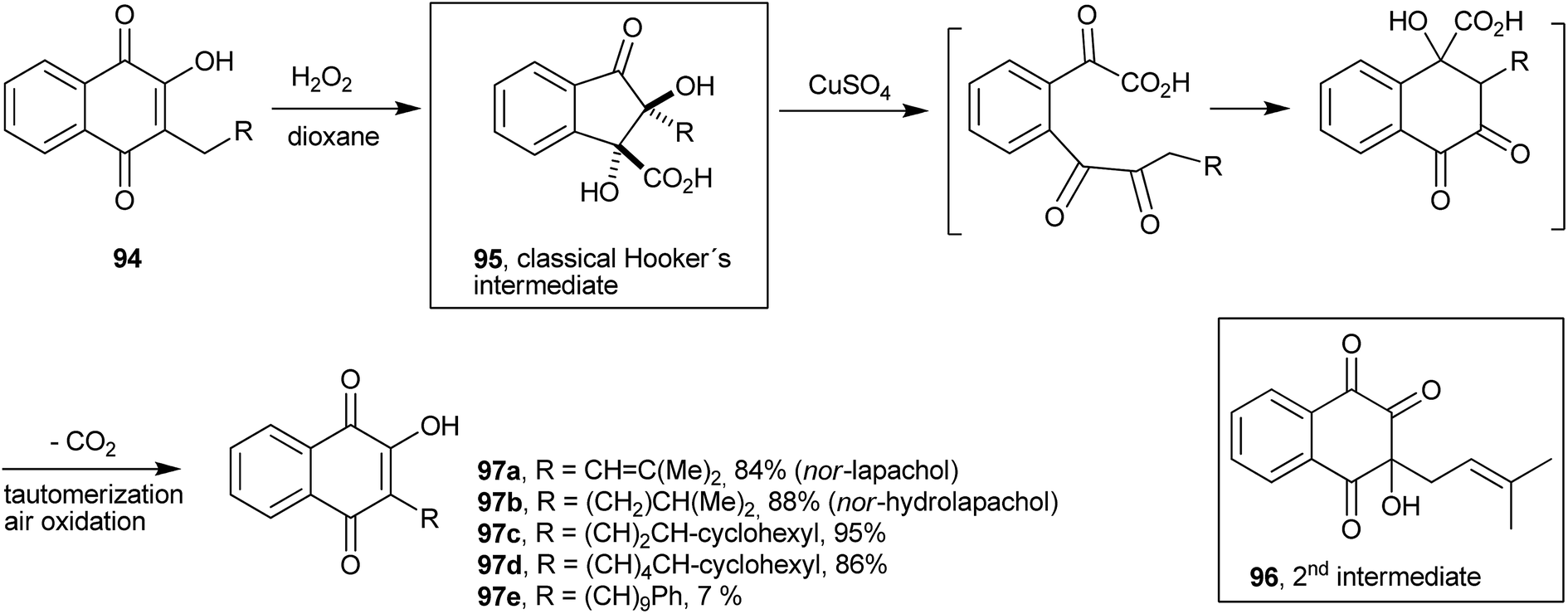 | ||
| Scheme 24 | ||
Schwalbe et al. have synthesized various 2-hydroxy-3-alkyl-1,4-naphthoquinones containing a variety of alkyl side chains, some of which were obtained using the Hooker oxidation method. The compounds were tested as inhibitors against the membrane-embedded protein quinol/fumarate reductase (QFR) from Wolinella succinogenes, which is a target related to QFRs from the human pathogens Helicobacter pylori and Campylobacter jejuni. Several naphthoquinones showed IC50 values in the low micromolar range. Systematic elongation of the alkyl side chain in the hydroxyl series resulted in increased inhibition. Furthermore, the presence of the hydroxyl group was proved crucial for the activity.200
3.6 One further step in the chemistry of lawsone (1)
As discussed above, the C-3 position of 1, next to the hydroxy group, is very reactive toward electrophiles. Thus, the reactions with, e.g., halogens, nitro derivatives201 and diazonium salts202 are easily performed in one step and yield important derivatives of 1 that can be used as starting reagents for the synthesis of other compounds (Scheme 25). For example, a series of 3-hydroxy-2-hydrazino-1,4-naphthoquinones 99 were synthesized from the nitroso derivative (97) by reduction with NaBH4/MeOH, followed by diazotization of the resulting 3-amine-2-hydroxy-1,4-naphthoquinone with sodium nitrite in hydrochloric acid, and then by reaction of the 3-diazo-naphthalene-1,2,4-trione (98) with 1,3-dicarbonyl enolates in the presence of K2CO3 in acetone. Overall yields of 99 from 1 varied from 16 to 20%. From the reactions of 1 with diazonium salts, various 3-(2-R-phenylhydrazono)-naphthalene-1,2,4-triones 100a–n were isolated in good yields. Their copper(II) complexes 101 showed improved antimalarial activity203 and anticancer properties204 compared to the free ligands.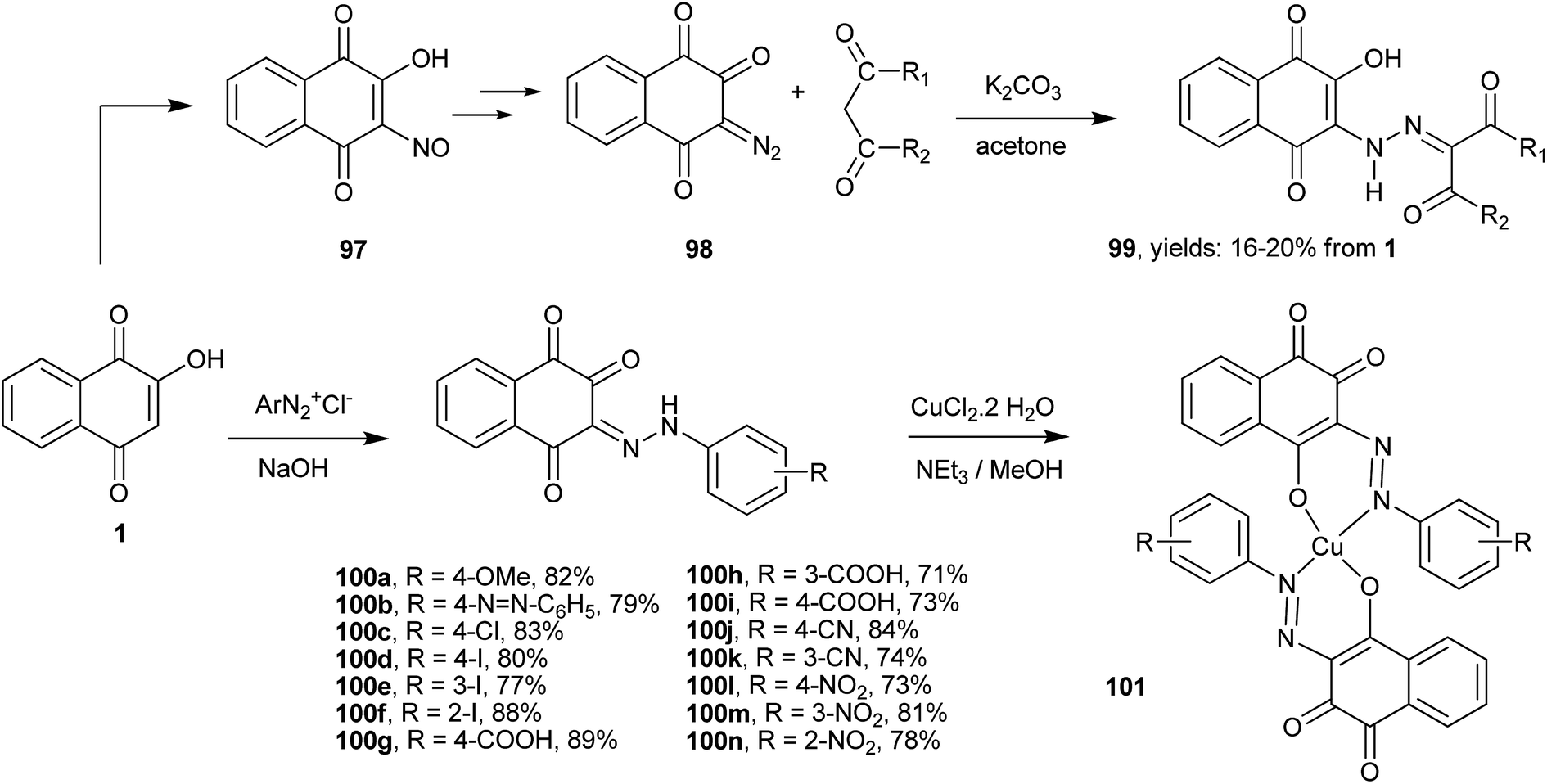 | ||
| Scheme 25 | ||
In this part of the review, emphasis will be given to useful compounds synthesized from the reactions of 1 with electrophiles and their further reactions to produce novel derivatives.
Before the discovery of cross coupling reactions, C(sp2)–C(sp2) coupling at position 3 of 1 was achieved through the condensation with aldehydes. Indeed, the Knoevenagel condensation of 1 with aldehydes forms the reactive o-quinone methides that, depending upon the experimental conditions and the structure of the aldehyde, can follow several reaction pathways. Fieser117 and Hooker205 used this reaction to produce alkenyl derivatives, as illustrated in Scheme 26 (compounds 102a–e). For instance, the reaction of 1 with isovaleraldehyde resulted in the formation of isolapachol (105). In a subsequent step, hydrogenation of the newly formed double bond of the side chain can lead to C-alkylated compounds (103a–e). These two reactions are generally applicable to the preparation of derivatives of 1 containing the corresponding C-alkenyl and C-alkyl groups at the quinone ring next to the hydroxyl group. Giles et al. also used this reaction for the preparation of the alkenyl derivative (105) as the key step in the total synthesis of dihydro-2H-benzo[g]chromene-5,10-dione via condensation of naphthoquinone (104) with 3-methyl butanal in good yield.206 In search of compounds with pesticidal properties, Jacobsen et al. prepared several molecules similar to the series 102a–e and 105 in Scheme 26, but using triethylamine to catalyze the condensation of 1 with aldehydes. These reaction conditions gave much poorer yields of the compounds.207
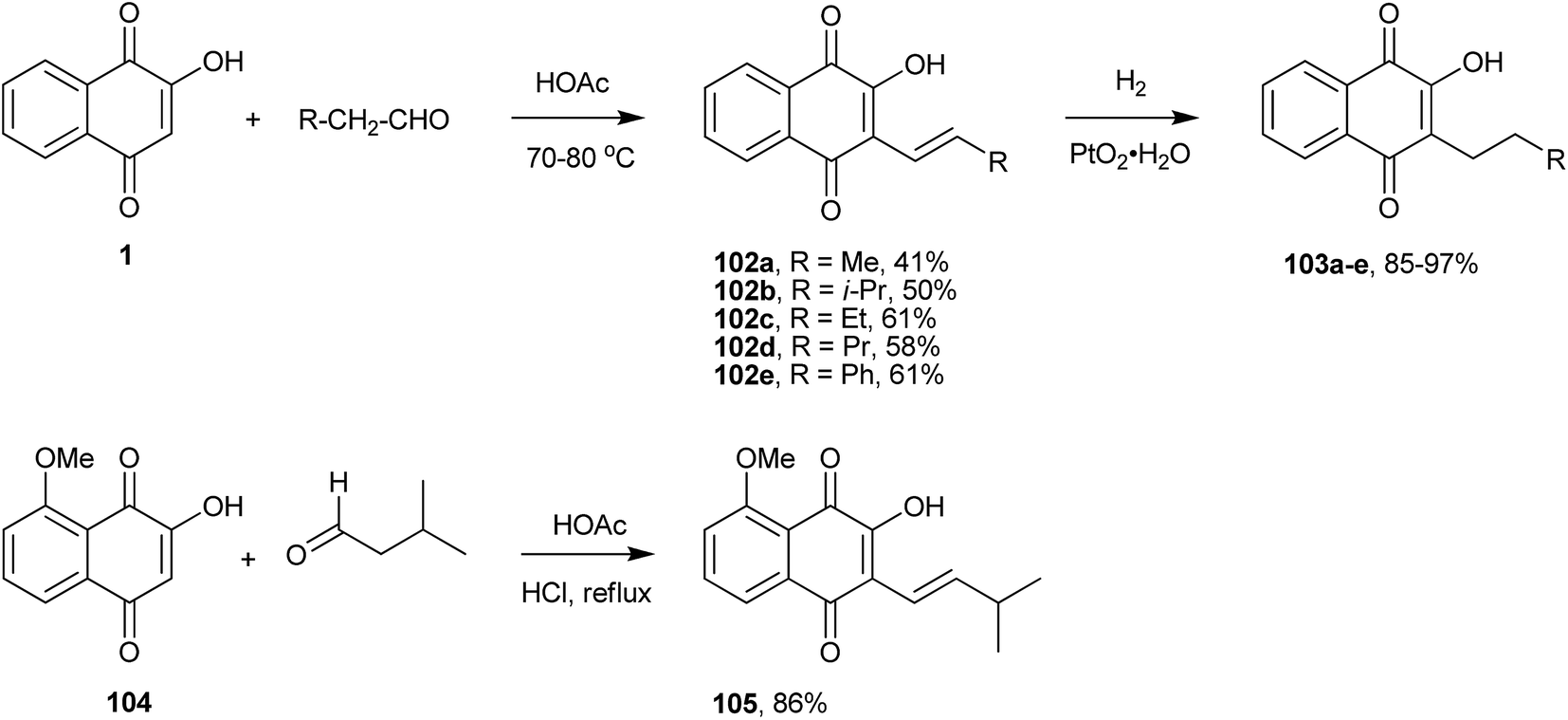 | ||
| Scheme 26 | ||
Advances in cross coupling reactions have made formation of C(sp2)–C(sp2) bonds involving 1 easier and more accessible than they have been in the past. The zwitterionic iodonium 3-phenyliodonium ylide (106) is easily obtained from 1 upon reaction with diacetoxyiodobenzene and other diacetoxyiodoarenes.208,209 This hypervalent iodine ylide is a suitable starting material for C-arylation and C-alkenylation reactions. It was used in Stille210,211 and Suzuki-type couplings. For instance, the Suzuki-type coupling of this ylide with aryl- and heteroarylboronates in DME/H2O produced C-aryl and C-alkenyl 2-hydroxy-naphthoquinones (107a–f) in fair to good yields (Scheme 27).212
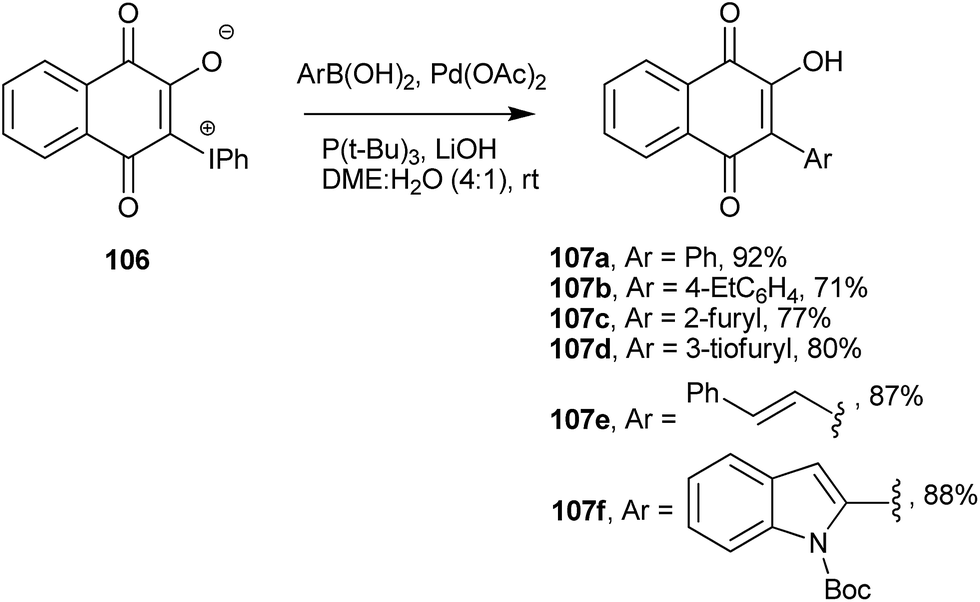 | ||
| Scheme 27 | ||
Ylide 106 also reacts with electron-rich arenes in the presence of BF3·Et2O as catalyst to afford the corresponding Suzuki-type C-arylated 1,4-naphthoquinones (107g–n).213 Furthermore, it also undergoes an unusual C-arylation–deformylation reaction with aromatic aldehydes activated by BF3·Et2O that also leads to Suzuki type 2-aryl-3-hydroxy-1,4-naphthoquinones (107g–n). This reaction is very useful because aromatic aldehydes are cheap and readily available (Scheme 28).
