
Catalyst-free formation of 1,4-diketones by addition of silyl enolates to oxyallyl zwitterions in situ generated from α-haloketones†
Juan Luoab,
Qihua Jianga,
Hao Chena and
Qiang Tang*ab
aDepartment of Medical Chemistry, College of Pharmacy, Chongqing Medical University, Chongqing 400016, P. R. China. E-mail: tangqiang@cqmu.edu.cn
bThe Faculty of Laboratory Medicine, Chongqing Medical University, Chongqing 401331, P. R. China
First published on 3rd August 2015
Abstract
Reported here is the exclusive formation of 1,4-diketones by the uncatalyzed reaction of silyl enolates and α-haloketones. Enolates I are inherently more likely to react with α-haloketones II at the carbonyl carbon to produce halohydrin derivatives III or 2-(2-oxoethyl)-oxiranes IV. Thus, a variety of metal-catalyzed coupling reactions have been developed to avoid the undesired reaction when attempting the preparation of 1,4-diketones. We found that the oxyallyl zwitterions in situ generated from α-haloketones enabled the addition of silyl enolates to the α-carbonyl position to exclusively form 1,4-diketones in weakly basic conditions. Various types of silyl enolates and α-haloketones were applied to the catalyst-free coupling.
Introduction
1,4-Diketones are common substructures of natural products and pharmaceuticals,1 like maoecrystal V1a and herquline,1d as well as highly useful synthetic building blocks of various carbocyclic and heterocyclic compounds, such as cyclopentenones,2 furans,3 thiophenes,4 pyrroles5 and pyridazine derivatives.6 Therefore, significant efforts have been directed toward the synthesis of those highly valuable synthons.7 The most straightforward method for their preparation would be the C–C bond formation between carbonylmethyl anion and cation equivalents.8 Although versatile enolates have been developed as carbonylmethyl anions, selecting appropriate candidates as carbonylmethyl cation units is still a challenging problem. α-Haloketones might be used as carbonylmethyl cation equivalents, but there is a regioselectivity problem resulting from the two reactive sites respectively located at the carbonyl carbon and the α-carbonyl position.9 Without metal catalysts, enolates I inherently are more likely to react with α-haloketones II at the carbonyl carbon to produce halohydrin derivatives III or 2-(2-oxoethyl)-oxiranes IV (Scheme 1, Path a).10 Thus, a variety of metal-catalyzed coupling reactions have been developed to avoid the undesired reaction for the preparation of 1,4-diketones V (Scheme 1, Path b).11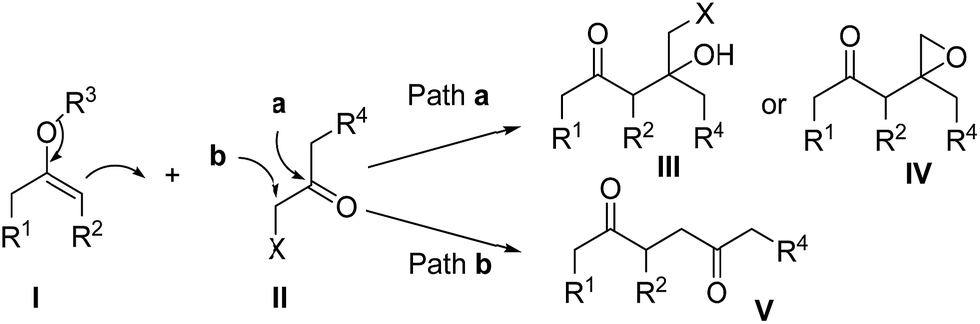 | ||
| Scheme 1 Reaction approaches of enolates with α-haloketones. | ||
Our interest in this chemistry stems from our work on the interrupted cycloadditions of oxyallyl zwitterions.12 We have recently reported that the direct coupling of unprotected indoles and α-halo ketones via in situ generated oxyallyl zwitterions provides α-indolylketones.13 Upon further exploration, we have also found an efficient catalytic-free method for the coupling of naphthols with oxyallyl zwitterions to produce α-naphtholylketones.14 Additionally, MacMillan's report has also demonstrated that oxyallyl zwitterions allow the addition of π-nucleophiles or even neutral heteroatoms to the α-carbonyl position under mild, weakly basic conditions.15
It is notable that the Harmata group has reported an efficient ene-like reaction between alkyl enol ethers VII and a special oxyallyl zwitterion VI yielded from a specific α-chloroketone (Scheme 2).12b It demonstrates that the leaving group, the Me2PhSi moiety substituted on the oxyallyl zwitterion VI, takes part in a crucial role in the hydride shifting process and finally benefits the reaction efficiency.
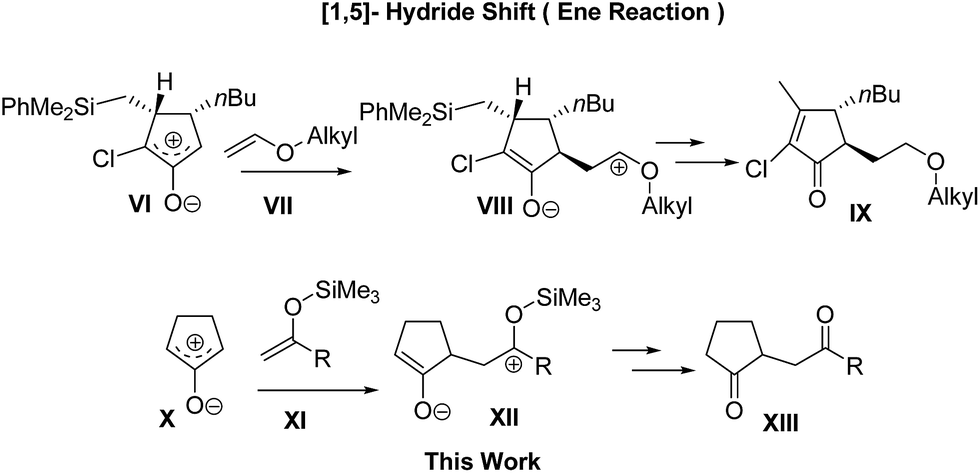 | ||
| Scheme 2 Reaction approaches of enolates with oxyallyl zwitterions. | ||