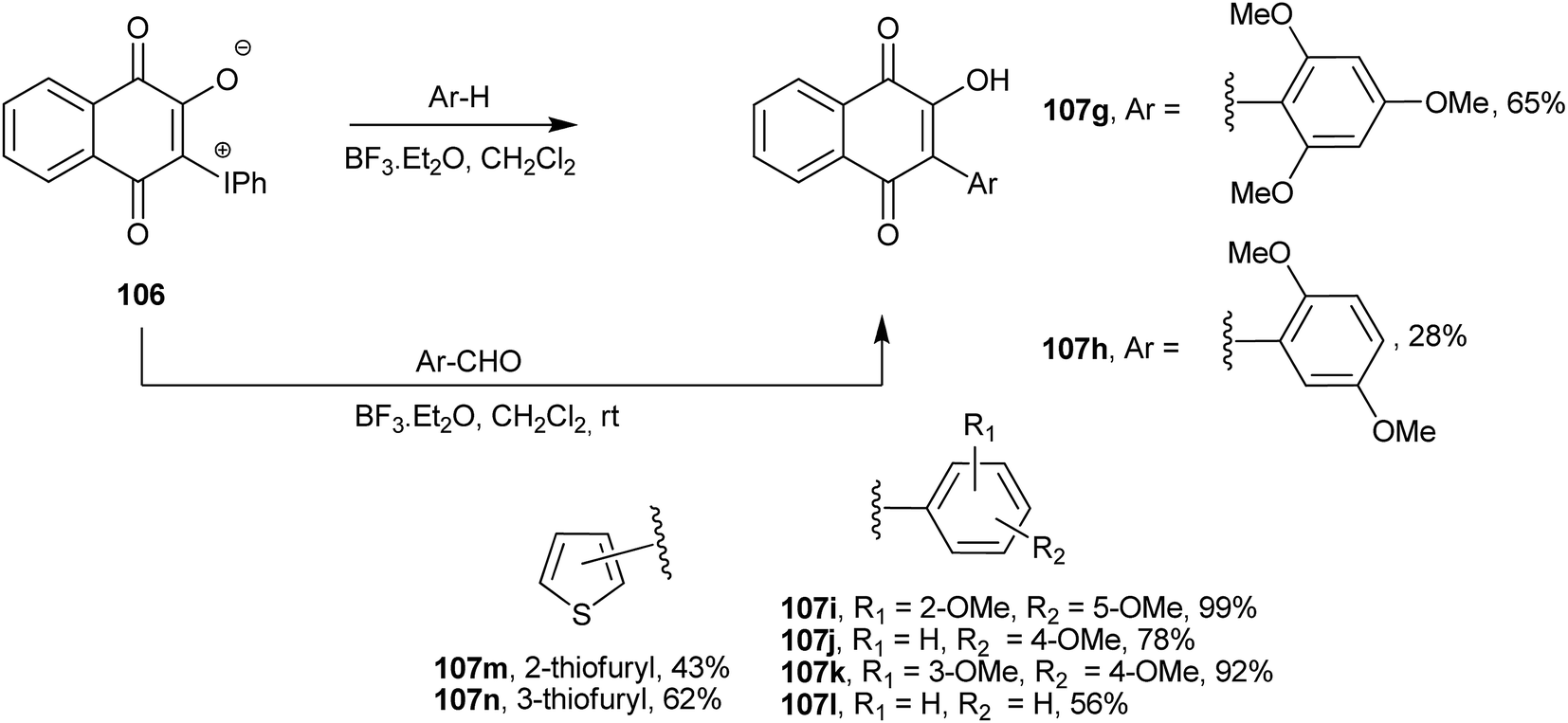 | ||
| Scheme 28 | ||
Another efficient C(sp2)–C(sp2) coupling involving 1 is the Heck reaction of 2-hydroxy-3-iodo-naphthoquinone (108) with electron-deficient alkenes (unsaturated acids and amides). Perez and coworkers214 described it as a simple methodology and with facile work-up to produce C-alkenyl products (109) in good to excellent yields (Scheme 29). It is worth noting that 2-hydroxy-3-bromo-1,4-naphthoquinone can be easily obtained from 1 by using several different reaction conditions such as morpholine/iodine,215 chloroform/H2SO4/H2O2/Br2 (92%), Br2/CH3CN,216 and NH4Br/oxone (99%).217
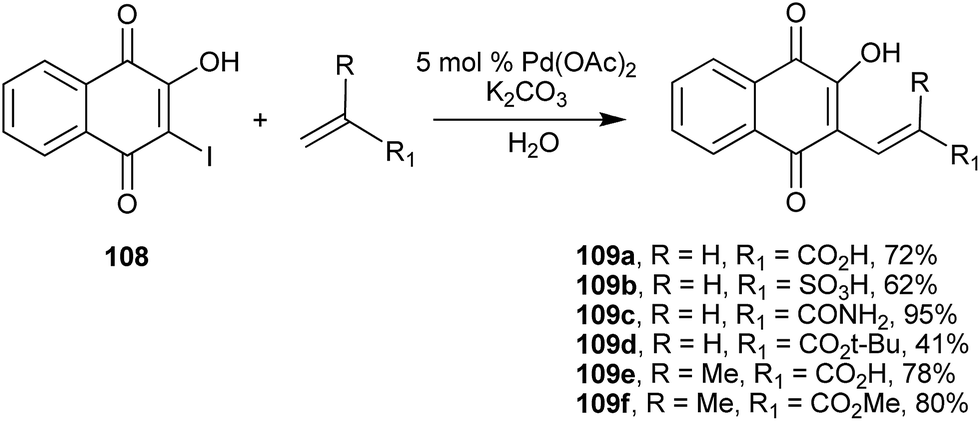 | ||
| Scheme 29 | ||
3.7 Synthesis of 2H-chromene- and chromane-fused naphthoquinones
A number of natural and synthetic heterocyclic naphthoquinones have important biological activities, and in the past few years, several research programs have been directed towards the synthesis of these compounds. Heterocycles are of great importance because many of the biochemical materials essential to life belong to this class of compounds, e.g., drugs, vitamins and active metabolites in plants and marine organisms. In this regard, numerous heterocyclic rings containing the naphthoquinone moiety have been synthesized, among which those bearing a chromene or a chromane ring fused with ortho- and para-quinones must be highlighted. They represent a special class of naturally occurring molecules present in many plants, fungi and insects.218 The most important substances of the chromane-fused-quinone class are α- (7) and β- (6) lapachones (Fig. 1). Thus, the development of reliable methods for the preparation 2H-chromene- and chromane-naphthoquinones will be valuable to access analogues and derivatives of these compounds.β-Lapachone (6) is a natural pyran-ortho-naphthoquinone originally obtained from the heartwood of various Lapacho trees that belong to the genus Tabebuia (Bigoniaceae) and that grow throughout South America. This compound has been demonstrated to exhibit many different biological activities, including a promising anticancer activity.219 Indeed, it presents significant antineoplastic220 activity against various human cancer cell lines from leukemia,221 prostate,222 malignant glioma,223 hepatoma,224 colon,225 breast,226 ovarian,227 lung228 and pancreatic229 tumors. Moreover, it has been intensely investigated as a possible prototype compound against Trypanosoma cruzi, the etiological agent of Chagas disease, in both acute and chronic infections.230–233 This compound inspired several researchers in search of new derivatives with better therapeutical index. Several heterocyclic derivatives, such as oxyranes,234,235 oxazoles,236 imidazoles237 and phenazines,238 have been synthesized from 6, and some have shown fewer side effects and improved trypanocidal activity.
A very useful route to new analogues of 6 involves the construction of 2H-chromene or chromane moieties adjacent to naphthoquinone 1, by using the o-quinone methide (o-QM) intermediate (110, Scheme 30)239 that undergoes a Knoevenagel condensation, followed by an electrocyclization process or a hetero Diels–Alder reaction to form the heterocyclic rings, respectively. Ferreira and coworkers240 reported the first reaction of 1 with α,β-unsaturated aldehydes producing exclusively chromene-naphthoquinones (111a–c) in 33–35% yield by the Knoevenagel reaction, followed by an intramolecular electrocyclization. The same synthetic route and conditions were used by Oliveira and coworkers241 for the preparation of the naturally occurring dihydro-α-caryopterone (112) and its isomer 6-hydroxy-dehydro-α-lapachone (113) from naphthoquinone (32).
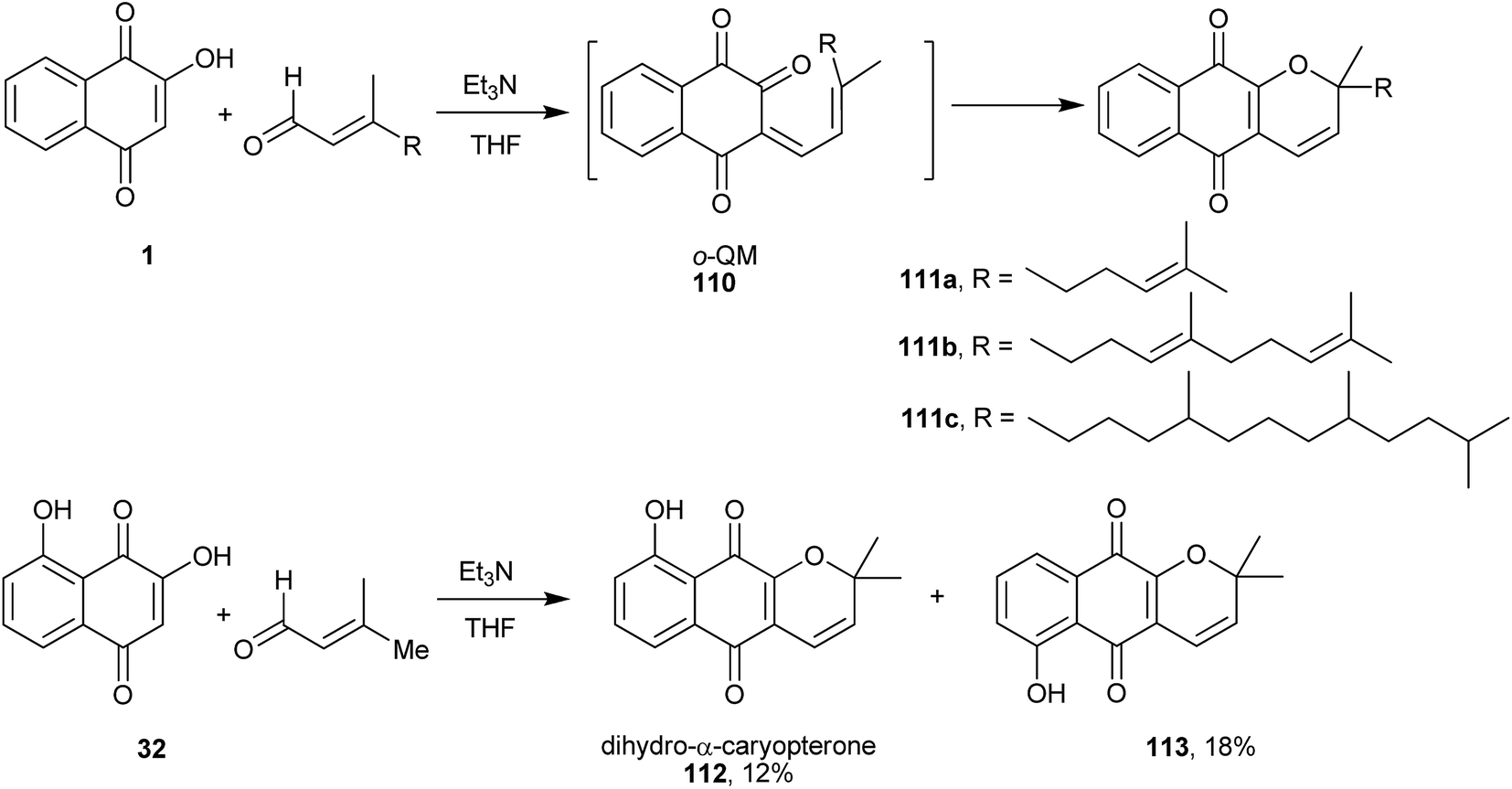 | ||
| Scheme 30 | ||
The same research group has also developed a novel preparation of the tetracyclic α- and β-pyran naphthoquinones (114 and 115), obtained in 70% overall yield by the reaction of citronellal,240 an aldehyde bearing a remote double bond, with 1, via the o-QM intermediate generated in situ (Scheme 31). Estévez-Braun and co-workers have performed the asymmetric version of this reaction with (S)-(−)-citronellal, producing compounds 114 and 115 in a 1:1 ratio and 94% overall yield.242
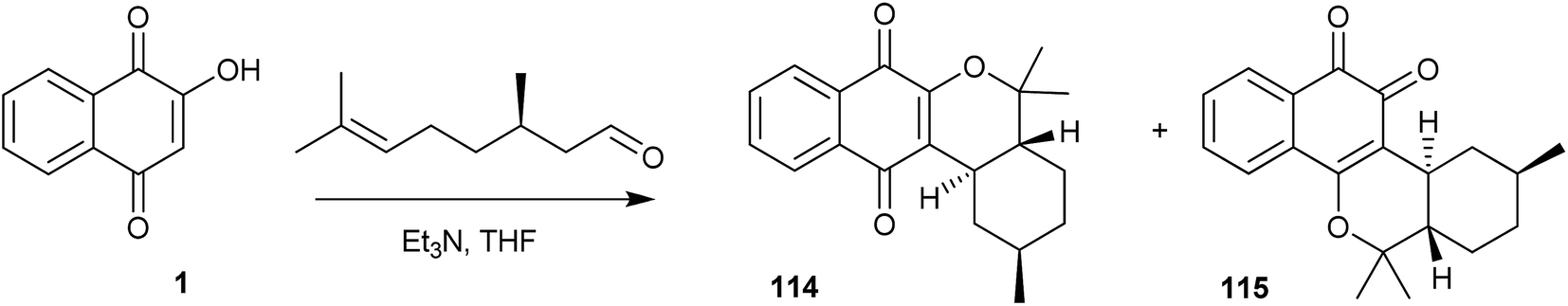 | ||
| Scheme 31 | ||
Nair and Treesa improved the later methodology by using an o-QM243 generated in situ from 1 and formaldehyde to perform the one-pot intermolecular Diels–Alder reaction with olefins. This new three-component protocol overcame the limitation regarding the use of appropriately substituted aldehydes in the tandem-Knoevenagel/intramolecular hetero Diels–Alder cycloaddition methodology. By this reaction, and using several different olefins, they prepared a wide variety of α- and β-lapachone derivatives (chromane-naphthoquinones) in moderate to good yields, examples of which are shown in Scheme 32.
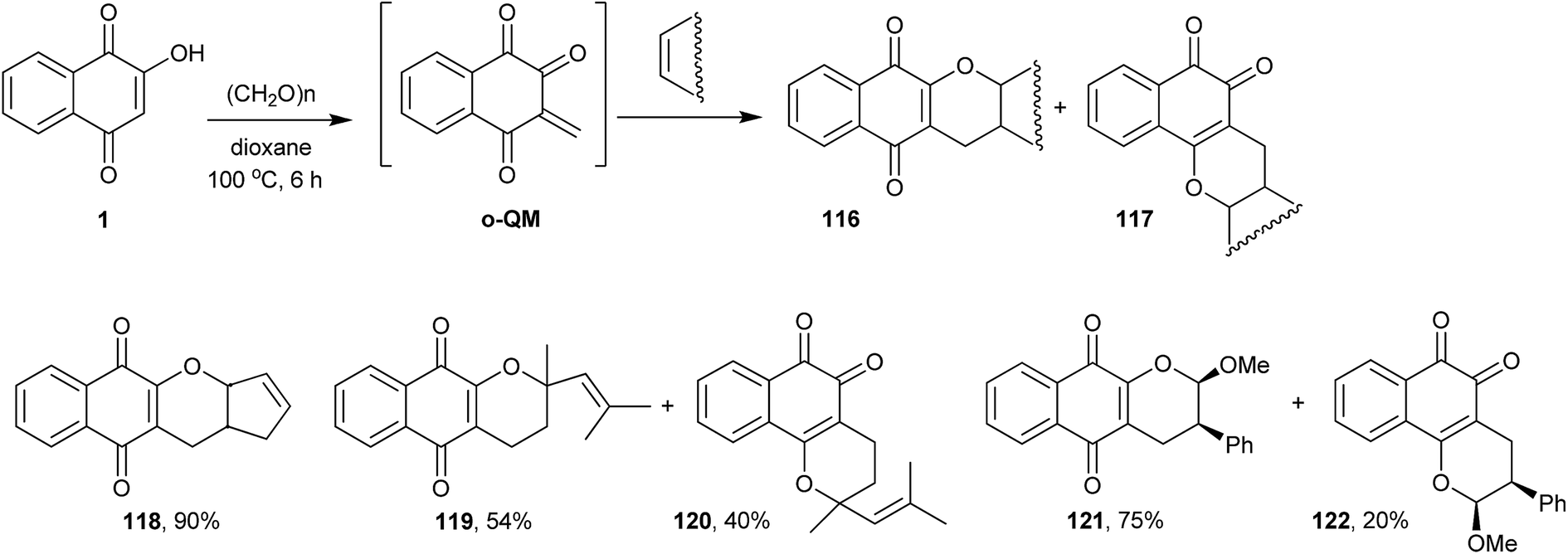 | ||
| Scheme 32 | ||
Despite the good performance achieved in the reactions with formaldehyde described above, the same reaction with aromatic aldehydes did not work. Nonetheless, by investigating solvent effects, Ferreira and coworkers found that the reaction was accelerated in ethanol/water (1:1) at reflux, leading to new substituted chromane-naphthoquinones.244 With this improved method, β-pyranonaphthoquinones were obtained more selectively, in better yields, with shorter reaction times and, most importantly, from any type of aldehyde, as illustrated in Scheme 33 and Table 1 (entries 1–8).
 | ||
| Scheme 33 | ||
| Entry | R1 | R2 | R3 | Conditions | Time (h) | Yield (%) | Ratio α/β (123:124) |
α (syn:anti) |
β (syn:anti) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | H | H | H | EtOH/H2O (1:1) |
6 | 94 | 3.1 | — | — |
| 2 | H | Me | H | EtOH/H2O (1:1) |
6 | 95 | 3.8 | — | — |
| 3 | H | Me | Me | EtOH/H2O (1:1) |
6 | 97 | 4.7 | — | — |
| 4 | 4-NO2-C6H4 | Me | H | EtOH/H2O (1:1) |
5 | 50 | 1.4 | 37:63 |
18:82 |
| 5 | 2-Thiophene | Me | H | EtOH/H2O (1:1) |
8 | 60 | 0.8 | 54:46 |
0:100 |
| 6 | Ph | Me | H | EtOH/H2O (1:1) |
8 | 52 | 0.6 | 20:80 |
12:88 |
| 7 | H | H | H | Dioxane/HOAc | 4 | 96 | 2.2 | — | — |
| 8 | 4-NO2-C6H4 | Me | H | Dioxane/HOAc | 5 | 55 | 1.1 | 35:65 |
17:83 |
This reaction protocol also allows the use of electron-rich olefins, such as silyl enol ethers, in the preparation of chromane-naphthoquinones (Scheme 34).245 In this way, a series of siloxy-containing naphtho[2,3-b]pyran-5,10-dione derivatives were obtained in moderate to high yields, with the reactions giving α-lapachone derivatives (125 and 126) regioselectively.
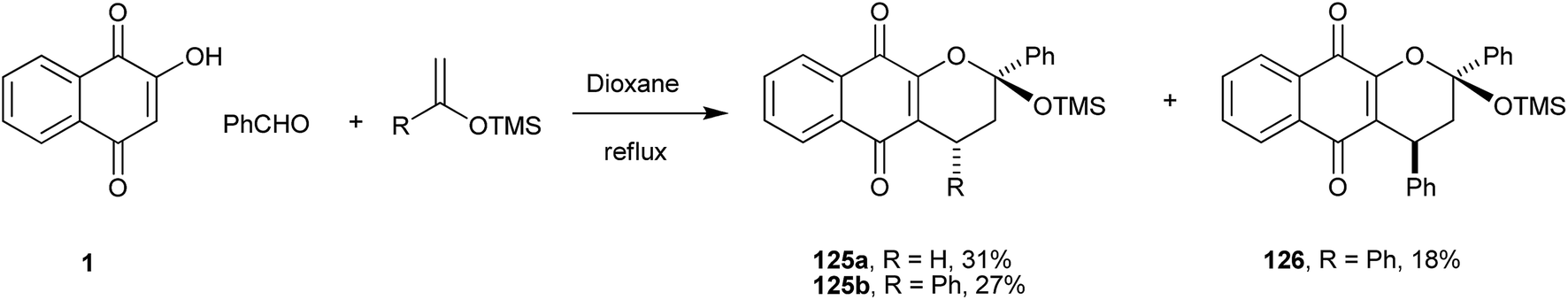 | ||
| Scheme 34 | ||
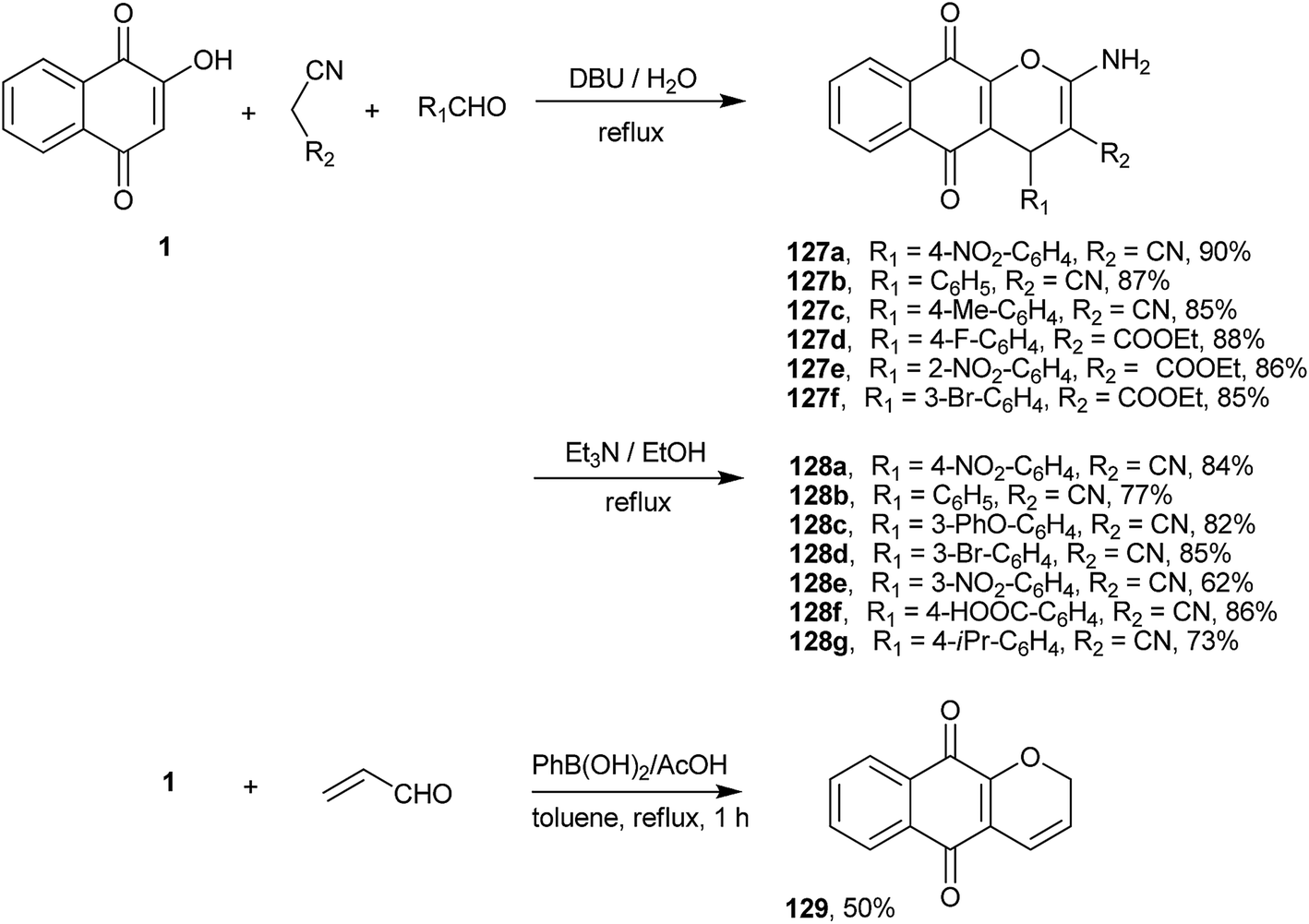
A new three-component route for the synthesis of 4H-chromene-naphthoquinones was developed by Saluja and coworkers246 involving the cyclo-condensation reaction of 1 with an aldehyde and malononitrile or ethyl cyanoacetate. The reaction produced excellent yields of chromene-naphthoquinones (127a–f and 128a–g) with a high atom economy (Scheme 35). The reaction with malononitrile and aromatic aldehydes was reinvestigated by Magedov et al. Their theoretical analysis using semiempirical and DFT calculations indicated that the formation of 1,4-naphthoquinones is favored over the 1,2-products both thermodynamically and kinetically. The antiproliferative properties of a number of chromene-naphthoquinones thus synthesized were investigated against a panel of cancer cell lines, and the apoptosis-inducing effects of the 3-substituted phenyl derivative (127b) was found to rival those of clinically relevant α-lapachone.247 An interesting one-pot preparation of 3H-chromene (129) was developed by Silva and coworkers248 that adapted the synthesis of chromenes from phenols developed by Snieckus et al.
 | ||
| Scheme 35 | ||
The reaction of 1 with α,β-unsaturated aldehydes usually leads to 2H-chromenes via tandem Knoevenagel electrocyclization. Under organocatalysis conditions, however, efficient, enantioselective cascade 1,4-addition/cyclization reactions take place (Scheme 36)249 and provide valuable chromane-naphthoquinones with aliphatic and aromatic α,β-unsaturated aldehydes in good yields and with excellent enantioselectivities (90–99% ee). Recently, Wang and coworkers studied a similar organocatalyzed reaction between 1 and β-unsaturated α-keto esters [(E)-ethyl-2-oxo-4-aryllbut-3-enoates] (Scheme 36).250 The most effective chiral organocatalyst for this reaction was a trans-indane thiourea (131), which allowed for quick construction of diversely functionalized naphthoquinones under mild reaction conditions, with excellent yields and high enantioselectivities.
 | ||
| Scheme 36 | ||
When the chromane-naphthoquinones are fused with a second naphthoquinone or with an aromatic ring, they belong to the class of xanthenes and benzoxanthenes, respectively. These compounds are of great interest because of their wide range of biological and pharmaceutical properties, such as antiviral251 and antibacterial properties.252 Several methods for their preparation have been described in the literature.
The one pot solvent-free condensation of 1 with β-naphthol and aromatic aldehydes in the presence of the heterogenous catalyst silica chloride affords the aryl-14H-dibenzo-xanthenes (133a–d) in short reaction times and excellent yields (Scheme 37).253,254 Recently, Du et al.255 performed this reaction to prepare xanthene derivatives from 1 and aromatic aldehydes, and by using 1,3-dicarbonyl compounds, they also produced a series of benzo-xanthenes derivatives (134a–f). The reactions are performed at 90 °C, in the ionic liquid [bmim+][Br−] that can be recycled and reused (Scheme 37). Similarly, Rahmati described the synthesis of various xanthenes from the condensation reactions of β-naphthol or dimedone with various aldehydes in the presence of trifluoroacetic acid as a catalyst in an ionic liquid.256 Li et al.257 also described a similar reaction, using β-naphthylamine instead of β-naphthol in [bmim+][BF4−] as solvent, from which they produced dibenzo-acridines (135a–g) in good yields (Scheme 37). These reactions were also studied by other research groups in the presence of various catalysts258–260 and ionic liquids.261
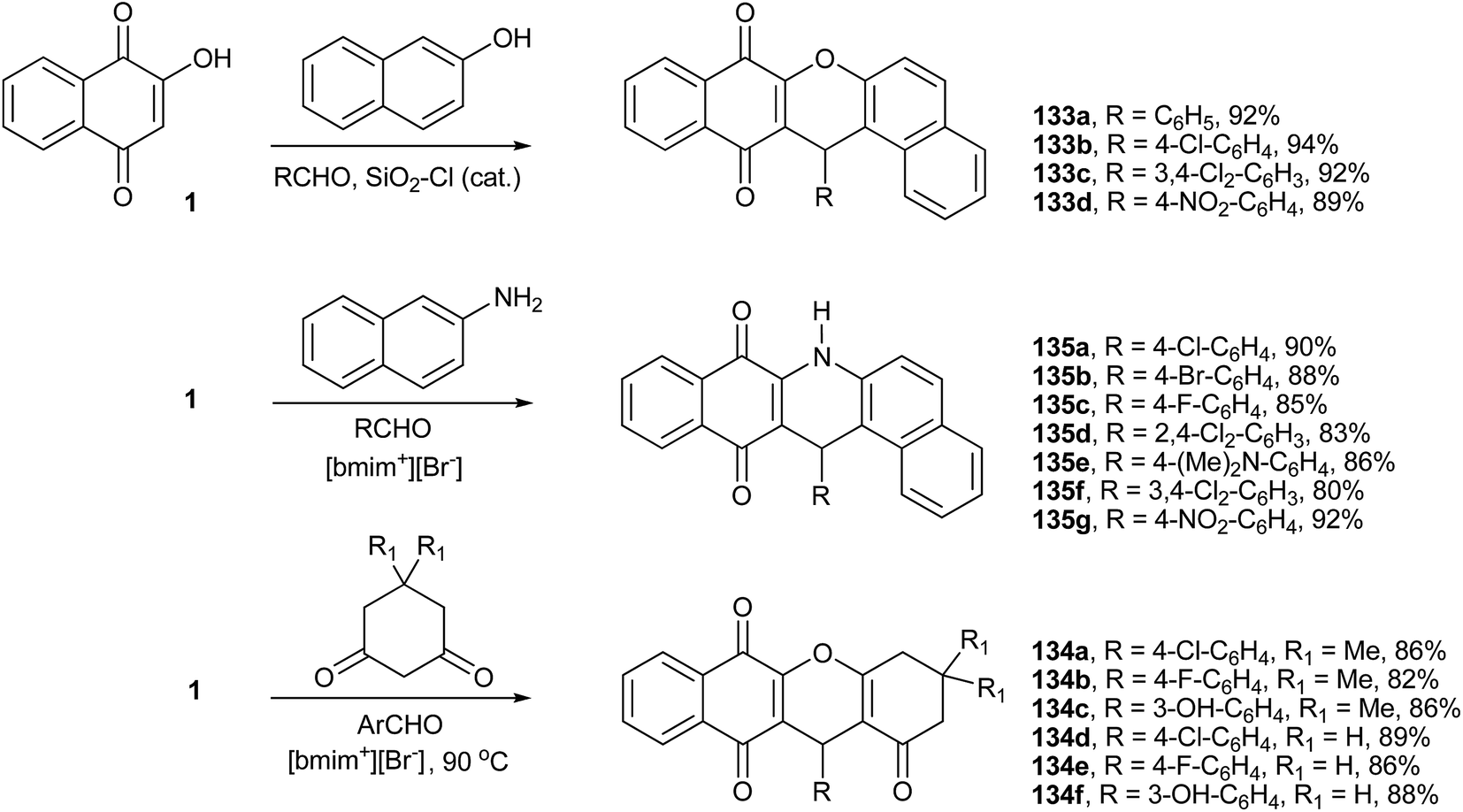 | ||
| Scheme 37 | ||
The dibenzo-acridines can also be prepared by the recently described multicomponent reactions262 developed by Dabiri et al., who reported the reactions of 1 with indoles, in place of aldehydes, and 1-H-pyrazol-5-amines in the presence of L-proline as catalyst. Several new spiro-oxindoles (138a–e) were obtained in good yields (Scheme 38).263 Perumal and coworkers developed a four-component domino reaction of 1, aromatic aldehydes, methyl or ethyl acetoacetate and ammonium acetate in ethanol, under heating or microwave irradiation, to produce tetrahydrobenzo[g]quinoline-5,10-diones (139a–g) in good yields (Scheme 38). This transformation probably proceeds via o-QM, followed by Michael addition of the enol of the methyl or ethyl acetoacetate and intramolecular regioselective annulation with ammonia available from the dissociation of ammonium acetate (Scheme 38).264
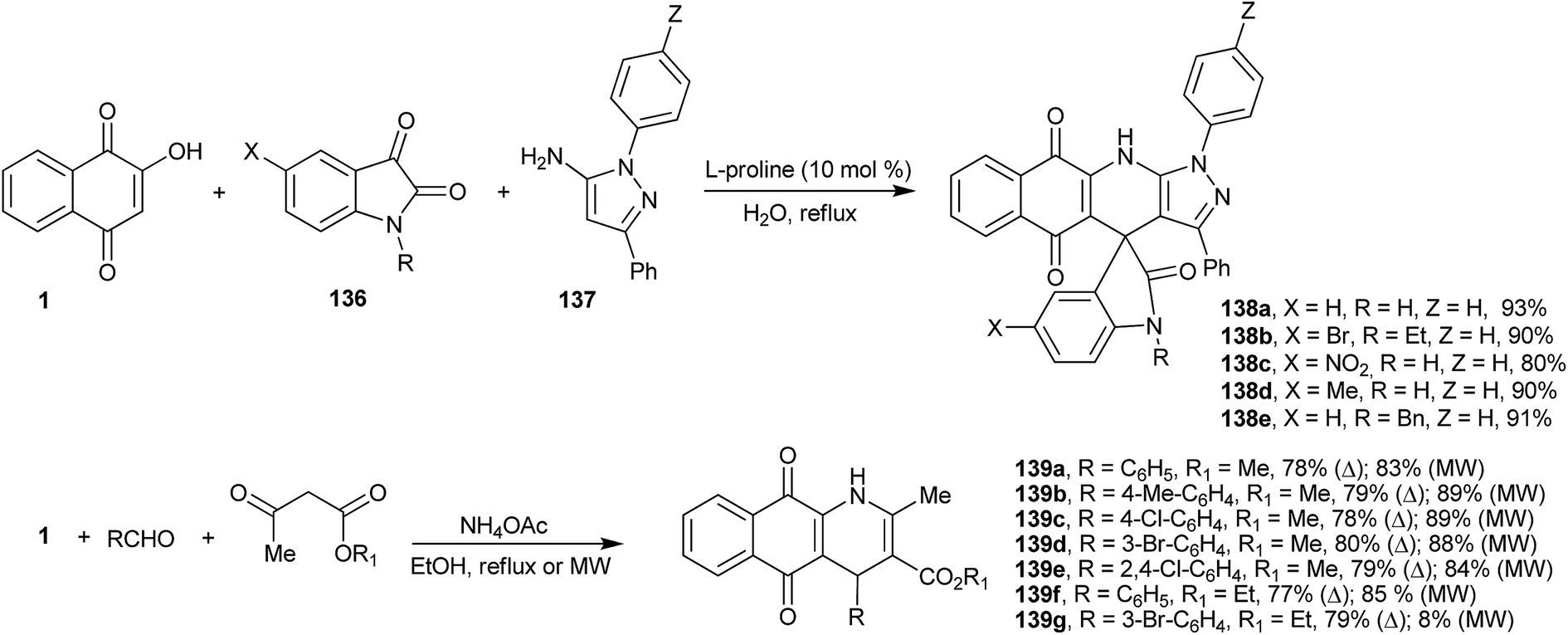 | ||
| Scheme 38 | ||
3.8 Synthesis of xanthenes via o-quinone methide
9H-Xanthenes (140, Fig. 5) constitute a general class of natural and synthetic heterocyclic compounds265–267 with many applications, such as in biological targets and fluorescent dyes.268 In the biological field, these heterocyclic compounds exhibit a broad spectrum of activities, such as trypanothione reductase inhibition,269 antiviral,270 antibacterial,271,272 antiplasmodial,273,274 anticancer,275–278 antimalarial,279 antifungal,280 antiinflammatory,281 and antihypertensive282 activities, which have been investigated for photodynamic therapy.283 Because of all of these biological properties, many methods for the preparation of 9H-xanthene derivatives have been described in the literature,284–287 mainly related to the two- or three-component reaction of β-naphthol and aldehydes.253,254,256,288–296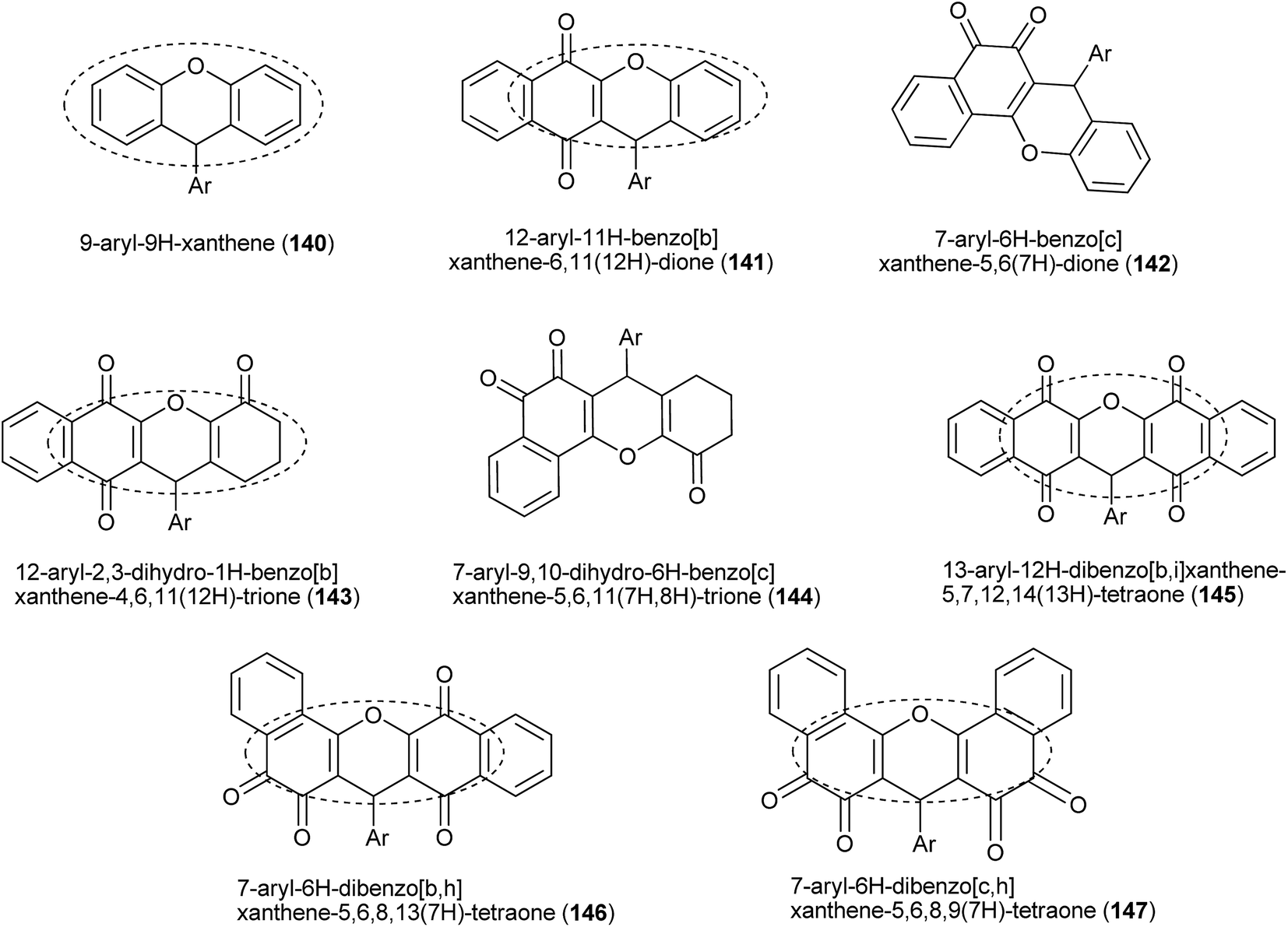 | ||
| Fig. 5 Naphthoquinones fused with xanthenes. | ||
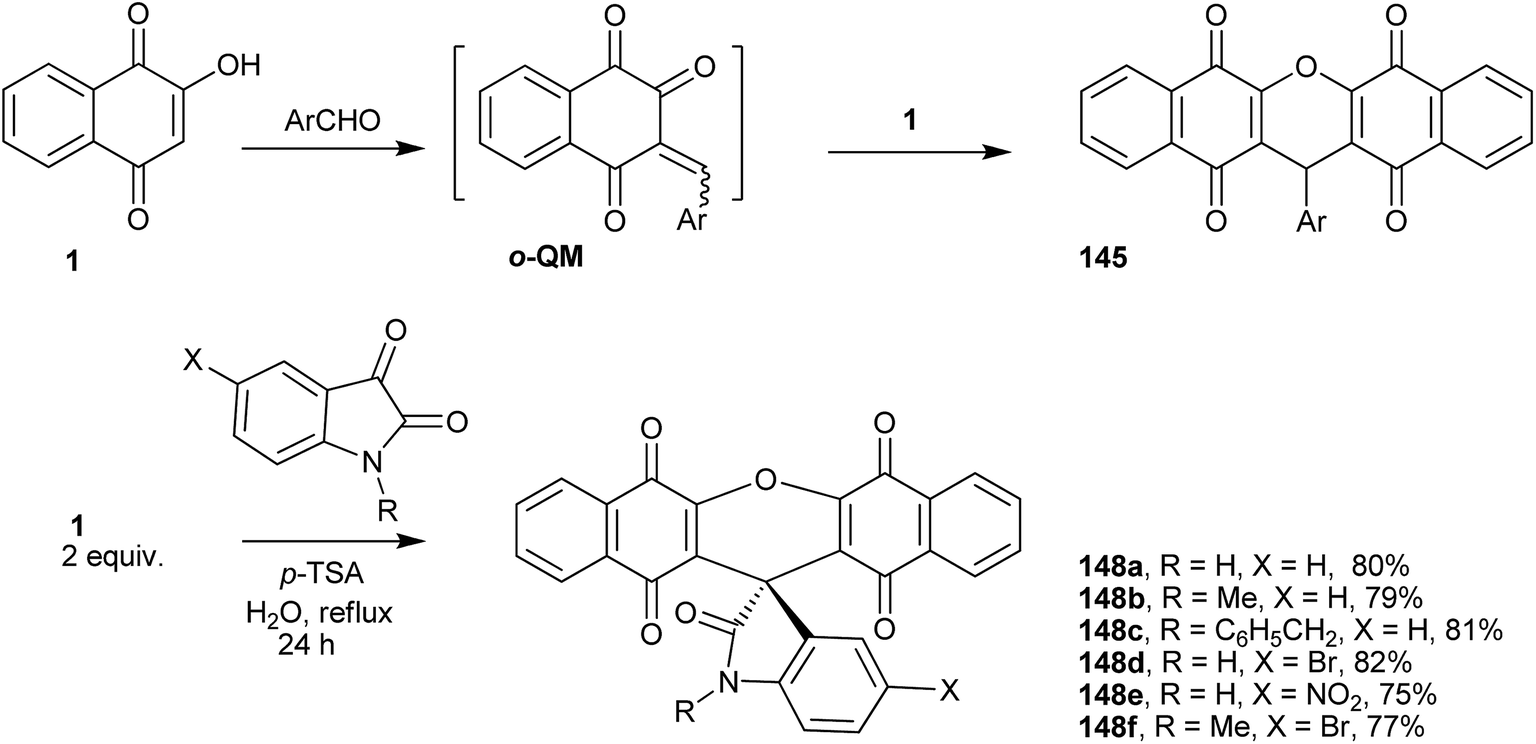
The preparations of various naphthoquinones fused with xanthene rings (Fig. 5) have been reported, e.g., benzoxanthenes with one naphthoquinone moiety and represented by compound 141 and its isomer 142; benzoxanthenetrione 143 and its isomer 144;255,297–303 and dibenzoxanthenes containing two naphthoquinone moieties attached to the central pyran ring and that can exist as one of the three isomers 145, 146 and 147. Tisseh et al. reported the first simple and efficient method for the preparation of dibenzo[b,i]xanthene derivatives 145 under solvent-free conditions (p-toluene sulfonic acid 10 mol%), but with very basic spectroscopic characterization (Scheme 39). In general, this o-quinone methide protocol works very well in the absence of an additional nucleophile, which allows another equivalent of 1 to add to the o-QM intermediate formed in situ, giving the desired product after cyclization with loss of a water molecule.304 Reactive carbonyl compounds, such as isatins, have also been used in this reaction in the presence of a catalytic amount of p-toluenesulfonic acid (p-TSA). The reactions proceeded smoothly in water, at reflux, for 24 h, to produce spiro[dibenzo[b,i]-xanthenes 148a–f adducts in good yields (Scheme 39).305
 | ||
| Scheme 39 | ||
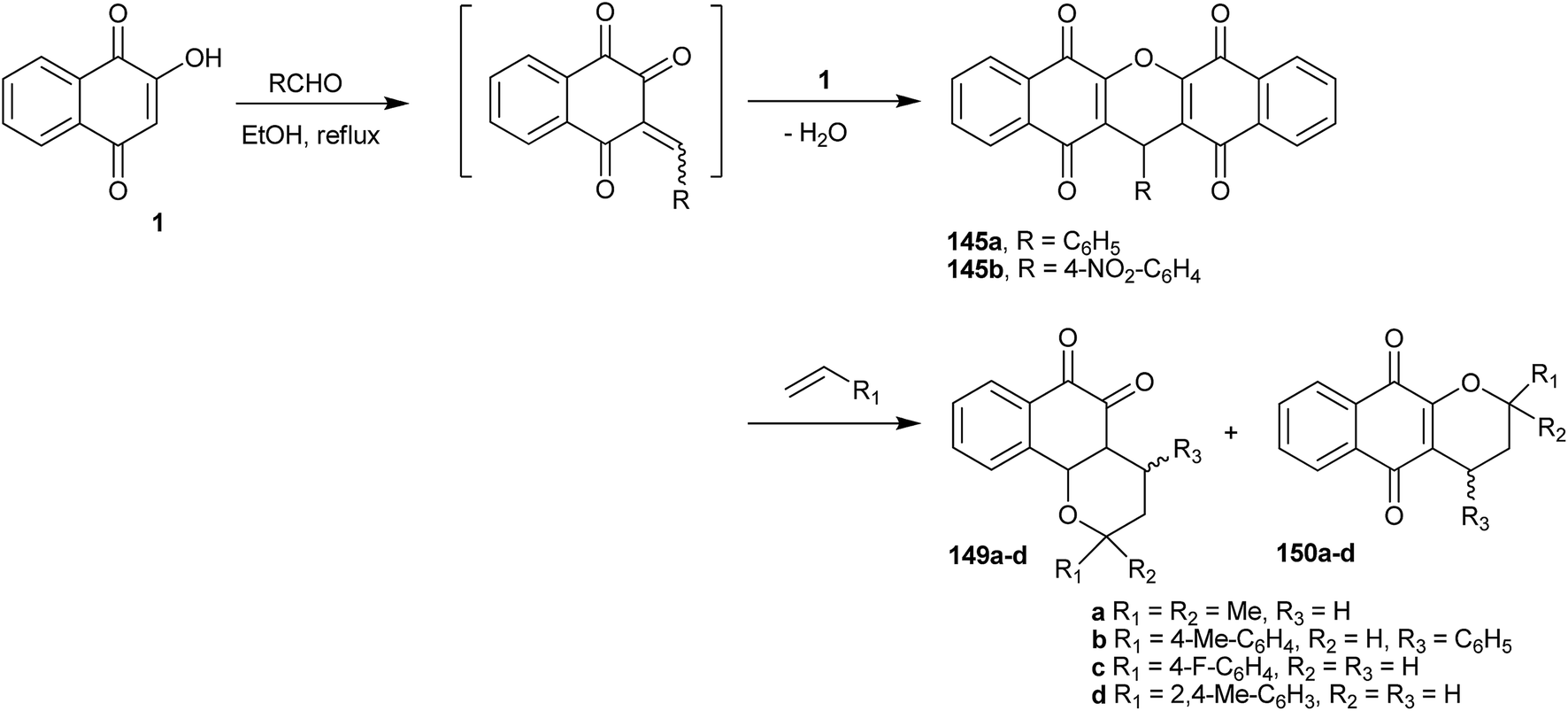
Costa and co-workers306 have described the syntheses of various pyranaphthoquinones by reacting 1 with an aldehyde (formaldehyde or arylaldehydes) that results in the in situ generation of the o-QM, which then undergoes hetero Diels–Alder reactions with the appropriate dienophiles. Xanthenes 149a–d and 150a–d were produced by nucleophilic addition of 1 on the o-QM intermediate, followed by dehydration. Some selected examples are shown in Scheme 40. All compounds were tested as inhibitors of the ATPase activity of the viral NS3 enzyme in in vitro assays and the replication of dengue virus in cultured cells. Two 1,4-pyranaphthoquinones inhibited dengue virus replication in mammal cells by 99.0%, and three others reduced the dengue virus ATPase activity of NS3.