Based on the above findings, we hypothesized that the course of the reaction might be changed if the leaving group is located on enolates XI, and that simple oxyallyl zwitterion X generated in weakly basic conditions might allow the exclusive addition of silyl enolates XI to the α-carbonyl position of X and finally form 1,4-diketones XIII after desilylation (Scheme 2). With the merits of the mildness of the desilylation process, the ease of preparation and the cleanliness of reactions, silyl enolates have long been known as weak nucleophiles in the Mukaiyama aldol addition, Michael addition, and alkylation reaction.16 However, to our knowledge, this is the first report on their application in the synthesis of 1,4-diketones without any catalyst.17
Background
Oxyallyl zwitterions and cyclopentanone intermediates, which can be transformed to each other, were first proposed as transient electrophilic intermediates in the Favorskii rearrangement in 1894.18 More specifically, under strongly basic conditions, a variety of nucleophiles add to the carbonyl carbon of the incipient cyclopentanone intermediates induced from α-haloketones and subsequently produce carboxylic acids and their derivatives after bond migration.19 Furthermore, the oxyallyl zwitterions have also been known as dienophiles in [4 + 3] cycloadditions to construct seven-membered carbocycles across a wide range of unique applications.20To shed light on the reaction mechanism of nucleophiles with oxyallyls (cyclopentanone intermediates and oxyallyl zwitterions), different types of reactions are summarized in Scheme 3 and 4. Under typical Favorskii rearrangement conditions (Scheme 3),21 strong bases induce the formation of cyclopentanone intermediates XV from α-haloketones and then the ring-contracted product XVII,22 XVIII,23 or XIX![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 is produced depending on the p-nucleophile we used. Remarkably, treatment of 2-chlorocyclopentanone with the weak base (sodium carbonate) instead of the strong base (sodium hydroxide) affords an excellent yield of the substituted product XX. However, in the presence of a weaker base, such as sodium bicarbonate, or without any bases, most of the starting material remains unreacted, and only a trace of the substituted product XX is detected.14 Based on those observations, we proposed that the generation of oxyallyl zwitterions XVI (the valence tautomers of the cyclopropane intermediates XV) under the weakly basic conditions is the key step, which permits the subsequent addition of the p-nucleophiles to render the substituted products (XX–XXIII).25
24 is produced depending on the p-nucleophile we used. Remarkably, treatment of 2-chlorocyclopentanone with the weak base (sodium carbonate) instead of the strong base (sodium hydroxide) affords an excellent yield of the substituted product XX. However, in the presence of a weaker base, such as sodium bicarbonate, or without any bases, most of the starting material remains unreacted, and only a trace of the substituted product XX is detected.14 Based on those observations, we proposed that the generation of oxyallyl zwitterions XVI (the valence tautomers of the cyclopropane intermediates XV) under the weakly basic conditions is the key step, which permits the subsequent addition of the p-nucleophiles to render the substituted products (XX–XXIII).25
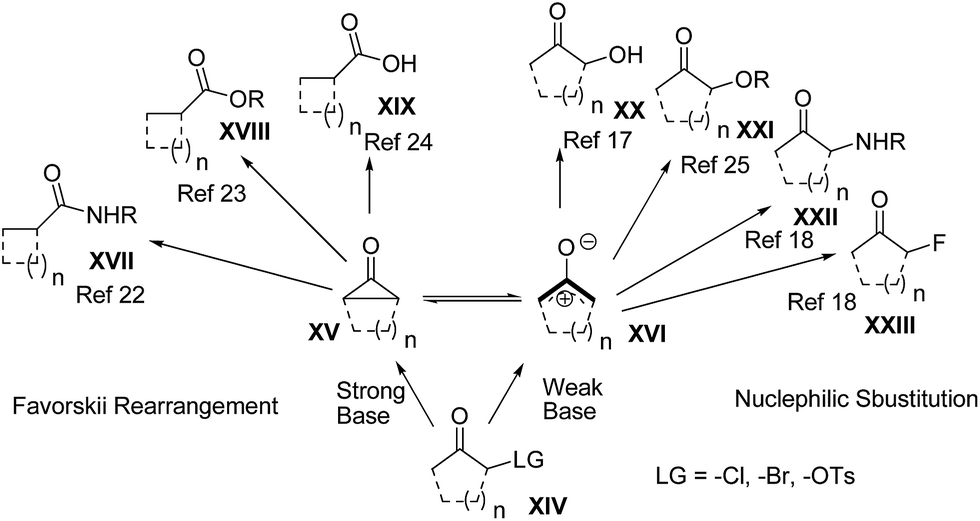 | ||
| Scheme 3 The reaction of oxyallyls with p-nucleophiles. | ||
 | ||
| Scheme 4 The reaction of oxyallyl zwitterions with π-nucleophiles using TFE or HFIP as solvent. | ||
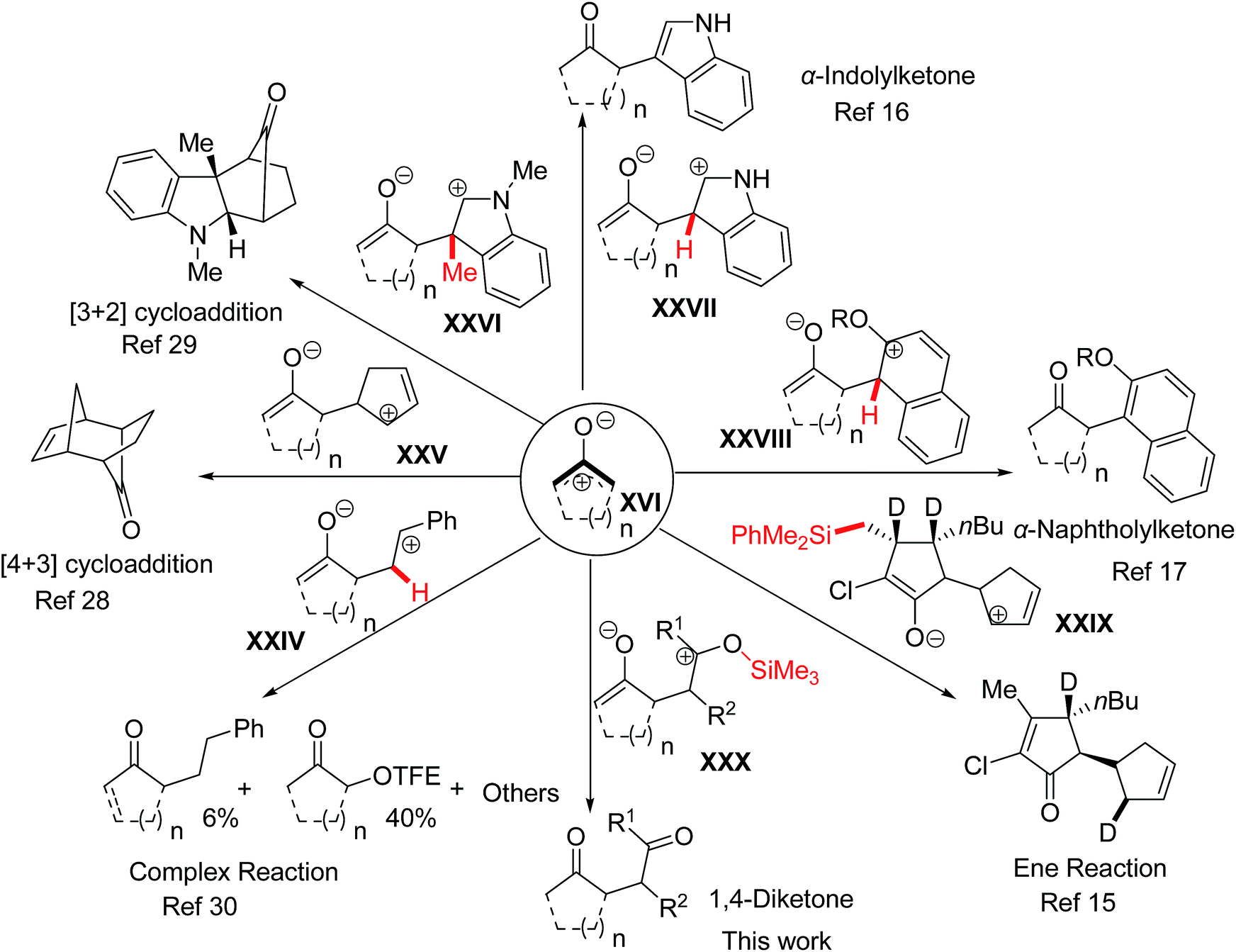
It should be noted that both oxyallyl zwitterions and cyclopentanone intermediates are formed in the reaction of α-haloketones with furan using sodium 2,2,2-trifluoroethoxide as a base in 2,2,2-trifluoroethanol (NaTFE/TFE), and that the two corresponding types of products, the ring-contracted products (XVIII, Scheme 3) and the [4 + 3] cycloadducts (Scheme 4) are eventually generated.26