 | ||
| Scheme 40 | ||
3.9 Synthesis of other heterocycles from lawsone (1)
Furonaphthoquinones constitute another important class of naturally occurring or synthetic five-membered heterocyclic fused naphthoquinones. Several synthetic routes to this class of compounds have been described in the literature, some of which use 1 as the starting material.One important and direct method for synthesizing furonaphthoquinones is the one-electron oxidation of 1 with ceric ammonium nitrate (CAN) and C–C bond formation with olefins. CAN, a well know one-electron oxidant,307,308 easily handled and soluble in various solvents, is very useful in many reactions.309–311 For instance, the reaction of 1 with CAN and cyclodienes in acetonitrile resulted in the formation of 1,4- and 1,2-furonaphthoquinones (151–154), as presented in Scheme 41.312 Additionally, manganese(III) acetate dihydrate can promote the radical cyclization of 1 with alkenes, leading selectively to 1,4-furonaphthoquinones (155a–d) in excellent yields (Scheme 41).313
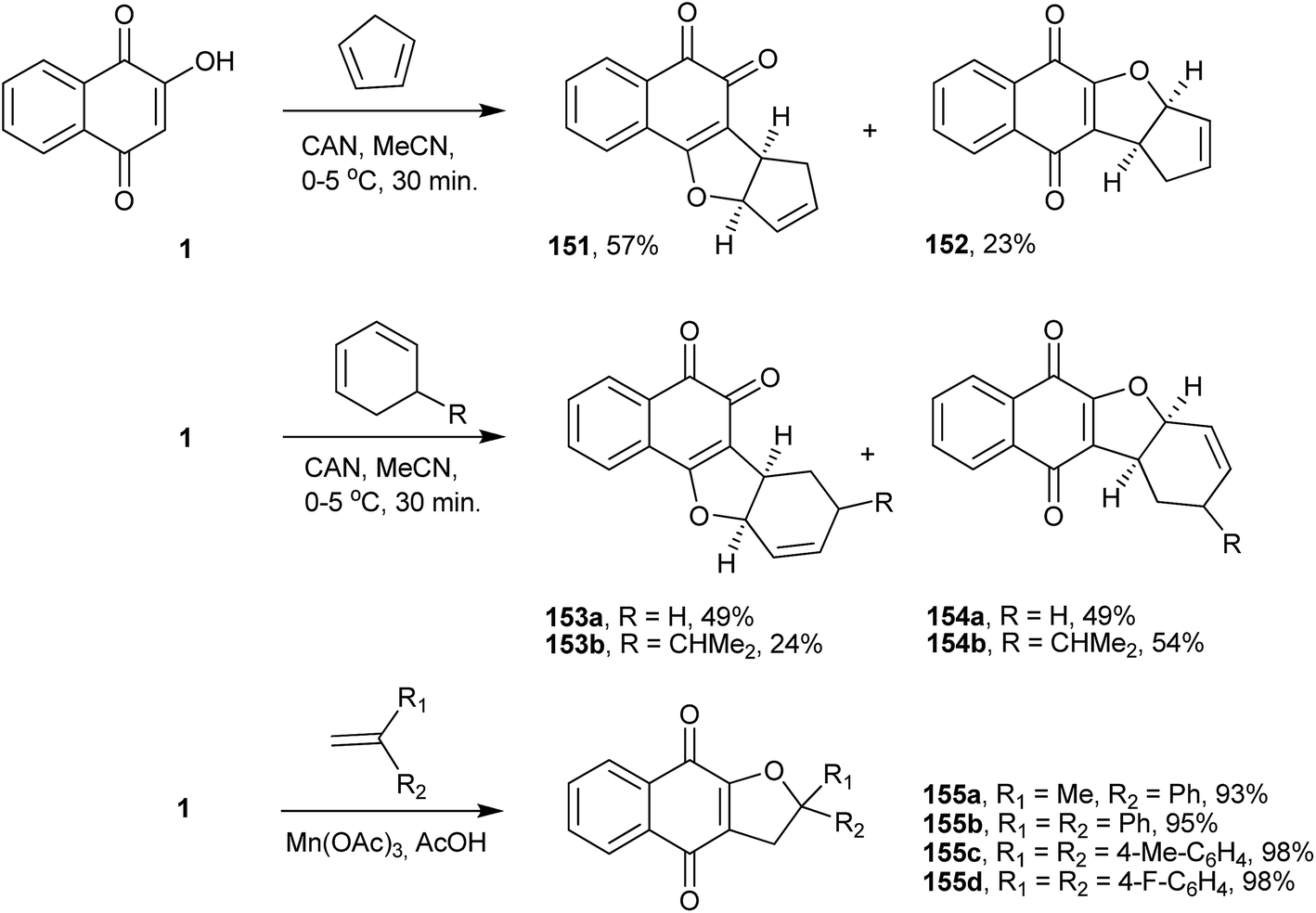 | ||
| Scheme 41 | ||
CAN also promotes the formation of a radical of 1 that adds to phenyl vinyl sulfide and ethyl vinyl ether, followed by intramolecular cyclization, producing dihydrofuronaphthoquinones (156, 157 and 158). These dihydrofurans can be transformed into furan tethered to 1,2- and 1,4-naphthoquinones (159a and 160a).314 It is worth noting that 1,4-furonaphthoquinone (159a) is a natural product isolated from Avicennia marina.315 Kobayashi and co-workers studied the same reaction between 2-hydroxy-1,4-naphthoquinone and phenylacetylene that yielded 159b and 160b (Scheme 42).316
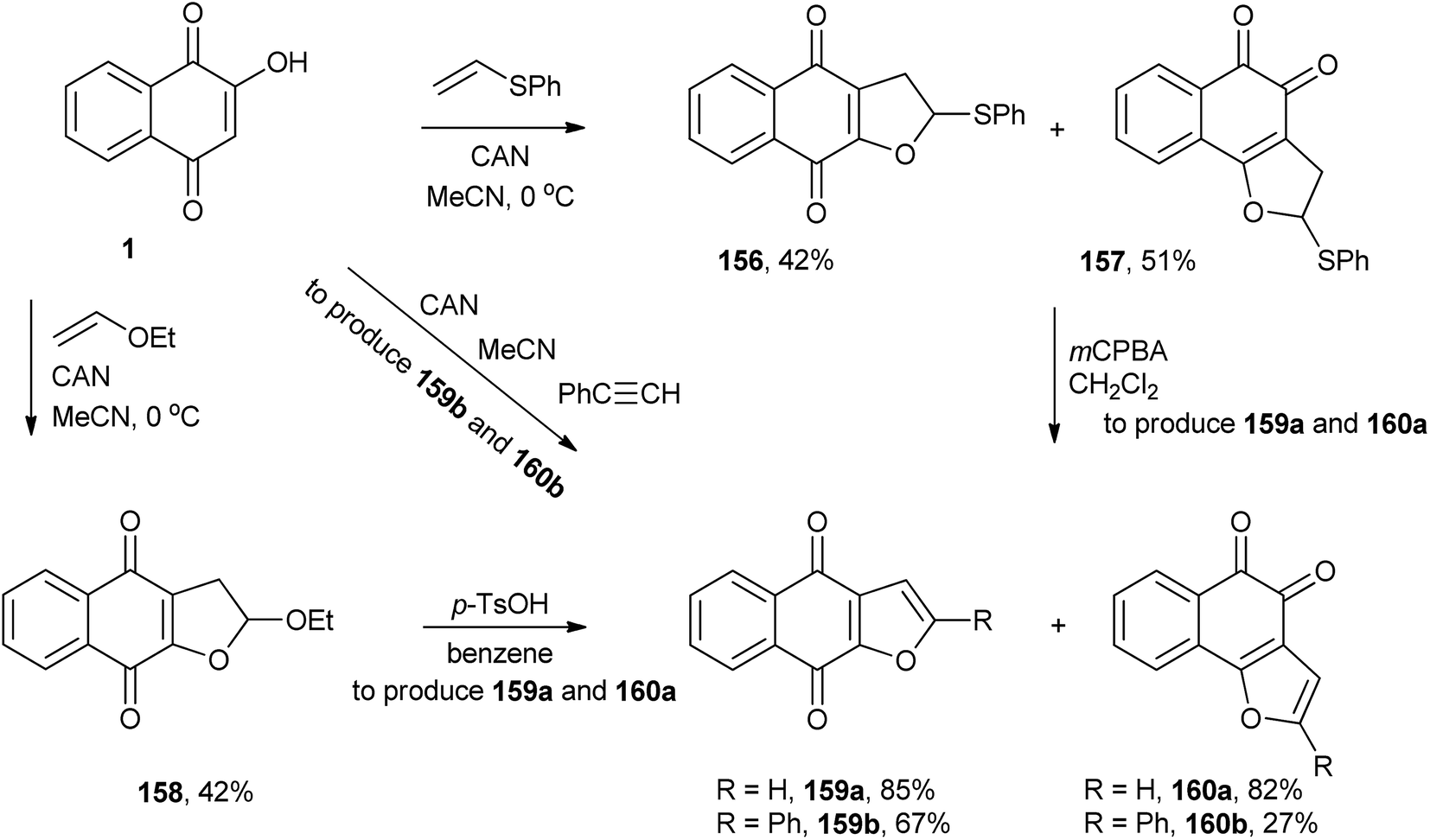 | ||
| Scheme 42 | ||
Teimouri and coworkers317 have developed an efficient and green method for the preparation of highly substituted linear naphtho[2,3-b]-furan-4,9-dione derivatives starting from 1, alkyl isocyanides and a variety of aldehydes. This one-step method furnishes regioselectively the linear 1,4-naphthoquinones (161a–f) in good yields (Scheme 43).
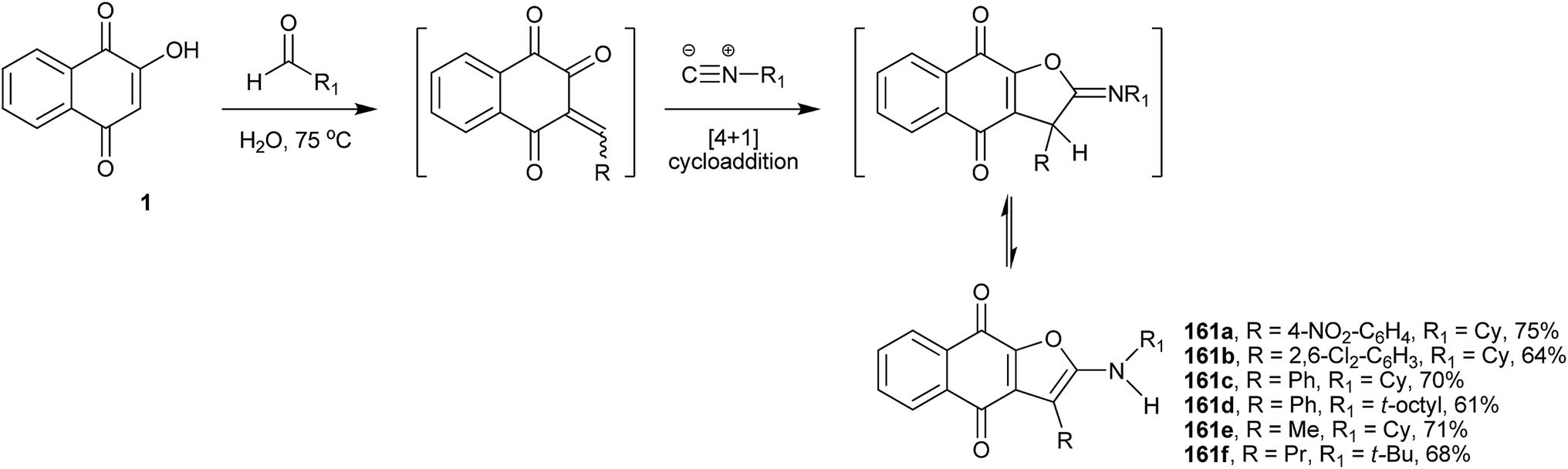 | ||
| Scheme 43 | ||
Wu and co-workers have described an efficient multicomponent method for the synthesis of benzo[h]pyrazolo[3,4-b]quinolines tethered to the 1,4-naphthoquinone nucleus. The syntheses were performed by condensation of 1 with 3-methyl-1-phenyl-1H-pyrazol-5-amine and various benzaldehydes using molecular iodine as catalyst in water. The most plausible mechanism involves a cascade sequence starting with Knoevenagel condensation to form the o-QM, followed by Michael addition of the amino-pyrazole, intramolecular nucleophilic cyclization and, finally, dehydration. This method is experimentally simple, uses inexpensive and readily available reagents and catalysts, and the products, 4-aryl-3-methyl-1-phenyl-1H-benzo[h]pyrazolo[3,4-b]quinoline-5,10-diones (162a–e), are formed in good yields and high atom economy (Scheme 44).318 The same multicomponent domino annulation reaction was studied by Li and coworkers under modified experimental conditions. Upon treatment with acetic acid under microwave irradiation, the benzo[h] isoxazolo[3,4-b]quinolines (163a–g) were formed regioselectivily, in which the β-isomer of the naphthoquinone is tethered to benzo[h]pyrazolo[3,4-b]quinolines (Scheme 44).319 Recently, another multi-component protocol was described for the synthesis of novel polyheterocyclic systems containing a benzo[h]quinoline-5,6-dione nucleus (164a–f) from 1 in aqueous medium. In this four-component process, L-proline catalyses the Michael addition of phenylhydrazine to 3-aminocrotononitrile with annulation via the elimination of ammonia to give 3-methyl-1-phenyl-1H-pyrazol-5-amine in situ, as well as the Knoevenagel reaction of the aldehyde and 1 (Scheme 44).320
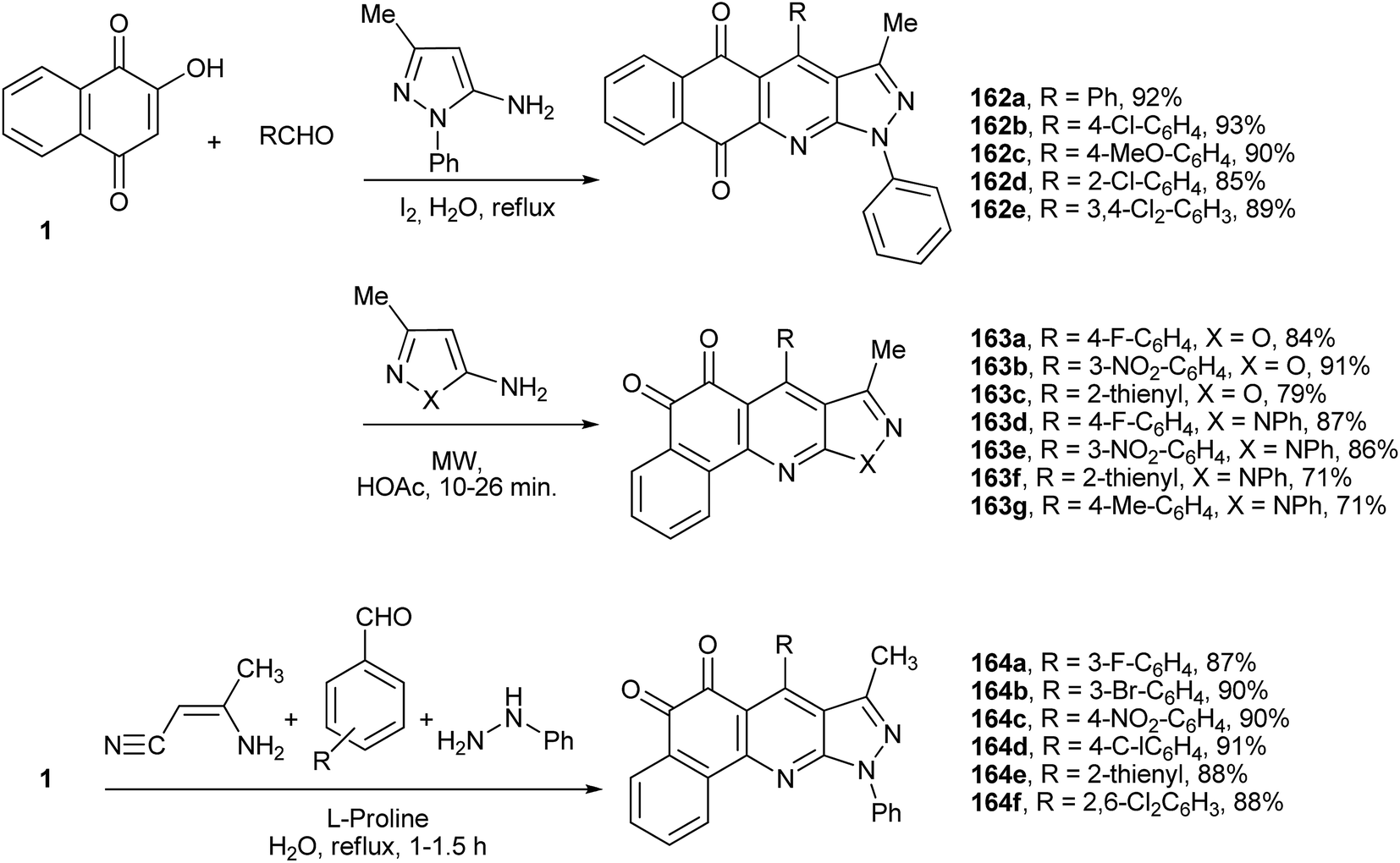 | ||
| Scheme 44 | ||
Perumal et al. reported a very interesting three-component one-pot synthetic sequence to prepare quinolines fused with the naphthoquinone moiety. In the first step, 1 reacts with the appropriate aniline, replacing the hydroxyl group to give product 165, which reacts with propargyl bromides furnishing N-propargylaminonaphthoquinones 166. Intramolecular 6-endo-dig cyclization catalyzed by copper(II) triflate furnishes the naphthoquinone-quinolines (167a–g) in good yields (Scheme 45).321
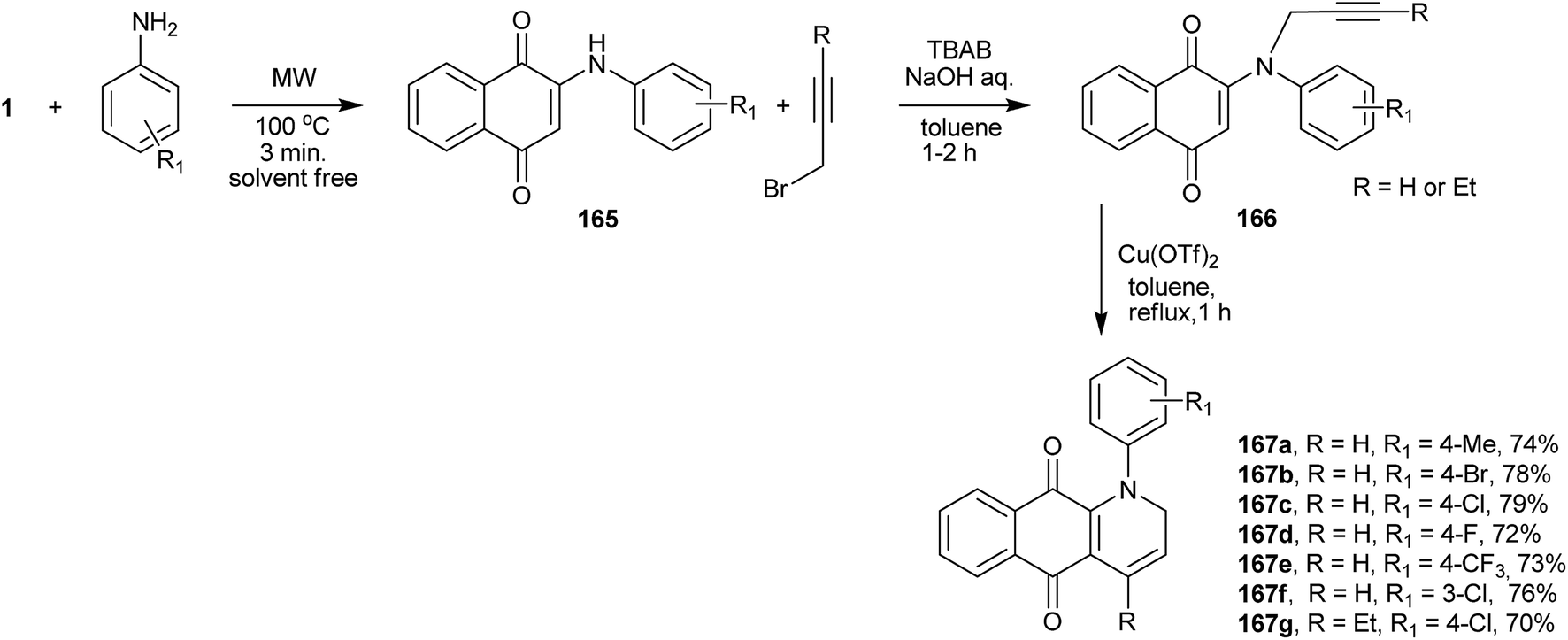 | ||
| Scheme 45 | ||
Recently, Jain et al.322 performed the reaction of 1 with 1,2-diaminobenzenes 168a–b and obtained the corresponding tetracyclic phenazine derivative (169a), whereas the reaction with 2,3-diaminotoluene gave a mixture of the regioisomers (169b) and (169c) (Scheme 46).
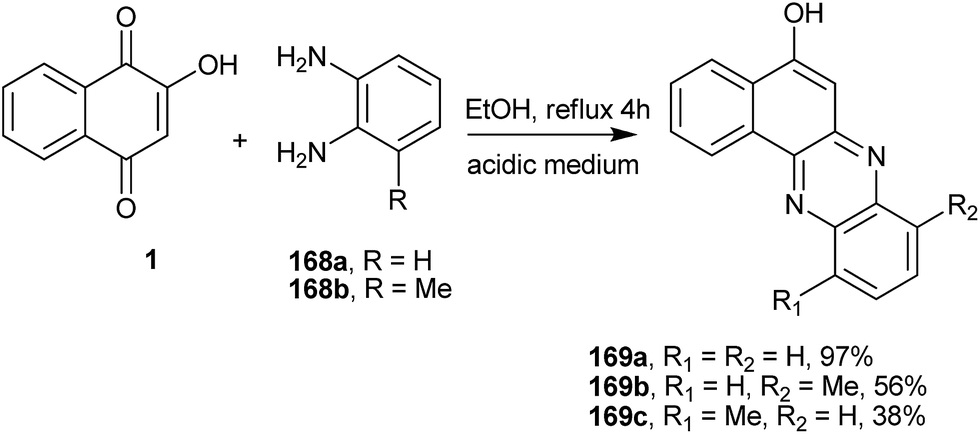 | ||
| Scheme 46 | ||
Song and coworkers323 have described an efficient one-pot synthesis of 5H-spiro[benzo[7,8]chromeno[2,3-c]pyrazole-7,30-indoline]-20,5,6(9H)-trione derivatives via a four-component reaction of hydrazine hydrate, β-keto esters, isatins and lawsone catalyzed by MgCl2 in ethanol. This reaction provides the advantages of excellent yields, environmentally friendly solvent, simple workup procedure and short reaction times (Scheme 47).
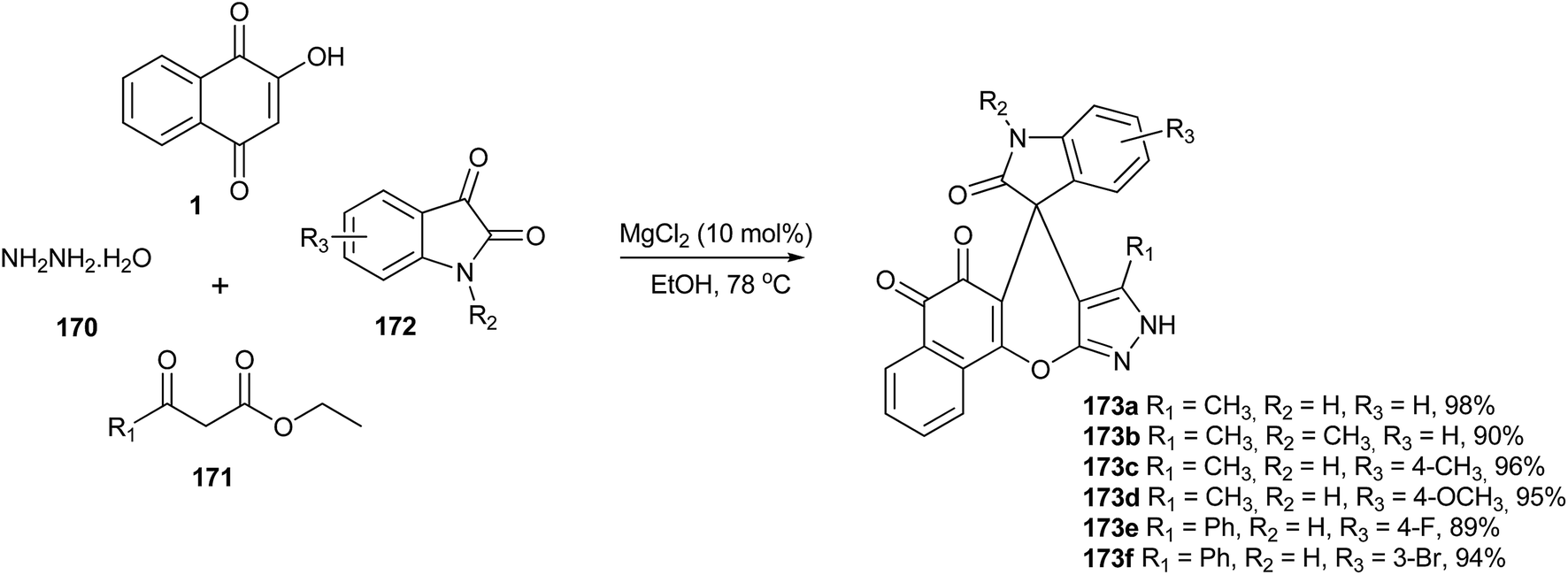 | ||
| Scheme 47 | ||
Very recently, Rocha and co-workers324 reported the synthesis of α-xyloidones 174a–j and oxepine 175 in moderate to good yields via the Knoevenagel condensation–electrocyclization reaction between 1 and α,β-unsaturated aldehydes in the presence of formic acid at reflux or microwave irradiation. Given the difficulty involved with synthesizing other xyloidone derivatives, such as 174i and 174j, an alternative synthetic method was investigated, involving the O- and C-alkylation of 1, followed by cyclization using ring-closing metathesis (RCM, Scheme 48 and Table 2).
 | ||
| Scheme 48 | ||
| Entry | Product | Δ | MW | Method |
|---|---|---|---|---|
| 1 | 174a, R1 = R2 = Me, R3 = H | 65% (1 h) | 73% (1.5 h, 80 °C) | (i) |
| 2 | 174b, R1 = Me, R2 = CH2-prenyl, R3 = H | 8% (1 h) | 70% (5.5 h, 80 °C) | (i) |
| 3 | 174c, R1 = R2 = H, R3 = Me | 26% (6 h) | 70% (1.2 h, 80 °C) | (i) |
| 4 | 174d, R1 = Me, R2 = R3 = H | 14% (1 h) | 54% (55 min, 80 °C) | (i) |
| 5 | 174e, R1 = R3 = H, R2 = Pr | 6% (1 h) | 46% (1.5 h, 80 °C) | (i) |
| 6 | 174f, R1 = R3 = H, R2 = furyl | 3% (1 h) | Degraded | (i) |
| 7 | 174g, R1 = R3 = H, R2 = phenyl | — | 10% (2 h, 80 °C) | (i) |
| 8 | 174h, R1 = R2 = R3 = H | — | — | (i) |
| Entry | Product | Steps | Method | ||
|---|---|---|---|---|---|
| a or b | c | d | |||
| 9 | 174i, R1 = R2 = R3 = H | 80% (a) | 66% | 73% | (ii) |
| 10 | 174j, R1 = R2 = H, R3 = CH(CH3)2 |
80% (a) | 45% | 88% | (ii) |
| 11 | 175 | 55% (b) | 40% | 54% | (ii) |
Cardoso and co-workers have also synthesized 18 new 1,2-furanonaphthoquinones tethered to 1,2,3-1H-triazoles from nor-lapacol (120b) and evaluated their activity against four different leukemia cell lines. Compounds 177q and 177r showed better results than the classical therapeutic drugs doxorubicin and cisplatin, with IC50 values for lymphoid cells (MOLT4) of 0.48 μM and 0.05 μM, respectively (Scheme 49).325
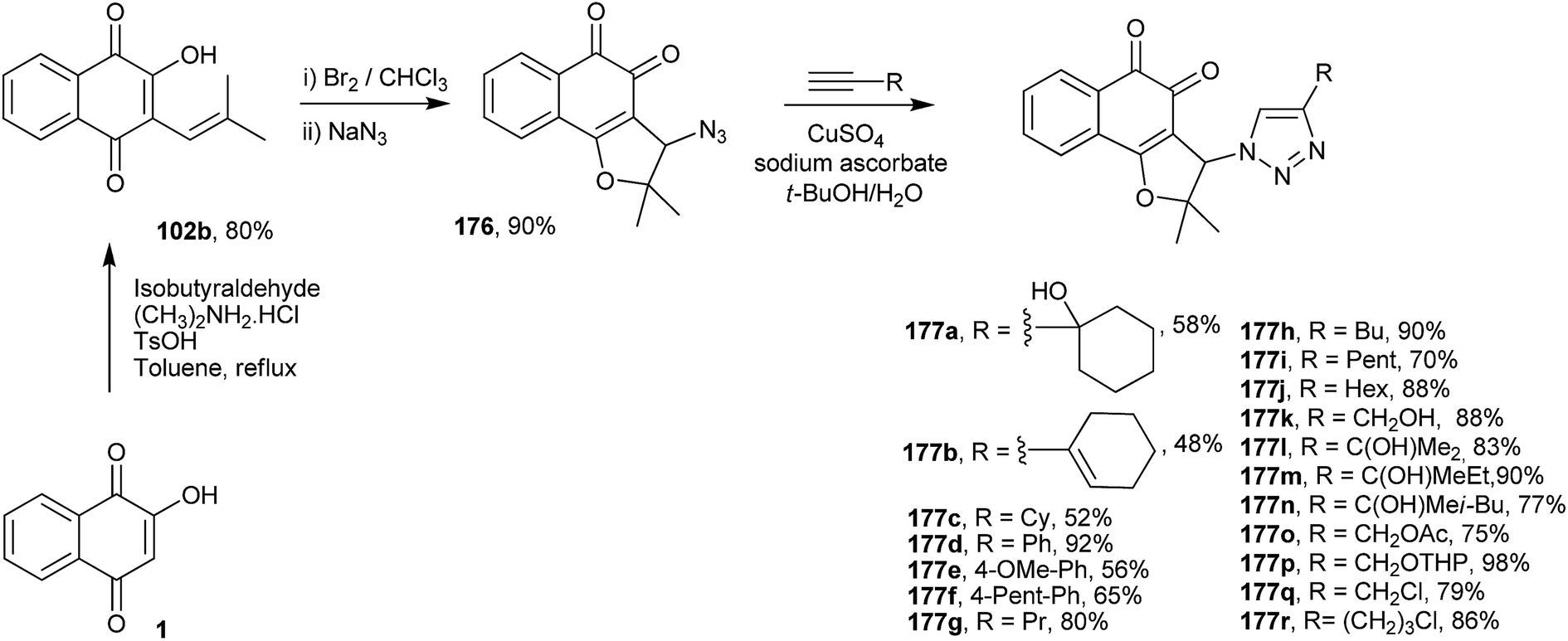 | ||
| Scheme 49 | ||
The same research group has also reported the synthesis of new nor-β-lapachones containing phenylthio groups at position C-3 of the furan ring (178a–g, Scheme 50). These compounds were screened against bloodstream trypomastigotes of Trypanosoma cruzi and showed a broad range of activities (IC50/24 h from 9.2 to 182.7 μM). The derivative 178b was 11 times more active than the standard drug, benznidazole (179).326
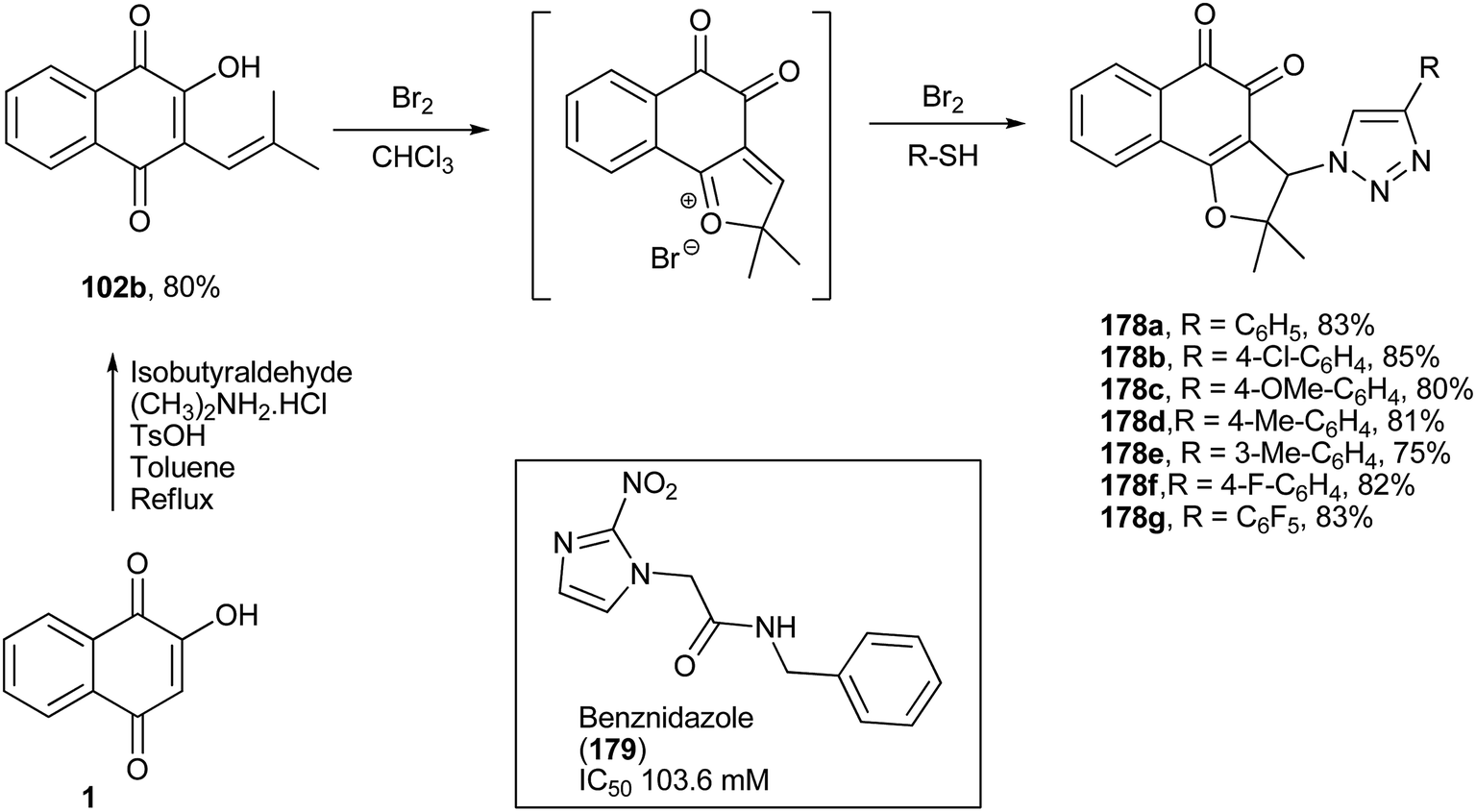 | ||
| Scheme 50 | ||
4. Conclusions
In spite of the controversy over the use of 1 as a hair and skin dye, due to its toxicological and biological side effects, there is no doubt that this molecule is an extremely useful starting material for organic synthesis. The reactions of lawsone (1), described in hundreds of publications, have allowed the preparation of a variety of biologically active compounds that are potentially useful for the treatment of several diseases. Thus, the chemistry of this molecule is sufficiently useful to justify it being reviewed.Acknowledgements
The authors would like to acknowledge the agencies that fund their research and their fellowships: CNPq (National Council of Research of Brazil), CAPES and FAPERJ, in particular, through PRONEX-FAPERJ (E-26/110.574/2010) and PRONEM-FAPERJ (E-26/110.157/2014).References
- R. H. Thompson, Naturally Occurring Quinones IV: Recents Advances, Champman & Hall, London, 1997 Search PubMed
.