It is well known that oxyallyl zwitterions27 tend to react with enes either in a stepwise or a concerted fashion so as to generate the [4 + 3]28 or [3 + 2]29 cycloadducts (Scheme 4). However, several valuable interrupted cycloadditions of oxyallyl zwitterions have been reported in recent years. The mechanism of the interrupted cycloadditions depends on (i) the nucleophilicity of π-nucleophiles, (ii) the positions of the leaving groups on the intermediates XXIV–XXX and (iii) the difficulty of removing the leaving groups from the intermediates XXIV–XXX. Compared with indole or silyl enolate, the low nucleophilicity of styrene and the difficult deprotonation of the intermediate XXIV (denoted in red color) result in the complexity of the reaction of styrene with 2-chlorocyclopentanone in TFE.30 Whereas, an efficient reaction takes place when the leaving groups become easy to release from the intermediates XXVII–XXX. Moreover, the course of the reaction is changed just by changing the positions of the leaving groups (the silyl group) on the intermediates, and thus the product of the intermediate XXX is 1,4-diketone, entirely different from the product of the intermediate XXIX.
Results and discussion
Optimization of reaction conditions
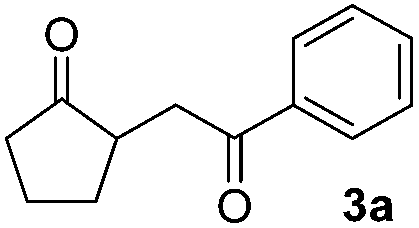
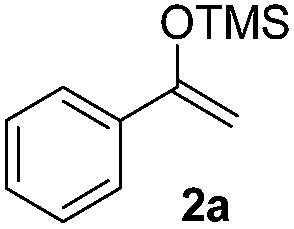
The proposed transformation was first examined using 1-phenyl-1-trimethylsiloxyethylene 2a and 2-chlorocyclopentanone 1a in the presence of sodium carbonate as a base, and trifluoroethanol (TFE) as a solvent (Table 1). To our delight, the desired 1,4-diketone 3a was obtained efficiently in one step without halohydrin III or cycloaddition compound being detected, thereby demonstrating the feasibility of our proposal (the structure of 3a was characterized by 2D NMR spectroscopy). However, an excess amount of silyl enolate was needed because of its instability in TFE (Table 1, entry 2). Moreover, three equivalents of silyl enolate were eventually found to be optimal in terms of yield (Table 1, entry 4), as a higher amount of silyl enolate could lead to the generation of quite a few byproducts containing the silyl group. Remarkably, we discovered that the basicity of bases shows a significant effect on the reaction cleanliness and yield. In fact, an organic base Et3N, could also effectively initiate the reaction (Table 1, entries 8). However, when employing a relatively weak base, i.e. sodium bicarbonate, only a trace of the desired product was detected (Table 1, entry 6). On the contrary, when a strong base, i.e. NaOH, was used, the reaction became messy (Table 1, entry 7).
|
|||||
|---|---|---|---|---|---|
| Entry | Base [1.2 eq.] | 1a [equiv.] | 2a [equiv.] | Time [h] | Yieldb [%] |
| a Reaction conditions: 1a (1.0 mmol), 2a (1.0–4.0 mmol), base (1.2 mmol) in TFE (2 mL) at 25 °C.b Isolated yield.c Silyl enolate was not detected by TLC after twelve hours. | |||||
| 1 | Na2CO3 | 1 | 1 | 6 | 42 |
| 2 | Na2CO3 | 0 | 1 | 12 | —c |
| 3 | Na2CO3 | 1 | 2 | 12 | 61 |
| 4 | Na2CO3 | 1 | 3 | 12 | 70 |
| 5 | Na2CO3 | 1 | 4 | 12 | 65 |
| 6 | NaHCO3 | 1 | 3 | 24 | Trace |
| 7 | NaOH | 1 | 3 | 3 | Complex |
| 8 | Et3N | 1 | 3 | 12 | 60 |
Scope and generality of the substrates
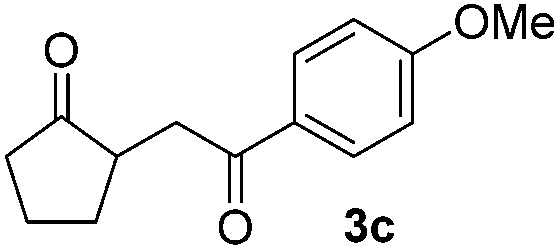
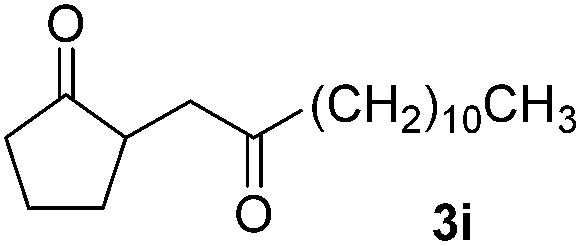
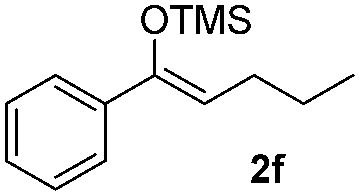
With the optimized conditions in hand, we next examined other silyl enolates in this new synthetic protocol. Gratifyingly, a variety of silyl enolates functioned well in this nucleophilic reaction. In general, since alkyl enol ethers (Table 2, entries 7–12) are more stable than aryl enol ethers (Table 2, entries 1–6) in TFE, a little excess amount of alkyl enol ethers led to better yields in the reaction system. No steric effect was observed for the terminal enol ethers (Table 2, entries 1–4 and 7–9) and the disubstituted enol ethers (Table 2, entries 5–6 and 10–12) showed nearly the same reaction efficiency as the monosubstituted enol ethers.