- P. T. Gallagher, Contemp. Org. Synth., 1996, 3, 433–446 RSC
- J. H. Lee, J. H. Cheong, Y. M. Park and Y. H. Choi, Pharmacol. Res., 2005, 51, 553–560 CrossRef CAS PubMed
- C. Asche, Mini-Rev. Med. Chem., 2005, 5, 449–467 CrossRef CAS
- E. B. Skibo, C. Xing and R. T. Dorr, J. Med. Chem., 2001, 44, 3545–3562 CrossRef CAS PubMed
- A. F. dos Santos, P. A. L. Ferraz, F. C. de Abreu, E. Chiari, M. O. F. Goulart and A. E. G. Sant'Ana, Planta Med., 2001, 67, 92–93 CrossRef CAS
- T. P. Barbosa, C. A. Camara, T. M. S. Silva, R. M. Martins, A. C. Pinto and M. D. Vargas, Bioorg. Med. Chem., 2005, 13, 6464–6469 CrossRef CAS PubMed
- V. F. Ferreira, A. Jorqueira, A. M. T. Souza, M. N. da Silva, M. C. B. V. de Souza, R. M. Gouvea, C. R. Rodrigues, A. V. Pinto, H. C. Castro, D. O. Santos, H. P. Araújo and S. C. Bourguignon, Bioorg. Med. Chem., 2006, 14, 5459–5466 CrossRef CAS PubMed
- A. Jorqueira, R. M. Gouvêa, V. F. Ferreira, M. N. da Silva, M. C. B. V. de Souza, A. A. Zuma, D. F. B. Cavalcanti, H. P. Araújo and S. C. Bourguignon, Parasitol. Res., 2006, 99, 429–433 CrossRef PubMed
- E. N. Silva-Jr, R. F. S. Menna-Barreto, M. C. F. R. Pinto, R. S. F. Silva, D. V. Teixeira, M. C. B. V. de Souza, C. A. Simone, S. L. de Castro, V. F. Ferreira and A. V. Pinto, Eur. J. Med. Chem., 2008, 43, 1774–1780 CrossRef PubMed
- M. J. Teixeira, Y. M. de Almeida, J. R. Viana, J. G. Holanda, T. P. Rodrigues, J. R. C. Prata, I. V. B. Coelho, V. S. Rao and M. M. L. Pompeu, Phytother. Res., 2001, 15, 44–48 CrossRef CAS
- E. R. de Almeida, A. A. S. Filho, E. R. dos Santos and C. A. C. Lopes, J. Ethnopharmacol., 1990, 29, 239–241 CrossRef CAS
- S. Gafner, J. L. Wolfender, M. Nianga, H. Stoeckli-Evans and K. Hostettmann, Phytochemistry, 1996, 42, 1315–1320 CrossRef CAS
- M. Tabata, M. Tsukada and H. Fukui, Planta Med., 1982, 44, 234–236 CrossRef CAS PubMed
- C. N. Pinto, A. P. Dantas, K. C. G. de Moura, F. S. Emery, P. F. Polequevitch, M. D. Pinto, S. L. Castro and A. V. Pinto, Arzneim. Forsch., 2000, 50, 1120–1128 CAS
- A. V. Pinto and S. L. Castro, Molecules, 2009, 14, 4570–4590 CrossRef PubMed
- C. O. Salas, M. Faúndez, A. Morello, J. D. Maya and R. A. Tapia, Curr. Med. Chem., 2011, 18, 144–161 CrossRef CAS
- L. Salmon-Chemin, E. Buisine, V. Yardley, S. Kohler, M.-A. Debreu, V. Landry, C. Sergheraert, S. L. Croft, R. L. Krauth-Siegel and E. Davioud-Charvet, J. Med. Chem., 2001, 44, 548–565 CrossRef CAS PubMed
- T. Müller, L. Johann, B. Jannack, M. Brückner, D. A. Lanfranchi, H. Bauer, C. Sanchez, V. Yardley, C. Deregnaucourt, J. Schrevel, M. Lanzer, R. H. Schirmer and E. Davioud-Charvet, J. Am. Chem. Soc., 2011, 133, 11557–11571 CrossRef PubMed
- E. V. M. Santos, J. W. M. Carneiro and V. F. Ferreira, Bioorg. Med. Chem., 2004, 12, 87–93 CrossRef PubMed
- J. A. Hickman, Curr. Opin. Genet. Dev., 2002, 12, 67–72 CrossRef CAS
- A. Esteves-Souza, D. V. Figueiredo, A. Esteves, C. A. Camara, M. D. Vargas, A. C. Pinto and A. Echevarria, Braz. J. Med. Biol. Res., 2007, 40, 1399–1402 CrossRef CAS PubMed
- L. I. L. López, S. D. N. Flores, S. Y. S. Belmares and A. S. Galindo, Vitae Revista de La Facultad De Química Farmacéutica, 2014, 21, 2145–2660 Search PubMed
- L. M. Hughes, C. A. Lanteri, M. T. O'Neil, J. D. Johnson, G. W. Gribble and B. L. Trumpower, Mol. Biochem. Parasitol., 2011, 177, 12–19 CrossRef CAS PubMed
- G. Chaudhary, S. Goyal and P. Poonia, Int. J. Pharm. Sci. Drug Res., 2010, 2, 91–98 Search PubMed
- M. Kamal and T. Jawaid, Int. J. Biomed. Res., 2010, 1, 62–68 Search PubMed
- S. Rajwar and P. Khatri, Asian Journal of Pharmaceutical Sciences and Clinical Research, 2011, 1, 22–40 Search PubMed
- V. K. Jiny, K. S. Silvipriya, S. Resmi and C. I. Jolly, Inventi Impact: Cosmeceuticals, 2010, Cc/3/10 Search PubMed
- V. A. Motz, C. P. Bowers, L. M. Young and D. H. Kinder, J. Ethnopharmacol., 2012, 143, 314–318 CrossRef PubMed
- A. C. Dweek, Int. J. Cosmet. Sci., 2002, 24, 287–302 CrossRef PubMed
- K. Murakami, M. Haneda, S. Iwata and M. Yoshino, Toxicol. in Vitro, 2010, 24, 905–909 CrossRef CAS PubMed
- R. C. Montenegro, A. J. Araújo, M. T. Molina, J. D. B. Marinho Filho, D. D. Rocha, E. Lopéz-Montero, M. O. F. Goulart, E. S. Bento, A. P. N. N. Alves, C. Pessoa, M. O. de Moraes and L. V. Costa-Lotufo, Chem.-Biol. Interact., 2010, 184, 439–448 CrossRef CAS PubMed
- T. J. Monks, R. P. Hanzlik, G. M. Cohen, D. Ross and D. G. Graham, Toxicol. Appl. Pharmacol., 1992, 112, 2–16 CrossRef CAS
- J. Koyama, I. Morita, N. Kobayashi, T. Osakai, H. Hotta, J. Takayasu, H. Nishino and H. Tokuda, Cancer Lett., 2003, 201, 25–30 CrossRef CAS
- A. C. Dweck, in SCS Symposium on “Haircare-beyond the fringe”, Chepstow, S. Wales, UK, 26–27th, March 2001 Search PubMed
- D. C. Mcmillan, S. D. Sarvate, J. E. Oatis Jr. and D. J. Jollow, Toxicol. Sci., 2004, 2, 647–655 CrossRef PubMed
- J. Araldi and S. S. Guterres, Infarma, 2005, 17, 78–83 Search PubMed
- R. Pradhan, P. Dandawate, A. Vyas, S. Padhye, B. Biersack, R. Schobert, A. Ahmad and F. H. Sarkar, Curr. Drug Targets, 2012, 13, 1777–1798 CrossRef CAS
- A. E. Musa, B. Madhan, W. Madhulatha, J. R. Rao, G. A. Gasmelseed and S. Sadulla, J. Am. Leather Chem. Assoc., 2009, 104, 183–190 CAS
- M. Dogru, Z. Baysal and C. Aytekin, Prep. Biochem. Biotechnol., 2006, 36, 215–221 CrossRef CAS PubMed
- S. Ali, T. Hussain and R. Nawaz, J. Cleaner Prod., 2009, 7, 61–66 CrossRef PubMed
- A. Ashnagar and A. Shiri, Int. J. ChemTech Res., 2011, 3, 1941–1944 CAS
- D. Anju, S. Kavita, G. Jugnu, G. Munish and S. Asha, J. Pharm. Sci. Innovation, 2012, 1, 17–20 Search PubMed
- N. Rahmoun, Z. Boucherit-Otmani, K. Boucherit, M. Benabdallah and N. Choukchou-Braham, Pharm. Biol., 2013, 51, 131–135 CrossRef PubMed
- O. Habbal, S. S. Hasson, A. H. El-Hag, Z. Al-Mahrooqi, N. Al-Hashmi, Z. Al-Bimani, M. S. Al-Balushi and A. A. Al-Jabri, Asian Pac. J. Trop. Biomed., 2011, 1, 173–176 CrossRef CAS
- M. N. Rahmoun, M. Benabdallah, D. Villemin, K. Boucherit, B. Mostefa-Kara, C. Ziani-Cherif and N. Choukchou-Braham, Der Pharma Chemica, 2010, 2, 320–326 CAS
- V. K. Sharma, Tubercle, 1990, 71, 293–295 CrossRef CAS
- H. S. Muhammad and S. Muhammad, Afr. J. Biotechnol., 2005, 4, 934–937 Search PubMed
- G. J. Nohynek, R. Fautz, F. Benech-Kieffer and H. Toutain, Food Chem. Toxicol., 2004, 42, 517–543 CrossRef CAS PubMed
- R. Munday, B. L. Smith and C. M. Munday, Chem.-Biol. Interact., 1995, 98, 185–192 CrossRef CAS
- I. M. L. Majoie and D. P. Bruynzeel, Am. J. Contact Dermatitis, 1996, 7, 38–40 CrossRef CAS
- To access the report SCCNFP/0583/02 see http://ec.europa.eu/food/fs/sc/sccp/out177_en.pdf.
- A. N. Kok, M. V. Ertekin, V. Ertekin and B. Avci, Int. J. Clin. Pract., 2004, 58, 530–532 CrossRef CAS PubMed
- M. E. Kraeling, R. L. Bronaugh and C. T. Jung, Cutaneous Ocul. Toxicol., 2007, 26, 45–56 CrossRef CAS PubMed
- D. C. McMillan, S. D. Sarvate, J. E. Oatis Jr. and D. J. Jollow, Toxicol. Sci., 2004, 82, 647–655 CrossRef CAS PubMed
- S. Katar, C. Devecioglu, M. N. Özbek and S. Ecer, Clin. Exp. Dermatol., 2007, 32, 235–236 CrossRef CAS PubMed
- H. H. Kandil, M. M. Al-Ghanem, M. A. Sarwat and F. S. Al-Thallab, Ann. Trop. Paediatr., 1996, 16, 287–291 CAS
- N. M. Rahmoun, Z. Boucherit-Otmani, K. Boucherit, M. Benabdallah, D. Villemin and N. Choukchou-Braham, Med. Mal. Infect., 2012, 42, 270–275 CrossRef CAS PubMed
- N. Sritrairat, N. Nukul, P. Inthasame, A. Sansuk, J. Prasirt, T. Leewatthanakorn, U. Piamsawad, A. Dejrudee, P. Panichayupakaranant, K. Pangsomboon, N. Chanowanna, J. Hintao, R. Teanpaisan, W. Chaethong, P. Yongstar, N. Pruphetkaew, V. Chongsuvivatwong and W. Nittayananta, J. Oral Pathol. Med., 2011, 40, 90–96 CrossRef PubMed
- T. C. Fischer, C. Gosch, B. Mirbeth, M. Gselmann, V. Thallmair and K. Stich, J. Agric. Food Chem., 2012, 60, 12074–12081 CrossRef CAS PubMed
- D. Marzin and D. Kirkland, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2004, 560, 41–47 CrossRef CAS PubMed
- V. Klaus, T. Hartmann, J. Gambini, P. Graf, W. Stahl, A. Hartwig and L. O. Klotz, Arch. Biochem. Biophys., 2010, 496, 93–100 CrossRef CAS PubMed
- This report can be accessed in http://www.vkm.no/dav/d63ccd25cf.pdf.
- A. C. Watkinson, K. R. Brian, K. A. Walters and J. Hadgraft, Int. J. Cosmet. Sci., 1992, 14, 265–275 CrossRef CAS PubMed
- R. R. Korać and K. M. Khambholja, Pharmacogn. Rev., 2011, 5, 164–173 CrossRef PubMed
- A. Kosmalska, M. Zaborski and A. Masek, Przem. Chem., 2010, 89, 420–424 CAS
- A. Ostovari, S. M. Hoseinieh, M. Peikari, S. R. Shadizadeh and S. J. Hashemi, Corros. Sci., 2009, 51, 1935–1949 CrossRef CAS PubMed
- A. Y. El-Etre, M. Abdallah and Z. E. El-Tantawy, Corros. Sci., 2005, 47, 385–395 CrossRef CAS PubMed
- R. Andoong and H. Chihchiang, Environ. Sci. Technol., 2005, 39, 7460–7468 CrossRef
- R.-A. Doong and Y.-J. Lai, Water Res., 2005, 39, 2309–2318 CrossRef CAS PubMed
- R. Jelly, S. W. Lewi, C. Lennard, K. F. Limw and J. Almog, Chem. Commun., 2008, 3513–3515 RSC
- Y. M. Hijji, B. Barare and Y. Zhang, Sens. Actuators, B, 2012, 169, 106–112 CrossRef CAS PubMed
- A. Mangini, Gazz. Chim. Ital., 1931, 61, 820–826 CAS
- F. L. S. Bustamante, M. M. P. Silva, W. A. Alves, C. B. Pinheiro, J. A. L. C. Resende and M. Lanznaster, Polyhedron, 2012, 42, 43–49 CrossRef CAS PubMed
- H. Kido, W. C. Fernelius and C. G. Haas Jr., Anal. Chim. Acta, 1960, 23, 116–123 CAS
- I. Casanova, A. Sousa-Pedrares, J. Viqueira, M. L. Durán, J. Romero, A. Sousa and J. A. García-Vázquez, New J. Chem., 2013, 37, 2303–2316 RSC
- S. Salunke-Gawali, L. Kathawate, Y. Shinde, V. G. Puranik and T. Weyhermüller, J. Mol. Struct., 2012, 1010, 38–45 CrossRef CAS PubMed
- G. Valle-Bourrouet, V. M. Ugalde-Saldívar, M. Gómez, L. A. Ortiz-Frade, I. González and C. Frontana, Electrochim. Acta, 2010, 55, 9042–9050 CrossRef CAS PubMed
- C. G. Lima, F. Flávio, T. Araujo, A. DuFresne and J. M. Knudsen, Inorg. Nucl. Chem. Lett., 1971, 7, 513–517 CrossRef
- S. Padhye, P. Garge and M. P. Gupta, Inorg. Chim. Acta, 1988, 152, 37–40 CrossRef CAS
- P. Garge, R. Chikate, S. Padhye, J.-M. Savariault, P. de Loth and J.-P. Tuchagues, Inorg. Chem., 1990, 29, 3315–3320 CrossRef CAS
- M. E. Bodini, P. E. Bravo and M. V. Aranciba, Polyhedron, 1994, 13, 497–503 CrossRef CAS
- S. Salunke-Gawali, S. Y. Rane, V. G. Puranik, C. Guyard-Duhayon and F. Varret, Polyhedron, 2004, 23, 2541–2547 CrossRef CAS PubMed
- S. Oramas-Royo, C. Torrejón, I. Cuadrado, R. Hernández-Molina, S. Hortelano, A. Estévez-Braun and B. de Las Heras, Bioorg. Med. Chem., 2013, 21, 2471–2477 CrossRef CAS PubMed
- A. M. El-Hendawy, Polyhedron, 1991, 10, 2511–2518 CrossRef CAS
- R. Sauriasari, D.-H. Wang, Y. Takemura, K. Tsutsui, N. Masuoka, K. Sano, M. Horita, B.-L. Wang and K. Ogino, Toxicology, 2007, 235, 103–111 CrossRef CAS PubMed
- F. L. S. Bustamante, J. M. Metello, F. A. V. de Castro, C. B. Pinheiro, M. D. Pereira and M. Lanznaster, Inorg. Chem., 2013, 52, 1167–1169 CrossRef CAS PubMed
- Y. Yasin, N. M. Ismail, M. Z. Hussein and N. Aminudin, J. Biomed. Nanotechnol., 2011, 7, 486–488 CrossRef CAS PubMed
- V. D. Kelkar, R. R. Gokhale and H. R. Gholap, Synth. React. Inorg. Met.-Org. Chem., 1998, 28, 1253–1264 CrossRef CAS PubMed
- S. Spyroudis, Molecules, 2000, 5, 1291–1330 CrossRef CAS PubMed
- S. C. Hooker, J. Chem. Soc., 1892, 61, 611–650 RSC
- Note: Hooker did not reported de yield of lawsone in this reference.
- J. M. Heerding and H. W. Moore, J. Org. Chem., 1991, 56, 4048–4050 CrossRef CAS
- K. H. Lee and H. W. Moore, Tetrahedron Lett., 1993, 34, 235–238 CrossRef CAS
- C. R. Solorio-Alvarado, E. Peña-Cabrera and C. Alvarez-Toledano, ARKIVOC, 2004, 64–70 CrossRef CAS
- L. F. Fieser, J. Am. Chem. Soc., 1948, 70, 3165–3174 CrossRef CAS
- L. F. Fieser, Org. Synth., 1943, 2, 430 Search PubMed
- A. Inoue, N. Kuroki and K. Konishi, Bull. Univ. Osaka Prefect., Ser. A, 1959, 8, 31–55 Search PubMed
- J. B. Conant and B. B. Corson, Org. Synth., 1937, 17, 9 CrossRef CAS
- L. F. Fieser, Org. Synth., 1943, 2, 42 Search PubMed
- L. F. Fieser and E. L. Martin, Org. Synth., 1955, 3, 465–467 Search PubMed
- A. S. Amarasekara and W. W. Pathmasiri, J. Chem. Soc., Perkin Trans. 1, 1995, 1653–1658 RSC
- A. B. de Oliveira, D. T. Ferreira and D. S. Raslan, Tetrahedron Lett., 1988, 29, 155–158 CrossRef CAS
- Y. Yan, E.-H. Kang, K.-E. Yang, S.-L. Tong, C.-G. Fang, S.-J. Liu and F.-S. Xiao, Catal. Commun., 2004, 5, 387–390 CrossRef CAS PubMed
- K. Yang, S.-L. Tong, Y. Yan, E.-H. Kang, F.-S. Xiao, I. Liq, X. Chang and C.-G. Fang, Chem. Res. Chin. Univ., 2005, 21, 326–333 CrossRef CAS PubMed
- J. Zhao, B. Gao and Y. Gong, Catal. Lett., 2011, 141, 1808–1813 CrossRef CAS
- M. de Min, S. Croux, C. Tournaire, M. Hocquaux, B. Jacquet, E. Oliveros and M. T. Maurette, Tetrahedron, 1992, 48, 1869–1882 CrossRef CAS
- D. Vidril-Robert, M. T. Maurette and E. Oliveros, Tetrahedron Lett., 1984, 25, 529–532 CrossRef CAS
- K. N. Singh, R. Kumar and A. K. Shukla, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2007, 46, 1347–1351 Search PubMed
- D. Villemin, M. Hammadi and M. Hachemi, Synth. Commun., 2002, 32, 1501–1515 CrossRef CAS PubMed
- K. Osowska-Pacewicka and H. Alper, J. Org. Chem., 1988, 53, 808–810 CrossRef CAS
- M. Hocquaux, B. Jacquet, D. Vioril-Robert, M. T. Maurette and E. Oliveros, Tetrahedron Lett., 1984, 25, 533–536 CrossRef CAS
- H. A. Anderson, J. Smith and K. H. Thomson, J. Chem. Soc., 1965, 2141–2144 RSC
- A. C. Baillie and R. H. Thomson, J. Chem. Soc. C, 1966, 2184 RSC
- C. Wright and G. V. Ullas, J. Labelled Compd. Radiopharm., 2002, 45, 1265–1268 CrossRef CAS PubMed
- R. Yang, D. Kizer, H. Wu, E. Volckova, X. Miao, S. M. Ali, M. Tandon, R. E. Savage, T. C. K. Chan and M. A. Ashwell, Bioorg. Med. Chem., 2008, 16, 5635–5643 CrossRef CAS PubMed
- L. F. Fieser, J. Am. Chem. Soc., 1927, 49, 857–861 CrossRef CAS
- L. F. Fieser, J. Am. Chem. Soc., 1926, 48, 3201–3214 CrossRef CAS
- J. S. Sun, A. H. Geisel and B. A. Frydman, Tetrahedron Lett., 1998, 39, 8221–8224 CrossRef CAS
- B. P. S. Khambay, D. Batty, M. Cahill and I. Denholm, J. Agric. Food Chem., 1999, 47, 770–775 CrossRef CAS PubMed
- B. P. S. Khambay, D. Batty, D. G. Beddie, I. Denholm and M. R. Cahill, Pestic. Sci., 1997, 50, 291–296 CrossRef CAS
- R. G. Cooke, Aust. J. Sci. Res., 1950, A3, 481–486 Search PubMed
- J. S. Sun, A. H. Geiser and B. Frydman, Tetrahedron Lett., 1998, 39, 8221–8224 CrossRef CAS
- N. Kongkathip, B. Kongkathip, P. Siripong, C. Sangma, S. Luangkamin, M. Niyomdecha, S. Pattanapa, S. Piyaviriyagul and P. Kongsaeree, Bioorg. Med. Chem., 2003, 11, 3179–3191 CrossRef CAS
- X. Lu, A. Altharawi, E. N. Hansen and T. E. Long, Synthesis, 2012, 3225–3230 CAS
- P. M. García-Barrantes, G. V. Lamoureux, A. L. Pérez, R. N. García-Sánchez, A. R. Martínez and A. San Feliciano, Eur. J. Med. Chem., 2013, 70, 548–557 CrossRef PubMed
- E. L. Bonifazi, C. Ríos-Luci, L. G. León, G. Burton, J. M. Padrón and R. I. Misico, Bioorg. Med. Chem., 2010, 18, 2621–2630 CrossRef CAS PubMed
- A. L. Perez, G. Lamoureux and A. Sánchez-Kopper, Tetrahedron Lett., 2007, 48, 3735–3738 CrossRef CAS PubMed
- L. W. Bieber, P. J. R. Neto and R. M. Generino, Tetrahedron Lett., 1999, 40, 4473–4476 CrossRef CAS
- S. B. Ferreira, D. R. da Rocha, J. W. M. Carneiro, W. C. Santos and V. F. Ferreira, Synlett, 2011, 1551–1554 CrossRef CAS
- A. Sharma, I. O. Santos, P. Gaur, V. F. Ferreira, C. R. S. Garcia and D. R. da Rocha, Eur. J. Med. Chem., 2013, 59, 48–53 CrossRef CAS PubMed
- E. G. Pinto, I. O. Santos, T. J. Schmidt, S. E. T. Borborema, V. F. Ferreira, D. R. Rocha and A. G. Tempone, PLoS One, 2014, 9, e105127 Search PubMed
- Y. E. Sabutskii, S. G. Polonik, V. A. Denisenko and P. S. Dmitrenok, Synthesis, 2014, 46, 2763–2770 CrossRef CAS
- M. V. de Araújo, P. S. O. de Souza, A. C. de Queiroz, C. B. B. da Matta, A. B. Leite, A. E. da Silva, J. A. A. de França, T. M. S. Silva, C. A. Camara and M. S. Alexandre-Moreira, Molecules, 2014, 19, 15180–15195 CrossRef PubMed
- M. T. Leffler and R. T. Hathaway, J. Am. Chem. Soc., 1948, 70, 3222–3223 CrossRef CAS
- C. E. Dalgliesh, J. Am. Chem. Soc., 1949, 71, 1697–1702 CrossRef CAS
- N. M. F. Lima, C. S. Correia, P. A. L. Ferraz, A. V. Pinto, M. C. R. F. Pinto, A. E. G. Santana and M. O. F. Goulart, J. Braz. Chem. Soc., 2002, 13, 822–829 CrossRef CAS
- A. Baramee, A. Coppin, M. Mortuaire, L. Pelinski, S. Tomavo and J. Brocard, Bioorg. Med. Chem., 2006, 14, 1294–1302 CrossRef CAS PubMed
- R. G. Fiorot, J. F. Allochio Filho, T. M. C. Pereira, V. Lacerda Jr., R. B. dos Santos, W. Romão and S. J. Greco, Tetrahedron Lett., 2014, 55, 4373–4377 CrossRef CAS PubMed
- M. Dabiri, Z. N. Tisseh and A. Bazgir, Dyes Pigm., 2011, 89, 63–69 CrossRef CAS PubMed
- H. R. Shaterian and M. Mohammadnia, J. Mol. Liq., 2013, 117, 353–360 CrossRef PubMed
- M. Ayaz and B. Westermann, Synlett, 2010, 10, 1489–1492 Search PubMed
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed
- A. P. Neves, C. C. Barbosa, S. J. Greco, M. D. Vargas, L. C. Visentin, C. B. Pinheiro, A. S. Mangrich, J. P. Barbosa and G. L. Costa, J. Braz. Chem. Soc., 2009, 20, 712–727 CrossRef CAS
- A. P. Neves, M. D. Vargas, C. A. Téllez Soto, J. M. Ramos, L. C. Visentin, C. B. Pinheiro, A. S. Mangrich and E. I. P. de Rezende, Spectrochim. Acta, Part A, 2012, 94, 152–163 CrossRef CAS PubMed
- A. P. Neves, K. C. B. Maia, M. D. Vargas, L. C. Visentin, A. Casellato, M. A. Novak and A. S. Mangrich, Polyhedron, 2010, 29, 2884–2891 CrossRef CAS PubMed
- G. B. Silva, A. P. Neves, M. D. Vargas, W. A. Alves, J. D. B. Marinho-Filho, C. Pessoa, M. O. Moraes and L. V. Costa-Lotufo, J. Braz. Chem. Soc., 2013, 24, 675–684 CrossRef
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC
- A. P. Neves, G. B. da Silva, M. D. Vargas, C. B. Pinheiro, L. C. Visentin, J. D. Filho, A. J. Araújo, L. V. Costa-Lotufo, C. Pessoa and M. O. de Moraes, Dalton Trans., 2010, 39, 10203–10216 RSC
- A. P. Neves, M. X. G. Pereira, E. J. Peterson, R. Kipping, M. D. Vargas, F. P. Silva-Jr, J. W. M. Carneiro and N. P. Farrell, J. Inorg. Biochem., 2013, 119, 54–64 CrossRef CAS PubMed
- A. M. V. Pinto, J. P. G. Leite, A. P. Neves, G. B. da Silva, M. D. Vargas and I. C. P. N. Paixão, Arch. Virol., 2014, 159, 1827–1833 CAS
- H. E. Zaugg, J. Am. Chem. Soc., 1949, 71, 1890–1891 CrossRef CAS
- C. Salas, R. A. Tapia, K. Ciudad, V. Armstrong, M. Orellana, U. Kemmerling, J. Ferreira, J. D. Maya and A. Morello, Bioorg. Med. Chem., 2008, 16, 668–674 CrossRef CAS PubMed
- R. Cassis, R. Tapia and J. A. Valderrama, J. Heterocycl. Chem., 1984, 21, 865–868 CrossRef CAS PubMed
- J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315–3326 CrossRef CAS PubMed
- R. G. Soengas, J. C. Estévez, R. J. Estevez and M. A. Maestro, Tetrahedron: Asymmetry, 2003, 14, 1653–1658 CrossRef CAS
- D. K. Barange, V. Kavala, B. R. Raju, C.-W. Kuo, C. Tseng and Y.-C. Tu, Tetrahedron Lett., 2009, 50, 5116–5119 CrossRef CAS PubMed
- J. M. Otero, J. C. Barcia, C. O. Salas, P. Thomas, J. C. Estévez and R. J. Estévez, Tetrahedron, 2012, 68, 1612–1621 CrossRef CAS PubMed
- W.-M. Zhou, H. Liu and D.-M. Du, Org. Lett., 2008, 10, 2817–2820 CrossRef CAS PubMed
- Y.-F. Wang, W. Zhang, S.-P. Luo, G.-C. Zhang, A.-B. Xia, X.-S. Xu and D.-Q. Xu, Eur. J. Org. Chem., 2010, 4981–4985 CrossRef CAS PubMed
- T. Liu, Y. Wang, G. Wu, H. Song, Z. Zhou and C. Tang, J. Org. Chem., 2011, 76, 4119–4124 CrossRef CAS PubMed
- M. Matsuura, Y. Yamada, K. Sakai and T. Sato, Jpn. Kokai Tokkyo Koho JP 53015352 A 19780213, 1978
- K. Chandrasenan and R. H. Thomson, Tetrahedron, 1971, 27, 2529–2539 CrossRef CAS
- B. Hazra, S. Acharya, R. Ghosh, A. Patra and A. Banerjee, Synth. Commun., 1999, 29, 1571–1576 CrossRef CAS PubMed
- C. G. de Lima and A. DuFresne, Inorg. Nucl. Chem. Lett., 1971, 7, 843–846 CrossRef CAS
- F. L. S. Bustamante, J. M. Metello, F. A. V. de Castro, C. B. Pinheiro, M. D. Pereira and M. Lanznaster, Inorg. Chem., 2013, 52, 1167–1169 CrossRef CAS PubMed
- F. L. S. Bustamante, F. S. Miranda, F. A. V. de Castro, J. A. L. C. Resende, M. D. Pereira and M. Lanznaster, J. Inorg. Biochem., 2014, 132, 37–44 CrossRef CAS PubMed
- G. J. Kapadia, V. Balasubramanian, H. Tokuda, H. Konoshima, M. Takasaki, J. Koyama, K. Tagahaya and H. Nishino, Cancer Lett., 1997, 113, 47–53 CrossRef CAS
- N. Lall, M. das Sarma, B. Hazra and J. J. M. Meyer, J. Antimicrob. Chemother., 2003, 51, 435–438 CrossRef CAS PubMed
- L. Han, Y. Zhou and W. Zhao, Cryst. Growth Des., 2008, 8, 2052–2054 CAS
- Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001, vol. 1 and 2 Search PubMed
- B. Quiclet-Sire and S. Z. Zard, Beilstein J. Org. Chem., 2013, 9, 557–576 CrossRef CAS PubMed
- L. F. Fieser, E. Berliner, F.
J. Bondhus, F. C. Chang, W. G. Dauben, M. G. Ettlinge, G. Fawaz, M. Fields, M. Fieser, C. Heidelberger, H. Heymann, A. M. Seligman, W. R. Vaughan, A. G. Wilson, E. Wilson, M. I. Wu, M. T. Leffler, K. E. Hamlin, R. J. Hathaway, E. J. Matson, E. E. Moore, M. B. Moore, R. T. Rapala and H. E. Zaugg, J. Am. Chem. Soc., 1948, 70, 3151–3155 CrossRef CAS
- L. F. Fieser, E. Berliner, F. Bondhus, F. C. Chang, W. G. Dauben, M. G. Ettlinger, G. Fawaz, M. Fields, C. Heidelberger, H. Heymann, W. R. Vaughan, A. G. Wilson, E. Wilson, M. I. Wu, M. T. Leffler, K. E. Hamlin, E. J. Mataon, E. E. Moore, M. B. Moore and H. E. Zaugg, J. Am. Chem. Soc., 1948, 70, 3174–3215 CrossRef CAS
- N. Jacobsen and K. Torssell, Liebigs Ann. Chem., 1972, 763, 135–147 CrossRef CAS PubMed
- N. Jacobsen and K. Torssell, Acta Chem. Scand., 1973, 27, 3211–3215 CrossRef CAS
- N. Jacobsen, Org. Synth., 1988, 6, 890–892 Search PubMed
- G. A. Kraus and P. K. Choudhury, Tetrahedron Lett., 2001, 42, 6649–6650 CrossRef CAS
- B. L. Gu and J. Zhang, Recl. Trav. Chim. Pays-Bas, 1991, 110, 99–103 Search PubMed
- D. Tauraité, V. Razumas and E. Butkus, Chem. Phys. Lipids, 2009, 159, 45–50 CrossRef PubMed
- P. Boehm, K. Cooper and A. Hudson, J. Med. Chem., 1981, 24, 295–299 CrossRef CAS
- A. O. da Silva, R. S. Lopes, R. V. de Lima, C. S. S. Tozatti, M. R. Marques, S. de Albuquerque, A. Beatriz and D. P. de Lima, Eur. J. Med. Chem., 2013, 60, 51–56 CrossRef PubMed
- E. Paternò, Gazz. Chim. Ital., 1882, 12, 337–392 Search PubMed
- E. Paternò and G. Minunni, Gazz. Chim. Ital., 1889, 19, 601 Search PubMed
- S. C. Hooker, J. Am. Chem. Soc., 1936, 58, 1190–1197 CrossRef CAS
- S. C. Hooker, J. Chem. Soc., 1892, 61, 611–650 RSC
- S. C. Hooker, J. Chem. Soc., 1896, 68, 1355–1381 RSC
- S. C. Hooker, J. Am. Chem. Soc., 1936, 58, 1174–1179 CrossRef CAS
- S. C. Hooker, J. Am. Chem. Soc., 1936, 58, 1179–1189 CrossRef CAS
- S. C. Hooker and A. Steyermark, J. Am. Chem. Soc., 1936, 58, 1198–1201 CrossRef CAS
- H. W. Moore and R. J. Wikholm, Rearrangements of Quinones, in The Chemistry of Quinonoid compounds Part 1, ed. S. Patai, John Wiley & Sons, NY, 1974 Search PubMed
- A. S. Cunha, E. L. S. Lima, A. C. Pinto, A. Esteves-Souza, A. Echevarria, C. A. Camara, M. D. Vargas and J. C. Torres, J. Braz. Chem. Soc., 2006, 17, 439–442 CrossRef CAS
- L. F. Fieser, The Scientific method. A personal account of unusual projects in war and in peace, Reinhold Publishing Corporation, NY, 1964, p. 171 Search PubMed
- L. F. Fieser, L. Hartwell and A. M. Seligman, J. Am. Chem. Soc., 1936, 58, 1223–1228 CrossRef CAS
- L. F. Fieser and M. Fieser, J. Am. Chem.