|
|||
|---|---|---|---|
| Entry | Enolate | Product | Yieldb,c [%] |
| a Reaction conditions: 1a (1.0 mmol), silicon enolates (3.0 mmol in entries 1–3, 2.0 mmol in entries 4–11), Na2CO3 (1.2 mmol) in TFE (2 mL) at 25 °C.b Isolated yield.c The diastereomer ratio was determined by 1H NMR spectroscopic analysis of the crude material. | |||
| 1 | 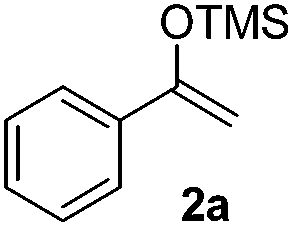 |
 |
70 |
| 2 |  |
 |
65 |
| 3 | 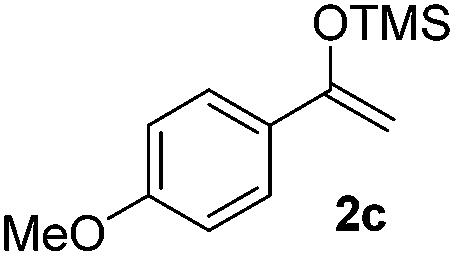 |
 |
78 |
| 4 | 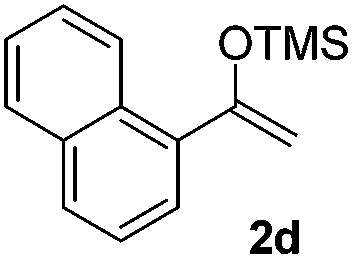 |
 |
69 |
| 5 | 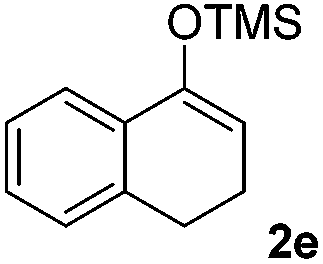 |
 |
73(1:1 dr) |
| 6 | 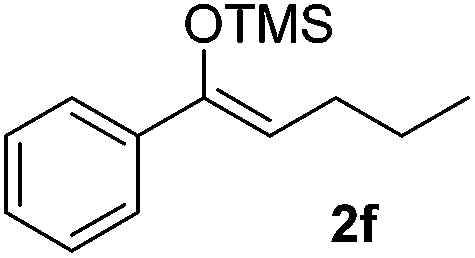 |
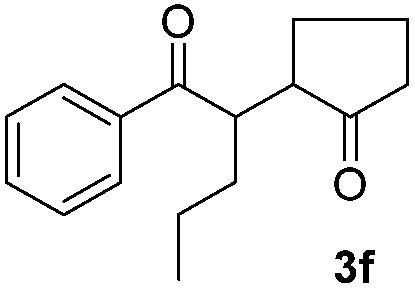 |
67(5:2 dr) |
| 7 | 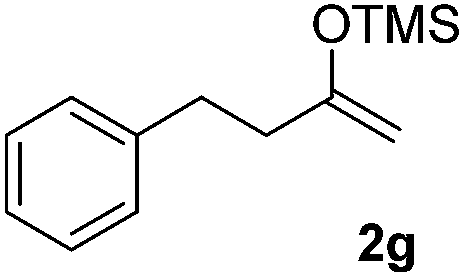 |
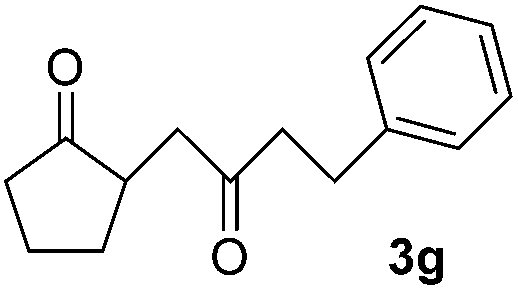 |
72 |
| 8 | 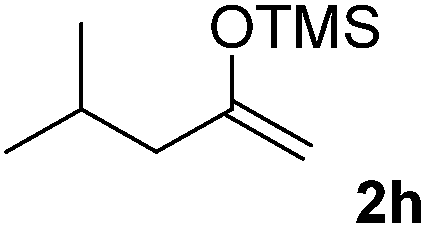 |
 |
73 |
| 9 | 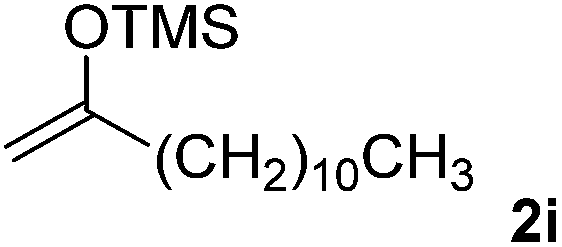 |
 |
76 |
| 10 | 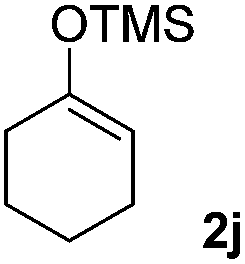 |
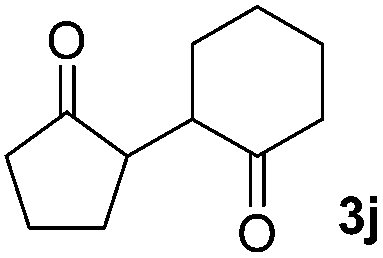 |
81(1:1 dr) |
| 11 | 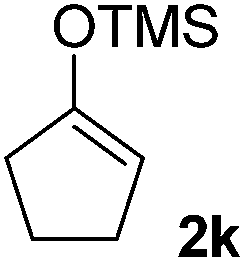 |
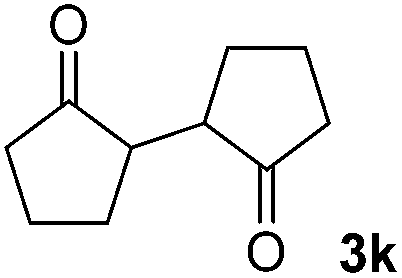 |
80(1:1 dr) |
| 12 | 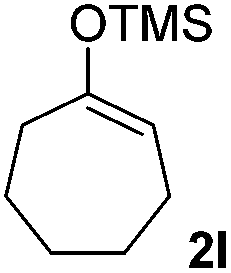 |
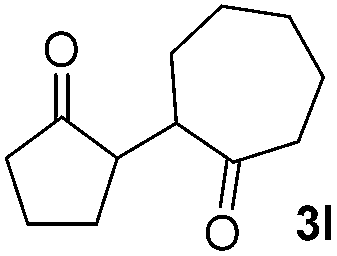 |
71(1:1 dr) |
In addition, we found that the electron density of silyl enolates plays an important role in the reaction efficiency. As a matter of fact, the reaction with silyl enolate (2c) bearing an electron-donating substitutent proceeded efficiently by this uncatalyzed system (Table 2, entry 3). On the contrary, no desired product was isolated for this reaction of 2-chlorocyclopentanone with silyl enolates bearing a strong electron-withdrawing substitutent, such as trimethyl((1-(4-nitrophenyl)vinyl)oxy)silane and 4-(1-((trimethylsilyl)oxy)vinyl)benzonitrile. Silyl enolates generated from aldehydes, such as trimethylsiloxyethylene, 2-phenyl-1-trimethylsiloxyethylene and 2-benzyl-1-trimethylsiloxyethyl-ene, yielded complicated reaction mixtures that were not studied further.
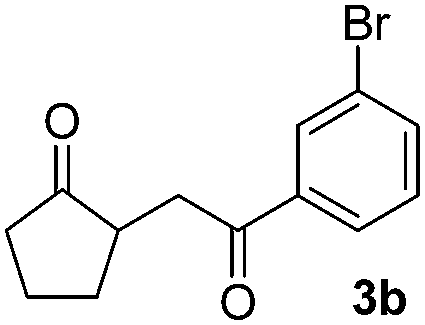
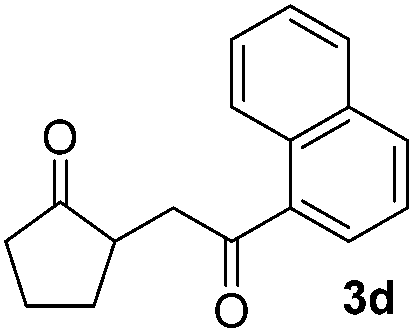
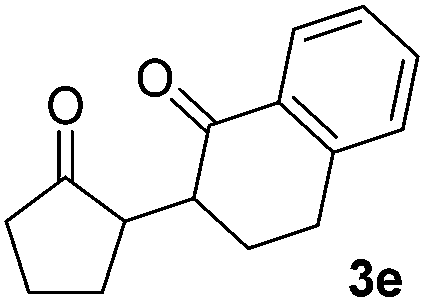
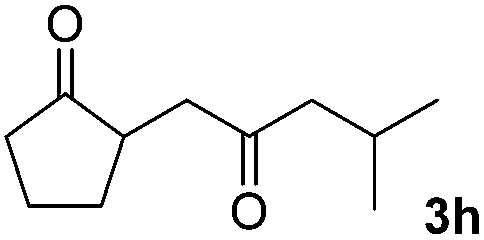
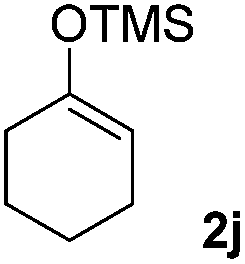
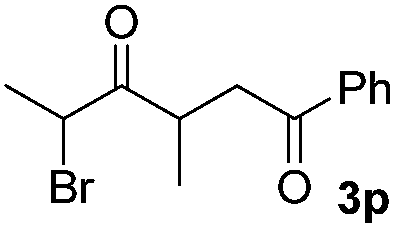
We next examined the scope of α-haloketones in the catalyst-free formation of 1,4-diketones. This transformation is not limited to five-membered rings since both six-membered rings and seven-membered rings are competent substrates (Table 3, entries 1–5). Additionally, both α-chloro- and α-bromocyclohexanones gave the same products with similar reaction rate and efficiency (Table 3, entries 1 and 2). For the reaction of α-iodocyclohexanone, the reaction rate was faster but the yield was lower in comparison with the other two halocyclohexanones. Use of acyclic α-haloketones afforded the corresponding 1,4-diketones in comparable yields (Table 3, entries 6–7). Furthermore, the reaction of dibromoketones (1h–1i) with an equimolar or excess molar amount of silyl enolates gave the monosubstituted products (Table 3, entries 8–9). The coupling reaction shows a high regioselectivity, as only one regioisomer (3q) was obtained, whose structure was confirmed by 2D NMR.
| Entry | Haloketone | Enolate | Product | Yieldb,c [%] |
|---|---|---|---|---|
| a Reaction conditions: α-haloketones (1.0 mmol), silicon enolates (3.0 mmol in entries 1,2,7,8; 2.0 mmol in entries 3–6), Na2CO3 (1.2 mmol) in TFE (2 mL) at 25 °C.b Isolated yield.c The diastereomer ratio was determined by 1H NMR spectroscopic analysis of the crude material. | ||||
| 1 |  |
 |
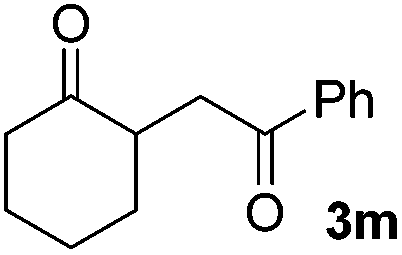 |
62 |
| 2 | 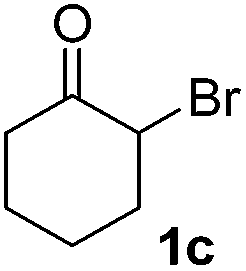 |
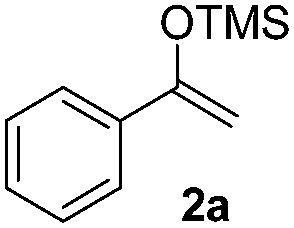 |
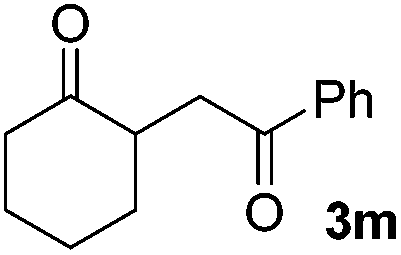 |
60 |
| 3 | 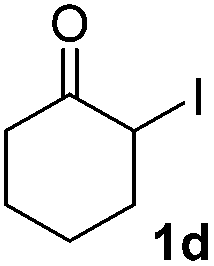 |
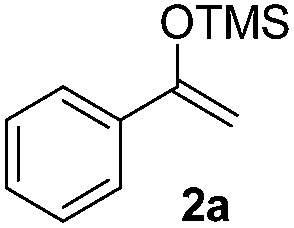 |
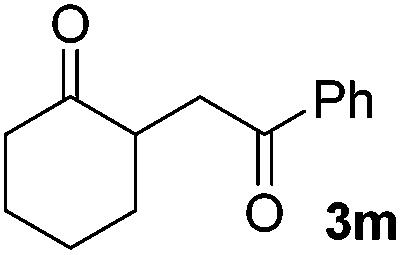 |
56 |
| 4 |  |
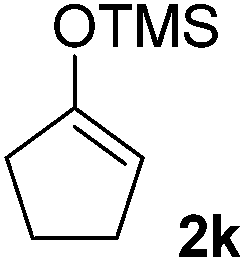
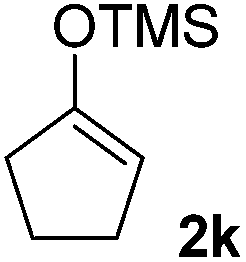 |
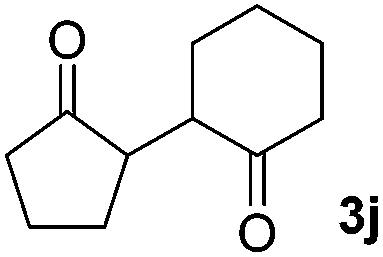 |
71(1:1 dr) |
| 5 | 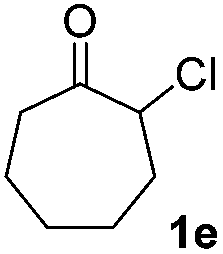 |
 |
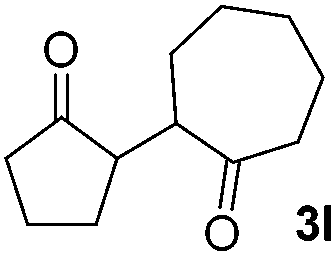 |
63(1:1 dr) |
| 6 | 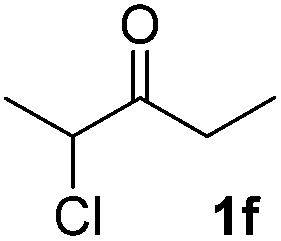 |
 |
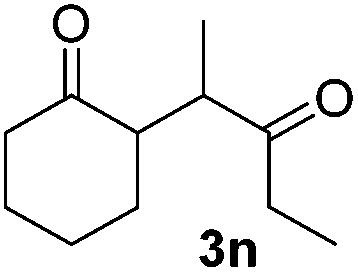 |
39 (5:1 dr) |
| 7 | 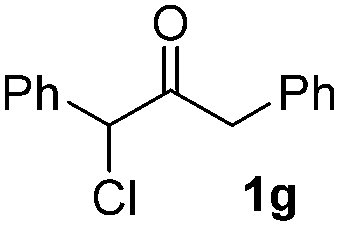 |
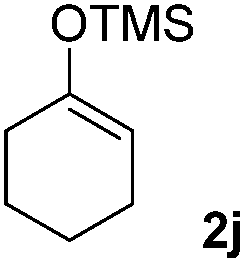 |
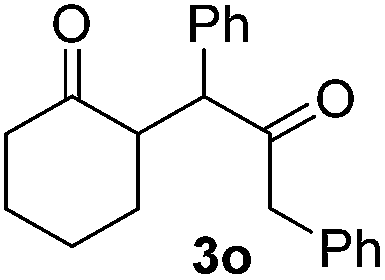 |
51(10:1 dr) |
| 8 | 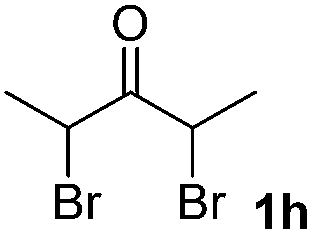 |
 |
 |
32(10:1 dr) |
| 9 | 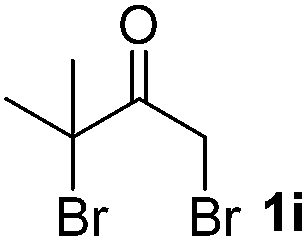 |
 |
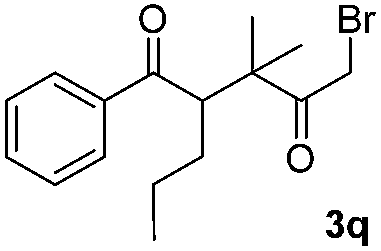 |
31 |
Experimental section
General information
Nuclear magnetic resonance spectra (1H and 13C) were recorded on 300, 400, and 500 MHz spectrometers with tetramethylsilane (TMS) as an internal standard. The splitting patterns are designated as singlet (s), doublet (d), triplet (t), quartet (q), dd (doublet of doublets); m (multiplets), and etc. All first-order splitting patterns were assigned on the basis of the appearance of the multiplet. Splitting patterns that could not be easily interpreted are designated as multiplet (m) or broad (br). High resolution mass spectral analysis (HRMS) was performed on ESI-QTOP mass spectrometer. Visualization was performed using a UV lamp or chemical stains like KMnO4 and 2,4-dinitrophenyl hydrazine solutions.Commercially available materials were used as received, except α-haloketones that were further purified via distillation or column chromatography over silica gel prior to use. Some of the α-chloroketones (2-bromocyclohexanone, 2-chlorocycloheptanone and 1,3-dibromo-3-methylbutan-2-one) were prepared using literature method.31
Typical procedure for catalyst-free coupling of silyl enolates with α-haloketones
A 4 mL vial equipped with a magnetic stir bar was charged with freshly distilled α-haloketone 1 (0.5 mmol), anhydrous Na2CO3 (0.6 mmol) and TFE (1.0 mL). Silyl enolate 2 (1.0–1.5 mmol) was added in portionwise to the reaction mixture at 25 °C. After completion of the reaction (about 12–24 h, monitored by TLC or crude 1H NMR analysis), the reaction was quenched with water (1.0 mL) and stirred for 30 min. Extraction with CH2Cl2 (3 × 10 mL), drying of the combined organic layers with Na2SO4, filtration, and evaporation of the solvent in vacuum gave a residue which was purified by column chromatography on silica gel with petroleum ether and ethyl acetate (v/v = 10:1 to 1:1) as the eluent to afford the desired product.
Conclusions
In conclusion, we have detailed a catalyst-free coupling reaction of silyl enolates with α-haloketones via in situ generated oxyallyl zwitterions in basic TFE. The reaction took place regioselectively at the α-carbonyl position and finally produced the useful 1,4-diketones. Further studies to define the enolate scope and the optimal catalyst are currently being pursued in our laboratory.Acknowledgements
We appreciate the financial support from the Fundamental and Advanced Research Projects of Chongqing City (No. cstc2014jcyjA10022), Starting Foundation for Talents Returning from Overseas of Ministry of Education, and Starting Foundation of College of Pharmacy, Chongqing Medical University.Notes and references
- (a) S.-H. Li, J. Wang, X.-M. Niu, Y.-H. Shen, H.-J. Zhang, H.-D. Sun, M.-L. Li, Q.-E. Tian, Y. Lu, P. Cao and Q.-T. Zheng, Org. Lett., 2004, 6, 4327 CrossRef CAS PubMed; (b) T. Fujisawa, K. Igeta, S. Odake, Y. Morita, J. Yasuda and T. Morikawa, Bioorg. Med. Chem., 2002, 10, 2569 CrossRef CAS; (c) M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735 CrossRef CAS PubMed; (d) A. Furusaki, T. Matsumoto, H. Ogura, H. Takayanagi, A. Hirano and S. Omura, J. Chem. Soc., Chem. Commun., 1980, 698a RSC.
- (a) E. Wenkert, Acc. Chem. Res., 1980, 13, 27 CrossRef CAS; (b) M. Miyashita, T. Yanami and A. Yoshikoshi, J. Am. Chem. Soc., 1976, 98, 4679 CrossRef CAS.
- (a) H. S. P. Rao and S. Jothilingam, J. Org. Chem., 2003, 68, 5392 CrossRef CAS PubMed; (b) B. W. Greatrex, M. C. Kimber, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2003, 68, 4239 CrossRef CAS PubMed; (c) P. Bosshard and C. H. Eugster, in Adv. Heterocycl. Chem., ed. A. R. Katritzky and A. J. Boulton, Academic Press, 1967, vol. 7 Search PubMed.
- (a) D. E. Wolf and K. Folkers, Org. React., 1951, 6, 410 Search PubMed; (b) M. A. Youtz and P. P. Perkins, J. Am. Chem. Soc., 1929, 51, 3511 CrossRef CAS.
- (a) E. O'Reilly, C. Iglesias, D. Ghislieri, J. Hopwood, J. L. Galman, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 2447 CrossRef PubMed; (b) C.-S. Li, Y.-H. Tsai, W.-C. Lee and W.-J. Kuo, J. Org. Chem., 2010, 75, 4004 CrossRef CAS PubMed; (c) E. Baltazzi and L. I. Krimen, Chem. Rev., 1963, 63, 511 CrossRef CAS.
- M. Tišler and B. Stanovnik, in Adv. Heterocycl. Chem., ed. A. R. Katritzky and A. J. Boulton, Academic Press, 1968, vol. 9 Search PubMed.
- (a) S. Mao, Y.-R. Gao, S.-L. Zhang, D.-D. Guo and Y.-Q. Wang, Eur. J. Org. Chem., 2015, 876 CrossRef CAS PubMed; (b) S. Huang, L. Kötzner, C. K. De and B. List, J. Am. Chem. Soc., 2015, 137, 3446 CrossRef CAS PubMed; (c) P. Setzer, G. Forcher, F. Boeda, M. S. M. Pearson-Long and P. Bertus, Eur. J. Org. Chem., 2014, 171 CrossRef CAS PubMed; (d) W. Zhao, D. M. Fink, C. A. Labutta and A. T. Radosevich, Org. Lett., 2013, 15, 3090 CrossRef CAS PubMed; (e) M. M. D. Wilde and M. Gravel, Angew. Chem., Int. Ed., 2013, 52, 12651 CrossRef CAS PubMed; (f) B. B. Parida, P. P. Das, M. Niocel and J. K. Cha, Org. Lett., 2013, 15, 1780 CrossRef CAS PubMed; (g) P. Setzer, A. Beauseigneur, M. S. M. Pearson-Long and P. Bertus, Angew. Chem., Int. Ed., 2010, 49, 8691 CrossRef CAS PubMed.
- (a) F. Guo, M. D. Clift and R. J. Thomson, Eur. J. Org. Chem., 2012, 4881 CrossRef CAS PubMed; (b) B. M. Casey and R. A. Flowers, J. Am. Chem. Soc., 2011, 133, 11492 CrossRef CAS PubMed; (c) J. Xie and Z.-Z. Huang, Chem. Commun., 2010, 46, 1947 RSC; (d) M. P. DeMartino, K. Chen and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 11546 CrossRef CAS PubMed; (e) P. S. Baran and M. P. DeMartino, Angew. Chem., Int. Ed., 2006, 45, 7083 CrossRef CAS PubMed.
- (a) A. Erian, S. Sherif and H. Gaber, Molecules, 2003, 8, 793 CrossRef CAS PubMed; (b) N. De Kimpe and R. Verhé, in α-Haloketones, α-Haloaldehydes and α-Haloimines (1988), John Wiley & Sons, Inc., 2010 Search PubMed.
- (a) I. Pri-Bar, P. S. Pearlman and J. K. Stille, J. Org. Chem., 1983, 48, 4629 CrossRef CAS; (b) M. Yasuda, T. Oh-hata, I. Shibata, A. Baba, H. Matsuda and N. Sonoda, Bull. Chem. Soc. Jpn., 1995, 68, 1180 CrossRef CAS; (c) F. Stauffer and R. Neier, Org. Lett., 2000, 2, 3535 CrossRef CAS PubMed.
- (a) C. Liu, Y. Deng, J. Wang, Y. Yang, S. Tang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 7337 CrossRef CAS PubMed; (b) M. Yasuda, S. Tsuji, I. Shibata and A. Baba, J. Org. Chem., 1997, 62, 8282 CrossRef CAS; (c) M. Kosugi, I. Takano, M. Sakurai, H. Sano and T. Migita, Chem. Lett., 1984, 1221 CrossRef CAS; (d) F. Zhang, P. Du, J. Chen, H. Wang, Q. Luo and X. Wan, Org. Lett., 2014, 16, 1932 CrossRef CAS PubMed; (e) M. Yasuda, S. Tsuji, Y. Shigeyoshi and A. Baba, J. Am. Chem. Soc., 2002, 124, 7440 CrossRef CAS PubMed; (f) A. Baba, M. Yasuda, K. Yano, I. Shibata and H. Matsuda, J. Chem. Soc., Perkin Trans. 1, 1990, 3205 RSC; (g) C. Chassin, E. A. Schmidt and H. M. R. Hoffmann, J. Am. Chem. Soc., 1974, 96, 606 CrossRef CAS.
- (a) X. Gao and M. Harmata, Tetrahedron, 2013, 69, 7675 CrossRef CAS PubMed; (b) M. Harmata, C. Huang, P. Rooshenas and R. Schreiner Peter, Angew. Chem., Int. Ed., 2008, 47, 8696 CrossRef CAS PubMed.
- Q. Tang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2012, 14, 1922 CrossRef CAS PubMed.
- J. Luo, H. Zhou, J. Hu, R. Wang and Q. Tang, RSC Adv., 2014, 4, 17370 RSC.
- M. N. Vander Wal, A. K. Dilger and D. W. C. MacMillan, Chem. Sci., 2013, 4, 3075 RSC.
- (a) G. C. Tay, C. Y. Huang and S. D. Rychnovsky, J. Org. Chem., 2014, 79, 8733 CrossRef CAS PubMed; (b) P. Brownbridge, Synthesis, 1983, 1 CrossRef CAS.
- (a) Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2012, 48, 5355 RSC; (b) K. Mikami, Y. Kawakami, K. Akiyama and K. Aikawa, J. Am. Chem. Soc., 2007, 129, 12950 CrossRef CAS PubMed.
- A. E. Favorskii, J. Russ. Phys.–Chem. Soc., 1894, 26, 590 Search PubMed.
- (a) D. Guijarro and M. Yus, Curr. Org. Chem., 2005, 9, 1713 CrossRef CAS; (b) B. Föhlisch, T. Franz and G. Kreiselmeier, Eur. J. Org. Chem., 2005, 4687 CrossRef PubMed; (c) M. Harmata, G. Bohnert, L. Kurti and C. L. Barnes, Tetrahedron Lett., 2002, 43, 2347 CrossRef CAS; (d) A. W. Fort, J. Am. Chem. Soc., 1962, 84, 4979 CrossRef CAS.
- (a) A. G. Lohse and R. P. Hsung, Chem.–Eur. J., 2011, 17, 3812 CrossRef CAS PubMed; (b) M. Harmata, Chem. Commun., 2010, 46, 8886 RSC; (c) M. Harmata, Chem. Commun., 2010, 46, 8904 RSC; (d) D. A. Foley and A. R. Maguire, Tetrahedron, 2010, 66, 1131 CrossRef CAS PubMed; (e) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676 RSC; (f) M. Harmata, Adv. Synth. Catal., 2006, 348, 2297 CrossRef CAS PubMed; (g) M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem.–Eur. J., 2006, 12, 3438 CrossRef CAS PubMed; (h) J. Huang and R. P. Hsung, Chemtracts, 2005, 18, 207 CAS; (i) Y. Tamaru, Eur. J. Org. Chem., 2005, 2647 CrossRef CAS PubMed; (j) B. Niess and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2005, 44, 26 CrossRef CAS PubMed; (k) I. V. Hartung and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2004, 43, 1934 CrossRef CAS PubMed; (l) M. Harmata and P. Rashatasakhon, Tetrahedron, 2003, 59, 2371 CrossRef CAS; (m) M. Harmata, Acc. Chem. Res., 2001, 34, 595 CrossRef CAS PubMed; (n) J. H. Rigby and F. C. Pigge, Org. React., 1997, 51, 351 CAS; (o) M. Harmata, Tetrahedron, 1997, 53, 6235 CrossRef CAS; (p) J. Mann, Tetrahedron, 1986, 42, 4611 CrossRef CAS; (q) T. T. Tidwell, Angew. Chem., Int. Ed., 1984, 23, 20 CrossRef PubMed; (r) H. M. R. Hoffmann, Angew. Chem., 1984, 96, 29 CrossRef CAS PubMed; (s) H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1984, 23, 1 CrossRef PubMed; (t) R. Noyori and Y. Hayakawa, Org. React., 1983, 29, 163 CAS; (u) R. Noyori, Acc. Chem. Res., 1979, 12, 61 CrossRef CAS; (v) H. Takaya, S. Makino, Y. Hayakawa and R. Noyori, J. Am. Chem. Soc., 1978, 100, 1765 CrossRef CAS; (w) H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1973, 12, 819 CrossRef PubMed.
- G. D. Hamblin, R. P. Jimenez and T. S. Sorensen, J. Org. Chem., 2007, 72, 8033 CrossRef CAS PubMed.
- F. G. Bordwell and J. Almy, Favorskii rearrangements. VII, J. Org. Chem., 1973, 38, 571 CrossRef CAS.
- F. G. Bordwell and J. G. Strong, Org. Chem., 1973, 38, 579 CrossRef CAS.
- P. J. Chenier, J. Chem. Educ., 1978, 55, 286 CrossRef CAS.
- B. Föhlisch and R. Joachimi, Chem. Ber., 1987, 120, 1951 CrossRef PubMed.
- B. Föhlisch, T. Franz and G. Kreiselmeier, Eur. J. Org. Chem., 2005, 4687 CrossRef PubMed.
- (a) C. J. Cramer and S. E. Barrows, J. Phys. Org. Chem., 2000, 13, 176 CrossRef CAS; (b) C. J. Cramer and S. E. Barrows, J. Org. Chem., 1998, 63, 5523 CrossRef CAS; (c) G. Kuzmanich, F. Spänig, C.-K. Tsai, J. M. Um, R. M. Hoekstra, K. N. Houk, D. M. Guldi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2011, 133, 2342 CrossRef CAS PubMed; (d) J. E. Antoline, E. H. Krenske, A. G. Lohse, K. N. Houk and R. P. Hsung, J. Am. Chem. Soc., 2011, 133, 14443 CrossRef CAS PubMed.
- S. T. Handy and M. Okello, Synlett, 2002, 0489 CrossRef CAS.
- H. Li, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2014, 136, 6288 CrossRef CAS PubMed.
- J. Leitich and I. Heise, Eur. J. Org. Chem., 2001, 2707 CrossRef CAS.
- (a) K. Tanemura, T. Suzuki, Y. Nishida, K. Satsumabayashi and T. Horaguchi, Chem. Commun., 2004, 470 RSC; (b) K. M. Brummond and K. D. Gesenberg, Tetrahedron Lett., 1999, 40, 2231 CrossRef CAS; (c) R. R. Fraser and F. Kong, Synth. Commun., 1988, 18, 1071 CrossRef CAS PubMed.
- H. Sun, C. Yang, F. Gao, Z. Li and W. Xia, Org. Lett., 2013, 15, 624 CrossRef CAS PubMed.
- H. Paul and I. Wendel, Chem. Ber., 1957, 90, 1342 CrossRef CAS PubMed.
- E. G. E. Hawkins and R. Large, J. Chem. Soc., Perkin Trans. 1, 1974, 280 RSC.
- (a) C. T. Avetta, L. C. Konkol, C. N. Taylor, K. C. Dugan, C. L. Stern and R. J. Thomson, Org. Lett., 2008, 10, 5621 CrossRef CAS PubMed; (b) H. M. R. Hoffmann, I. Münnich, O. Nowitzki, H. Stucke and D. J. Williams, Tetrahedron, 1996, 52, 11783 CrossRef CAS.
- P. B. Cranwell, M. O'Brien, D. L. Browne, P. Koos, A. Polyzos, M. Pena-Lopez and S. V. Ley, Org. Biomol. Chem., 2012, 10, 5774 CAS.
- D. Seebach and J. Goliński, Helv. Chim. Acta, 1981, 64, 1413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, and IR spectral data for compounds. See DOI: 10.1039/c5ra12244a |
| This journal is © The Royal Society of Chemistry 2015 |