Soc., 1947, 70, 3215–3222 CrossRef
- D. Cram, J. Am. Chem. Soc., 1949, 71, 3950–3952 CrossRef CAS
- K. Lee, P. Turnbull and H. W. Moore, J. Org. Chem., 1995, 60, 461–464 CrossRef CAS
- A. I. Franscisco, G. Q. Silveira, J. A. L. C. Resende, T. L. Balliano, V. R. S. Malta and A. V. Pinto, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o341 CAS
- K. O. Eyong, M. Puppala, P. S. Kumar, M. Lamshöft, G. N. Folefoc, M. Spiteller and S. Baskaran, Org. Biomol. Chem., 2013, 11, 459–468 CAS
- H. R. Nasiri, M. Madej, R. Panisch, M. Lafontaine, J. W. Bats, C. R. Lancaster and H. Schwalbe, J. Med. Chem., 2013, 56, 9530–9541 CrossRef CAS PubMed
- C. G. T. Oliveira, F. F. Miranda, V. F. Ferreira, C. C. Freitas, R. F. Rabello, J. M. Carballido and L. C. D. Corrêa, J. Braz. Chem. Soc., 2001, 12, 339–345 CrossRef CAS
- M. A. Gouda, H. F. Eldien, M. M. Girges and M. A. Berghot, Med. Chem., 2013, 3, 2228–2232 Search PubMed
- N. H. Gokhale, K. Shirisha, S. B. Padhye, S. L. Croft, H. D. Kendrick and V. Mckeec, Bioorg. Med. Chem. Lett., 2006, 16, 430–432 CrossRef CAS PubMed
- A. I. Francisco, M. D. Vargas, T. P. Fragoso, J. W. M. Carneiro, A. Casellato, F. C. da Silva, V. F. Ferreira, J. P. Barbosa, C. Pessoa, L. V. Costa-Lotufo, J. D. B. Marinho Filho, M. O. de Moraes and A. S. Mangrich, J. Braz. Chem. Soc., 2010, 21, 1293–1302 CrossRef
- S. C. Hooker, J. Am. Chem. Soc., 1936, 58, 1163–1167 CrossRef CAS
- R. G. F. Giles and G. H. P. Roo, J. Chem. Soc., Perkin Trans. 1, 1976, 1632–1635 RSC
- K. Bock, N. Jacobsen and B. Terem, J. Chem. Soc., Perkin Trans. 1, 1986, 659–664 RSC
- E. Malamidou-Xenikaki and S. Spyroudis, Synlett, 2008, 2725–2740 CAS
- E. Hatzigrigoriou, S. Spyroudis and A. Varvoglis, Liebigs Ann. Chem., 1989, 167–170 CrossRef CAS PubMed
- K. W. Stagliano and H. C. Malinakova, Tetrahedron Lett., 1997, 38, 6617–6620 CrossRef CAS
- K. W. Stagliano and H. C. Malinakova, J. Org. Chem., 1999, 64, 8034–8040 CrossRef CAS
- G. Kazantzi, E. Malamidou-Xenikaki and S. Spyroudis, Synlett, 2006, 2597–2600 CAS
- E. Glinis, E. Malamidou-Xenikaki, H. Skouros, S. Spyroudis and M. Tsanakopoulou, Tetrahedron, 2010, 66, 5786–5792 CrossRef CAS PubMed
- A. L. Perez, G. Lamoureux and B. Y. Zhen-Wu, Tetrahedron Lett., 2007, 48, 3995–3998 CrossRef CAS PubMed
- A. L. Perez, G. Lamoureux and A. Herrera, Synth. Commun., 2004, 3389–3397 CrossRef CAS PubMed
- V. K. Tandon, D. B. Yadav, R. V. Singh, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 5324–5328 CrossRef CAS PubMed
- A. K. Macharla, R. C. Nappunni, M. R. Marri, S. Peraka and N. Nama, Tetrahedron Lett., 2012, 53, 191–195 CrossRef CAS PubMed
- A. B. de Oliveira, D. S. Raslan, M. C. M. Miraglia, A. A. L. Mesquita, C. L. Zani, D. T. Ferreira and J. G. S. Maia, Quim. Nova, 1990, 13, 302–307 CAS
- A. B. Pardee, Y. Z. Li and C. J. Li, Curr. Cancer Drug Targets, 2002, 2, 227–242 CrossRef CAS
- E. N. da Silva Júnior, M. C. B. V. de Souza, A. V. Pinto, M. C. F. R. Pinto, M. O. F. Goulart, C. Pessoa, L. Costa-Lotufo, R. C. Montenegro, M. O. Moraes and V. F. Ferreira, Bioorg. Med. Chem., 2007, 15, 7035–7041 CrossRef PubMed
- S. M. Planchon, S. Wuerzberger, B. Frydman, D. T. Witiak, P. Hutson, D. R. Church, G. Wilding and D. A. Boothman, Cancer Res., 1996, 55, 3706–3711 Search PubMed
- C. L. Li, C. Wang and A. B. Pardee, Cancer Res., 1995, 55, 3712–3715 CAS
- M. Weller, S. Winter, C. Schmidt, P. Esser, A. Fontana, J. Dichgans and P. Groscurth, Int. J. Cancer, 1997, 73, 707–714 CrossRef CAS
- C. C. Lai, T. J. Liu, L. K. Ho, M. J. Don and Y. P. Chau, Histol. Histopathol., 1998, 13, 89–97 CAS
- H. J. Woo, K. Y. Park, C. H. Rhu, W. H. Lee, B. T. Choi, G. Y. Kim, Y. M. Park and Y. H. Choi, J. Med. Food, 2006, 9, 161–168 CrossRef CAS PubMed
- S. M. Wuerzberger, J. J. Pink, S. M. Planchon, K. L. Byers, W. G. Bornmann and D. A. Boothman, Cancer Res., 1998, 58, 1876–1885 CAS
- C. J. Li, Y. Z. Li, A. V. Pinto and A. B. Pardee, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13369–13374 CrossRef CAS
- E. A. Bey, M. S. Bentle, K. E. Reinicke, Y. Dong, C.-R. Yang, L. Girard, J. D. Minna, W. G. Bornmann, J. Gao and G. A. Boothman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11832–11837 CrossRef CAS PubMed
- Y. Li, C. J. Li, D. Yu and A. B. Pardee, Mol. Med., 2000, 6, 1008–1015 CAS
- M. Dubin, S. H. Fernadez Villamil and A. O. Stoppani, Medicina (Buenos Aires, Argent.), 2001, 61, 343–350 CAS
- A. G. Ravelo, A. Estévez-Braun, H. Chávez-Orellana, E. Pérez-Sacau and D. Mesa-Siverio, Curr. Top. Med. Chem., 2004, 4, 241–350 CrossRef CAS
- J. J. Marr and R. Docampo, Rev. Infect. Dis., 1986, 8, 884–903 CrossRef CAS PubMed
- S. G. Goijman and A. O. M. Stoppani, Arch. Biochem. Biophys., 1985, 240, 273–280 CrossRef CAS
- V. F. Ferreira, A. Jorqueira, A. M. T. Souza, M. N. da Silva, M. C. B. V. de Souza, R. M. Gouvêa, C. R. Rodrigues, A. V. Pinto, H. C. Castro, D. O. Santos, H. P. Araújo and S. C. Bourguignon, Bioorg. Med. Chem., 2006, 14, 5459–5466 CrossRef CAS PubMed
- A. Jorqueira, R. M. Gouvêa, V. F. Ferreira, M. N. da Silva, M. C. B. V. de Souza, A. A. Zuma, D. F. B. Cavalcanti, H. P. Araújo and S. C. Bourguignon, Parasitol. Res., 2006, 9, 429–433 CrossRef PubMed
- K. C. G. de Moura, F. S. Emery, C. Neves-Pinto, M. C. F. R. Pinto, A. P. Dantas, K. Salomão, S. L. de Castro and A. V. Pinto, J. Braz. Chem. Soc., 2001, 12, 325–338 CrossRef CAS
- K. C. G. de Moura, K. Salomão, R. F. S. Menna-Barreto, F. S. Emery, M. C. F. R. Pinto, A. V. Pinto and S. L. Castro, Eur. J. Med. Chem., 2004, 39, 639–645 CrossRef CAS PubMed
- C. Neves-Pinto, V. R. S. Malta, M. C. F. R. Pinto, R. H. A. Santos, S. L. de Castro and A. V. Pinto, J. Med. Chem., 2002, 45, 2112–2115 CrossRef CAS PubMed
- S. B. Ferreira, F. C. da Silva, A. C. Pinto, D. T. G. Gonzaga and V. F. Ferreira, J. Heterocycl. Chem., 2009, 46, 1080–1096 CrossRef CAS PubMed
- V. F. Ferreira, A. V. Pinto and L. C. M. Coutada, An. Acad. Bras. Cienc., 1980, 52, 477–479 Search PubMed
- A. B. de Oliveira, D. T. Ferreira and D. S. Raslan, Tetrahedron Lett., 1988, 29, 155–158 CrossRef CAS
- S. Jiménez-Alonso, H. C. Orellana, A. Estévez-Braun, A. G. Ravelo, E. Pérez-Sacau and F. Machín, J. Med. Chem., 2008, 51, 6761–6772 CrossRef PubMed
- V. Nair and P. M. Treesa, Tetrahedron Lett., 2001, 42, 4549–4551 CrossRef CAS
- F. C. da Silva, S. B. Ferreira, C. R. Kaiser, A. C. Pinto and V. F. Ferreira, J. Braz. Chem. Soc., 2009, 20, 1478–1482 CrossRef CAS
- D.-Q. Peng, Y. Liu, Z.-F. Lu, Y.-M. Shen and J.-H. Xu, Synthesis, 2008, 1182–1192 CAS
- J. M. Khurana, B. Nand and P. Saluja, Tetrahedron, 2010, 66, 5637–5641 CrossRef CAS PubMed
- I. V. Magedov, A. S. Kireev, A. R. Jenkins, N. M. Evdokimov, D. T. Lima, P. Tongwa, J. Altig, W. F. Steelant, S. van slambrouck, M. Y. Antipin and A. Kornienko, Bioorg. Med. Chem. Lett., 2012, 22, 5195–5198 CrossRef CAS PubMed
- A. J. M. da Silva, C. D. Buarque, F. V. Brito, L. Aurelian, L. F. Macedo, L. H. Malkas, R. J. Hickey, D. V. S. Lopes, F. Noel, Y. L. B. Murakami, N. M. V. Silva, P. A. Melo, R. R. B. Caruso, N. G. Castro and P. R. R. Costa, Bioorg. Med. Chem., 2002, 10, 2731–2738 CrossRef CAS
- M. Rueping, E. Sugiono and E. Merino, Angew. Chem., Int. Ed., 2008, 47, 3046–3049 CrossRef CAS PubMed
- Y. Gao, Q. Ren, S.-M. Ang and J. Wang, Org. Biomol. Chem., 2011, 9, 3691–3697 CAS
- J. J. Omolo, V. Maharaj, D. Naidoo, T. Klimkait, H. M. Malebo, S. Mtullu, H. V. M. Lyaruu and C. B. de Koning, J. Nat. Prod., 2012, 75, 1712–1716 CrossRef CAS PubMed
- C.-T. Yen, K. Nakagawa-Goto, T.-L. Hwang, S. L. Morris-Natschke, K. F. Bastow, Y.-C. Wu and K.-H. Lee, Bioorg. Med. Chem., 2012, 22, 4018–4022 CrossRef CAS PubMed
- L. Wu, J. Zhang, L. Fang, C. Yang and F. Yan, Dyes Pigm., 2010, 86, 93–96 CrossRef CAS PubMed
- L. Q. Wu, Y. F. Wu, C. H. Yang, L. M. Yang and L. Yang, J. Braz. Chem. Soc., 2010, 21, 941–945 CrossRef CAS
- B. Du, G. Cai, B. Zhao, X. Meng, X. Wang and Y. Li, Res. Chem. Intermed., 2013, 39, 1323–1333 CrossRef CAS
- A. A. Rahmati, Chin. Chem. Lett., 2010, 21, 761–764 CrossRef CAS PubMed
- Y. Li, B. Du, X. Xu, D. Shi and S. Ji, Chin. J. Chem., 2009, 27, 1510–1514 CrossRef CAS PubMed
- M. M. Amini, Y. Fazaeli, Z. Yassaee, S. Feizi and A. Bazgir, Open Catal. J., 2009, 2, 40–44 CrossRef CAS
- A. Bazgir, Z. N. Tisseh and P. Mirzaei, Tetrahedron Lett., 2008, 49, 5165–5168 CrossRef CAS PubMed
- H. K. Jardosh and M. P. Patel, Med. Chem. Res., 2013, 22, 2954–2963 CrossRef CAS
- H. R. Shaterian, M. Ranjbar and K. Azizi, J. Mol. Liq., 2011, 162, 95–99 CrossRef CAS PubMed
- P. N. Batalha, Rev. Virtual Quim., 2012, 4, 13–45 CAS
- M. Dabiri, Z. N. Tisseh, M. Nobarah and A. Bazgir, Helv. Chim. Acta, 2011, 94, 824–830 CrossRef CAS PubMed
- B. D. Bala, K. Balamurugan, S. Perumal and S. Facile, Tetrahedron Lett., 2011, 52, 4562–4566 CrossRef PubMed
- P. Moosophon, S. Kanokmedhakul, K. Kanokmedhakul and K. Soytong, J. Nat. Prod., 2009, 72, 1442–1446 CrossRef CAS PubMed
- A. E. Hay, M. C. Aumond, S. Mallet, V. Dumontet, M. Litaudon, D. Rondeau and P. Richomme, J. Nat. Prod., 2004, 67, 707–709 CrossRef CAS PubMed
- N. Hashim, M. Rahmani, M. A. Sukari, A. M. Ali, N. B. Alitheen, R. Go and H. B. M. Ismail, J. Asian Nat. Prod. Res., 2010, 12, 106–112 CrossRef CAS PubMed
- X. Li, H. Zhang, Y. Xie, Y. Hu, H. Sun and Q. Zhu, Org. Biomol. Chem., 2014, 12, 2033–2036 CAS
- K. Chibale, M. Visser, D. van Schalkwyk, P. J. Smith, A. Saravanamuthu and A. H. Fairlamb, Tetrahedron, 2003, 59, 2289–2296 CrossRef CAS
- J. M. Jamison, K. Krabill, A. Hatwalkar, E. Jamison and C. C. Tsai, Cell Biol. Int. Rep., 1990, 14, 1075–1084 CrossRef CAS
- O. Evangelinou, A. G. Hatzidimitriou, E. Velali, A. A. Pantazaki, N. Voulgarakis and P. Slanidis, Polyhedron, 2014, 72, 122–129 CrossRef CAS PubMed
- L. P. Yunnikova, V. Y. Gorokhov, T. V. Makhova and G. A. Aleksandrova, Pharm. Chem. J., 2013, 47, 139–141 CrossRef CAS
- A. G. Azebaze, M. Meyer, A. Valentin, E. L. Nguemfo, Z. T. Fomum and A. E. Nkengfack, Chem. Pharm. Bull., 2006, 54, 111–113 CrossRef CAS
- F. Zelefack, D. Guilet, N. Fabre, C. Bayet, S. V. Chevalley, S. R. Ngouela, B. N. Lenta, A. Valentin, E. Tsamo and M. G. Dijoux-Franca, J. Nat. Prod., 2009, 72, 954–957 CrossRef CAS PubMed
- S. J. Tao, S. H. Guan, W. Wang, Z. Q. Lu, G. T. Chen, N. Sha, Q. X. Yue, X. Liu and D. A. Guo, J. Nat. Prod., 2009, 72, 117–124 CrossRef CAS PubMed
- G. W. Rewcastle, G. J. Atwell, L. Zhuang, B. C. Baguley and W. A. Denny, J. Med. Chem., 1991, 34, 217–222 CrossRef CAS
- S. L. Niu, Z. L. Li, F. Ji, G. Y. Liu, N. Zhao, X. Q. Liu, Y. K. Jing and H. M. Hua, Phytochemistry, 2012, 77, 280–286 CrossRef CAS PubMed
- K. H. Lee, H. B. Chai, P. A. Tamez, J. M. Pezzuto, G. A. Cordell, K. K. Win and M. Tin-Wa, Phytochemistry, 2003, 64, 535–541 CrossRef CAS
- S. Laphookhieo, J. K. Syers, R. Kiattansakul and K. Chantrapromma, Chem. Pharm. Bull., 2006, 54, 745–747 CrossRef CAS
- G. L. Djoufack, K. M. Valant-Vetschera, J. Schinnerl, L. Brecker, E. Lorbeer and W. Robien, Nat. Prod. Commun., 2010, 5, 1055–1060 CAS
- J. P. Poupelin, G. S. Ruf, O. F. Blanpin, G. Narcisse, G. U. Ernouf and R. Lacroix, Eur. J. Med. Chem., 1978, 13, 67–71 CAS
- T. F. Diniz, A. C. Pereira, L. S. A. Capettini, M. H. Santos, T. J. Nagem, V. S. Lemos and S. F. Cortes, Planta Med., 2013, 79, 1495–1500 CrossRef CAS PubMed
- R. M. Ion, A. Planner, K. Wiktorowicz and D. Frackowiak, Acta Biochim. Pol., 1998, 833–845 CAS
- A. Jha and J. Beal, Tetrahedron Lett., 2004, 45, 8999–9001 CrossRef CAS PubMed
- D. W. Knight and P. B. Little, J. Chem. Soc., Perkin Trans. 1, 2001, 1771–1777 RSC
- D. W. Knight and P. B. Little, Synlett, 1998, 1141–1143 CrossRef CAS
- M. A. Pasha and V. P. Jayashankara, Bioorg. Med. Chem. Lett., 2007, 17, 621–623 CrossRef CAS PubMed
- Y. Cao, C. Yao, B. Qin and H. Zhang, Res. Chem. Intermed., 2013, 39, 3055–3062 CrossRef CAS
- B. Das, B. Ravikanth, R. Ramu, K. Laxminarayana and B. Vittal Rao, J. Mol. Catal. A: Chem., 2006, 255, 74–77 CrossRef CAS PubMed
- S. Yadav, B. Nand and J. M. Khurana, Can. J. Chem., 2013, 91, 698–703 CrossRef CAS
- E. Soleimani, M. M. Khodaei and A. T. K. Koshvandi, Chin. Chem. Lett., 2011, 22, 927–930 CrossRef CAS PubMed
- J. Safaei-Ghomi and M. A. Ghasemzadeh, S. Afr. J. Chem., 2014, 67, 27–32 Search PubMed
- H. Zang, Y. Zhang, B. Cheng, W. Zhang, X. Xu and Y. Ren, Chin. J. Chem., 2012, 30, 362–366 CrossRef CAS PubMed
- H. Singh, S. Kumari and J. M. Khurana, Chin. Chem. Lett., 2014, 25, 1336–1340 CrossRef CAS PubMed
- C.-D. Huo, X.-Z. Bao, D.-C. Hu, X.-D. Jia, C.-G. Sun and C. Wang, Chin. Chem. Lett., 2014, 25, 699–701 CrossRef CAS PubMed
- S. Kantevari, M. V. Chary, A. P. R. Das, S. V. N. Vuppalapati and N. Lingaiah, Catal. Commun., 2008, 9, 1575–1578 CrossRef CAS PubMed
- Y. Chen, S. Wu, S. Tu, C. Li and F. Shi, J. Heterocycl. Chem., 2008, 45, 931–934 CrossRef CAS PubMed
- L. Wu, Y. Wu, F. Yan and L. Fang, Monatsh. Chem., 2010, 141, 871–875 CrossRef CAS
- X. Yang, L. Yang and L. Wu, Bull. Korean Chem. Soc., 2013, 34, 1303 CrossRef CAS
- A. Rahmatpour, Monatsh. Chem., 2013, 144, 1205–1212 CrossRef CAS
- J. M. Khurana, A. Lumb, A. Chaudhary and B. Nand, Synth. Commun., 2013, 43, 2147–2154 CrossRef CAS PubMed
- A. Olyaei and M. G. Alidoust, Synth. Commun., 2015, 45, 94–104 CrossRef CAS PubMed
- M. Dabiri, Z. N. Tisseh and A. Bazgir, J. Heterocycl. Chem., 2010, 47, 1062–1065 CrossRef CAS PubMed
- Z. N. Tisseh, S. C. Azimi, P. Mirzaei and A. Bazgir, Dyes Pigm., 2008, 79, 273–275 CrossRef CAS PubMed
- A. Bazgir, Z. N. Tisseh and P. Mirzaei, Tetrahedron Lett., 2008, 49, 5165–5168 CrossRef CAS PubMed
- E. C. B. da Costa, R. Amorin, F. C. da Silva, D. R. Rocha, M. P. Papa, L. B. de Arruda, R. Mohana-Borges, V. F. Ferreira, A. Tanuri, L. J. da Costa and S. B. Ferreira, PLoS One, 2013, 8, e82504–e82514 Search PubMed
- J. R. Hwu and K.-Y. King, Curr. Sci., 2001, 81, 1043–1053 CAS
- V. Sridharan and J. C. Menéndez, Chem. Rev., 2010, 110, 3805–3849 CrossRef CAS PubMed
- M. Zarei, A. Jarrahpour, E. Ebrahimi, A. Aye and S. A. T. Badrabady, Tetrahedron, 2012, 68, 5505–5512 CrossRef CAS PubMed
- N. P. Tale, A. V. Shelke and N. N. Karade, Synth. Commun., 2012, 42, 2959–2965 CrossRef CAS PubMed
- Y. S. Reddy, A. P. J. Pal, P. Gupta, A. A. Ansari and Y. D. Vankar, J. Org. Chem., 2011, 76, 5972–5984 CrossRef CAS PubMed
- V. Nair, P. M. Treesa, D. Maliakal and N. Rath, Tetrahedron, 2001, 57, 7705–7710 CrossRef CAS
- M. Yılmaz, M. Yakut and A. T. Pekel, Synth. Commun., 2008, 38, 914–927 CrossRef PubMed
- Y. R. Lee, B. S. Kim, Y. U. Jung, W. S. Koh, J. S. Cha and N. W. Kim, Synth. Commun., 2002, 32, 3099–3105 CrossRef CAS PubMed
- C. Ito, S. Katsuno, Y. Kondo, H. T.-W. Tan and H. Furukawa, Chem. Pharm. Bull., 2000, 48, 339–343 CrossRef CAS
- K. Kobayashi, T. Uneda, K. Tanaka, M. Mori, H. Tanaka, O. Morikawa and H. Konishi, Bull. Chem. Soc. Jpn., 1998, 71, 1691–1697 CrossRef CAS
- M. B. Teimouri and R. Bazhrang, Monatsh. Chem., 2008, 139, 957–961 CrossRef CAS
- L. Wu, L. Yang, F. Yan, C. Yang and L. Fang, Bull. Korean Chem. Soc., 2010, 31, 1051–1054 CrossRef CAS
- B. Jiang, G. Zhang, N. Ma, F. Shi, S.-J. Tu, P. Kaur and G. Li, Org. Biomol. Chem., 2011, 9, 3834–3837 CAS
- S. M. Rajesh, B. D. Bala, S. Perumal and J. C. Menéndez, Green Chem., 2011, 13, 3248–3254 RSC
- B. D. Bala, S. Muthusaravanan and S. Perumal, Tetrahedron Lett., 2013, 54, 3735–3739 CrossRef PubMed
- R. Jain, O. P. Agarwal and S. C. Jain, Asian J. Chem., 2013, 25, 1842–1844 CAS
- T. Shen, Z. Fu, F. Che, H. Dang, Y. Lin and Q. Song, Tetrahedron Lett., 2015, 56, 1072–1075 CrossRef CAS PubMed
- D. R. da Rocha, K. Mota, I. M. C. B. da Silva, V. F. Ferreira, S. B. Ferreira and F. C. da Silva, Tetrahedron, 2014, 70, 3266–3270 CrossRef CAS PubMed
- M. F. C. Cardoso, P. C. Rodrigues, M. E. I. M. Oliveira, I. L. Gama, I. M. C. B. da Silva, I. O. Santos, D. R. da Rocha, R. T. Pinho, V. F. Ferreira, M. C. B. V. de Souza, F. C. da Silva and F. P. Silva-Jr., Eur. J. Med. Chem., 2014, 84, 708–717 CrossRef CAS PubMed
- M. F. C. Cardoso, K. Salomão, A. C. Bombaça, D. R. da Rocha, F. C. da Silva, J. A. S. Cavaleiro, S. L. de Castro and V. F. Ferreira, Bioorg. Med. Chem., 2015, 23, 4763–4768 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |