 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toxicity of arsenite and thio-DMAV after long-term (21 days) incubation of human urothelial cells: cytotoxicity, genotoxicity and epigenetics
Marlies
Unterberg
ab,
Larissa
Leffers
a,
Florian
Hübner
c,
Hans-Ulrich
Humpf
ac,
Konstantin
Lepikhov
d,
Jörn
Walter
d,
Franziska
Ebert
b and
Tanja
Schwerdtle
*ab
aGraduate School of Chemistry, WWU Muenster, Wilhelm-Klemm-Straße 10, 48149 Muenster, Germany
bInstitute of Nutritional Sciences, University of Potsdam, Arthur-Scheunert-Allee 114-116, 14558 Nuthetal, Germany. E-mail: tanja.schwerdtle@uni-potsdam.de
cInstitute of Food Chemistry, WWU Muenster, Corrensstraße 45, 48149 Muenster, Germany
dDepartment of Genetics/Epigenetics, Campus Saarbruecken, Saarland University, 66123 Saarbruecken, Germany
First published on 30th May 2014
Abstract
This study aims to further mechanistically understand toxic modes of action after chronic inorganic arsenic exposure. Therefore long-term incubation studies in cultured cells were carried out, to display chronically attained changes, which cannot be observed in the generally applied in vitro short-term incubation studies. Particularly, the cytotoxic, genotoxic and epigenetic effects of an up to 21 days incubation of human urothelial (UROtsa) cells with pico- to nanomolar concentrations of iAsIII and its metabolite thio-DMAV were compared. After 21 days of incubation, cytotoxic effects were strongly enhanced in the case of iAsIII and might partly be due to glutathione depletion and genotoxic effects on the chromosomal level. These results are in strong contrast to cells exposed to thio-DMAV. Thus, cells seemed to be able to adapt to this arsenical, as indicated among others by an increase in the cellular glutathione level. Most interestingly, picomolar concentrations of both iAsIII and thio-DMAV caused global DNA hypomethylation in UROtsa cells, which was quantified in parallel by 5-medC immunostaining and a newly established, reliable, high resolution mass spectrometry (HRMS)-based test system. This is the first time that epigenetic effects are reported for thio-DMAV; iAsIII induced epigenetic effects occur in at least 8000 fold lower concentrations as reported in vitro before. The fact that both arsenicals cause DNA hypomethylation at really low, exposure-relevant concentrations in human urothelial cells suggests that this epigenetic effect might contribute to inorganic arsenic induced carcinogenicity, which for sure has to be further investigated in future studies.
Introduction
Among epigenetic modifications DNA methylation is one of the most widely studied epigenetic markers. It is the process of transferring methyl groups from S-adenosyl methionine (SAM) to cytosine nucleotides resulting in 5-methylcytosine (5-medC), predominantly occurring in cytosine-rich gene regions in mammals (called CpG islands) and is implemented through DNA methyltransferases (DNMTs). Several environmental factors are known to disrupt epigenetic programming and thereby cause adverse changes in gene expression predisposing the organism among others to cancer. Thereby, both DNA hyper- and hypomethylation are discussed to independently contribute to cancer development and progression.1–4Inorganic arsenic (iAs) is classified as human carcinogen causing tumors of the lung, skin and urinary bladder.5,6
At exposure-relevant concentrations iAs is neither directly DNA-reactive nor mutagenic. Proposed mechanisms of carcinogenicity include oxidative damage, interference with DNA damage repair and epigenetic effects.7,8 Thus, iAs has been shown to cause both DNA hyper- and/or hypomethylation, resulting in the activation of oncogene expression and silencing of tumor suppressor genes.9–13 These epigenetic findings fit to the perturbation of DNA methylation in cancer cells, frequently showing promoter hypermethylation accompanied with wide spread loss of DNA methylation.14 Moreover, epidemiological studies have linked chronic inorganic arsenic exposure to epigenetic controlled transcriptional deregulation and increased cancer incidences.15–18 Inorganic arsenic induced global DNA hypomethylation might result from SAM deficiency and/or reduction of DNMT activity.13
This study aims to further mechanistically understand toxic modes of action after chronic inorganic arsenic exposure. Therefore long-term incubation studies in cultured human urothelial cells (UROtsa) were carried out, to display chronically attained changes, which cannot be observed in the generally applied in vitro short-term incubation studies. UROtsa cells were incubated up to 21 days with iAsIII as well as with its highly toxic metabolite thio-dimethylarsinic acid (thio-DMAV).19–23 The impact of the arsenicals on global DNA methylation was quantified by an immunofluorescence-based24 assay and in parallel by a newly established high resolution mass spectrometry (HRMS)-based test system. In addition, we tried to draw a link between epigenetic effects, cytotoxic behaviour of the arsenicals as well as genomic instability to further understand and interpret possible crosstalk of mechanisms.
Materials and methods
Caution: Inorganic arsenic is classified as a human carcinogen. The following chemicals are hazardous and should be handled with care: sodium(meta)arsenite (iAsIII) and thio-dimethylarsinic acid (thio-DMAV).Materials
GSH (≥98%), GSSG (≥98%), GSH reductase (from S. cerevisiae), 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), ammonium formate (NH4FA), sodium succinate dibasic hexahydrate (sodium succinate), calcium chloride dehydrate (CaCl2), Triton™ X-100 solution, methanol (HPLC grade), hydrogen peroxide solution as well as 5-Aza-2′-deoxycytidine (5-Aza-dC) were obtained from Sigma Aldrich (Steinheim, Germany). Sodium(meta)arsenite (99%) and Alcian Blue were from Fluka (Buchs, Swiss). Trypsin, fetal calf serum (FCS) and penicillin–streptomycin solutions as well as bovine serum albumin (BSA) were bought from PAA (Pasching, Austria), Minimum essential medium Eagle (MEM) was obtained from Biochrom (Berlin, Germany). Dimethyl sulfoxide (DMSO), Tris-(hydroxymethyl)-aminomethane (Tris), paraformaldehyde, acridine orange and Giemsa stain solutions were purchased from Roth (Karlsruhe, Germany), HCl (normapur) was from VWR (Darmstadt, Germany). Tween® was from AppliChem (Darmstadt, Germany), the HNO3 (suprapur) from Merck (Darmstadt, Germany). Synthesis, purification and analytical characterisation of highly pure dimethylthioarsinic anhydride (precursor of thio-DMAV) were carried out as published before.19Cell culture, preparation of stock solutions and long-term incubation with the test compounds
As in vitro model, the non-tumorigenic urothelial cell line UROtsa was applied, which was kindly provided by Professor M. Stýblo (University of North Carolina, USA). UROtsa cells have proven to be a good model for the bladder epithelium.25 UROtsa cells were cultured as monolayer in MEM supplemented with penicillin (100 U mL−1), streptomycin (100 μg mL−1) and 10% fetal calf serum (FCS). Cultures were incubated under human cell culture standard conditions at 37 °C with 5% CO2 in air and 100% humidity.5-Aza-2′-deoxycytidine (5-Aza-dC) was dissolved in DMSO (100%); dilution was performed in sterile deionised water. iAsIII and thio-DMAV stock solutions were prepared in sterile deionised water shortly before each experiment.
For long-term incubation (7–21 days) with the arsenicals logarithmically growing cells were seeded (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 cells cm−2) on day 1 and incubated with the arsenic species after 24 h (day 2). On day 4 of the experiment, cells were trypsinised for the first time and detached with fresh, arsenic species containing medium. After counting (Casy TTC®, Roche Innovatis AG), cells were reseeded to ensure the appropriate cell density over the time period. This standard operating procedure was performed every three days until the respective endpoint were studied as indicated below. These in vitro long-term incubation conditions simulate a chronic exposure towards the arsenicals and exclude that cells are malignantly transformed.26
600 cells cm−2) on day 1 and incubated with the arsenic species after 24 h (day 2). On day 4 of the experiment, cells were trypsinised for the first time and detached with fresh, arsenic species containing medium. After counting (Casy TTC®, Roche Innovatis AG), cells were reseeded to ensure the appropriate cell density over the time period. This standard operating procedure was performed every three days until the respective endpoint were studied as indicated below. These in vitro long-term incubation conditions simulate a chronic exposure towards the arsenicals and exclude that cells are malignantly transformed.26
Cytotoxicity testing
Long-term cytotoxicity of the arsenicals in UROtsa cells was elucidated by quantifying their effects on cell number11 and colony forming ability.15 Briefly, after 2–21 days of incubation with the arsenicals cells were trypsinised and cell number was measured by an automatic cell counter (Casy TTC®, Roche Innovatis AG). To evaluate the impact on colony forming ability, after cell counting of each sample, 500 cells per dish were seeded again and after 6–7 days, colonies were fixed with EtOH, stained with Giemsa (25% in EtOH), counted and calculated as percent of control.Cellular glutathione and glutathione disulfide levels
The determination of total glutathione (GSH and its oxidised analogue GSSG) and GSSG was performed after 7, 14 and 21 days of incubation with the respective arsenic species as described previously.21Genotoxicity testing
Genotoxicity was assessed on the chromosomal level by investigating the induction of micronuclei as well as the formation of bi- or multinucleated cells after 7, 14 and 21 days of incubation with the arsenicals. Briefly, on incubation days 4, 11 or 18, cells were seeded in 6-well plates on Alcian Blue-coated glass cover slips. After the entire incubation of 7, 14 and 21 days with the arsenicals, cells were fixed and stained with acridine orange.11 Cover slips were coded and events per 1000 mononucleated cells were counted and categorised as follows: mononucleated, binucleated and multinucleated cells as well as with or without micronuclei. Experiments were performed without the application of cytochalasin B, because some arsenicals might interact with actin or the effect of the cytochalasin B itself.19Global DNA methylation
The optimisation of the assays was performed using cells, which were incubated for 24–120 h with the DNA-demethylating agent 5-Aza-dC (1 and 5 μM). Epigenetic effects of the arsenicals were quantified after 21 days of incubation with the respective arsenical.5-medC immunostaining
5-medC immunostaining was carried out after further optimisation of a method from Wossidlo et al.24 Briefly, cells were grown on Alcian Blue-coated glass cover slips. After a first fixation step of incubated cells with 1.37% paraformaldehyde in PBS for 25 min, cells were permeabilised with 0.5% Triton X-100 in phosphate buffered saline PBS (20 min) at room temperature (RT). Cells were treated with 4 M HCl solution (15 min, RT) and neutralised with Tris-Cl (10 min, pH 8, RT). After a second fixation step, cells were blocked over night at 4 °C (1% BSA, 0.1% Triton X-100 in PBS). The next morning, cover slips were detached and incubated with the primary, monoclonal anti-5-methylcytidine antibody (Eurogentex, BI-MECY-0100, Belgium) (1:500, in blocking solution) for 5 h at RT in humid atmosphere. Awaiting three washing steps with the blocking solution, cells were treated with the secondary, Alexa 488-conjugated anti-mouse antibody (Invitrogen, Eugene, Oregon, USA) (1:1000, in blocking solution with 0.1% Tween) for 1 h at RT in the dark. Cover slips were washed for three times, subsequently cells were mounted with Vectashield mounting medium containing 1 μg mL−1 DAPI (Vector Laboratories Inc., Burlingame, CA, USA) and analysed by fluorescence microscopy applying a Zeiss Axio ImagerM2 wide field fluorescence microscope (Zeiss, Jena, Germany). At least 300–400 cell nuclei per slide were selected by DAPI staining. In the selected areas the relative Alexa-488 fluorescence intensities were quantified using the Axio Vision (Version 4.5) imaging software (Zeiss, Jena, Germany).
High resolution mass spectrometry
For the HRMS-based quantification of global DNA methylation a new method was established. Briefly, after DNA isolation, DNA was enzymatically hydrolysed and subsequently analysed by HPLC-HRMS.In detail, cells were trypsinised, collected via centrifugation and washed with ice-cold PBS. DNA was isolated (Perfect Pure DNA Kit, 5 Prime, Hamburg, Germany), DNA content and purity were quantified photometrically and the sample was evaporated to dryness using a centrifugal vacuum evaporator. For enzymatic hydrolysis, samples were resolved in buffer (100 mM sodium succinate, 50 mM CaCl2, pH 6) and a micrococcale nuclease (0.4 U μL−1, from S. aureus) together with a phosphodiesterase (0.001 U μL−1, from bovine spleen) were applied overnight (at 37 °C), followed up by nuclease P1 (0.3 U μL−1, from P. citrinum) (all purchased from Sigma Aldrich, Steinheim, Germany) for 4 h. Enzymatic hydrolysis was terminated by centrifugation (17500g, 20 min) and evaporation of the supernatant. Finally, samples were resolved in water (deionised) to achieve a concentration of 0.25–0.5 μg μL−1 hydrolysed DNA.
HRMS analysis was carried out using a Thermo HPLC system (Accela LC with Accela Pump 60057-60010 and Accela Autosampler 60057-60020) coupled to a LTQ-Orbitrap-XL Fourier transform mass spectrometer equipped with heated ESI source (Thermo Scientific, Dreieich, Germany); UV absorbance was monitored at 277 nm27 (Variable Wavelength Monitor, Knauer, Bad Homburg, Germany). The samples were separated on a Synergi Fusion RP-80A C18 column (4 μm, 250 × 2 mm) attached to a Fusion RP security guard column (4 μm, 4 × 3 mm) (Phenomenex, Aschaffenburg, Germany). The flow rate was set to 0.3 mL min−1, with an injection volume of 10 μL. The samples were eluted with ammonium formate (50 mM, pH 5.4) (A) and methanol (B) using the following gradient: 0 min – 2% B, 30 min – 10% B, 31 min – 2% B, 36 min – 2% B (at RT). Ionisation was carried out with heated electrospray in positive mode with the following parameters: capillary temperature, 350 °C; vaporiser temperature, 350 °C; sheath gas flow, 35 arb; auxiliary gas flow, 10 arb; sweep gas flow, 0 arb; spray voltage, 3 kV; capillary voltage, 35 V; tube lens, 110 V. The scan event was programmed to perform a total ion scan of mass range from m/z 50 to m/z 500 at a resolution of 30000.
Data analysis was performed using the Xcalibur 2.07 SP1 software (Thermo Scientific, Dreieich, Germany). Hence, the base peaks of the DNA bases [M + H]+ were extracted with a width of ±10 ppm from the mass spectra and the ratio of peak areas (medC/(dC + medC)) was calculated by integrating the relative ionisation efficiency. The respective results were then compared to control cells for evaluation.
Statistical analysis
All experiments were at least carried out three times at three different days. As indicated in the respective figure legends, the mean standard deviation (SD) was calculated from the raw data and a statistical analysis was performed by using the ANOVA test (Dunnett and Tukey, respectively): *p < 0.05, **p < 0.01, ***p < 0.001.Results
Cytotoxicity testing
After 21 days incubation of UROtsa cells, iAsIII exerted stronger effects on cell number and on colony forming ability as compared to its metabolite thio-DMAV (Fig. 1A and B). Thus, 250 nM iAsIII significantly decreased both endpoints, whereas thio-DMAV exerted no significant cytotoxicity up to an incubation concentration of 500 nM. The cytotoxicity endpoint cell number was stronger affected by iAsIII than was colony forming ability. Furthermore, cytotoxicity of iAsIII clearly increased with incubation time, as demonstrated for the endpoint cell number (Fig. 1C).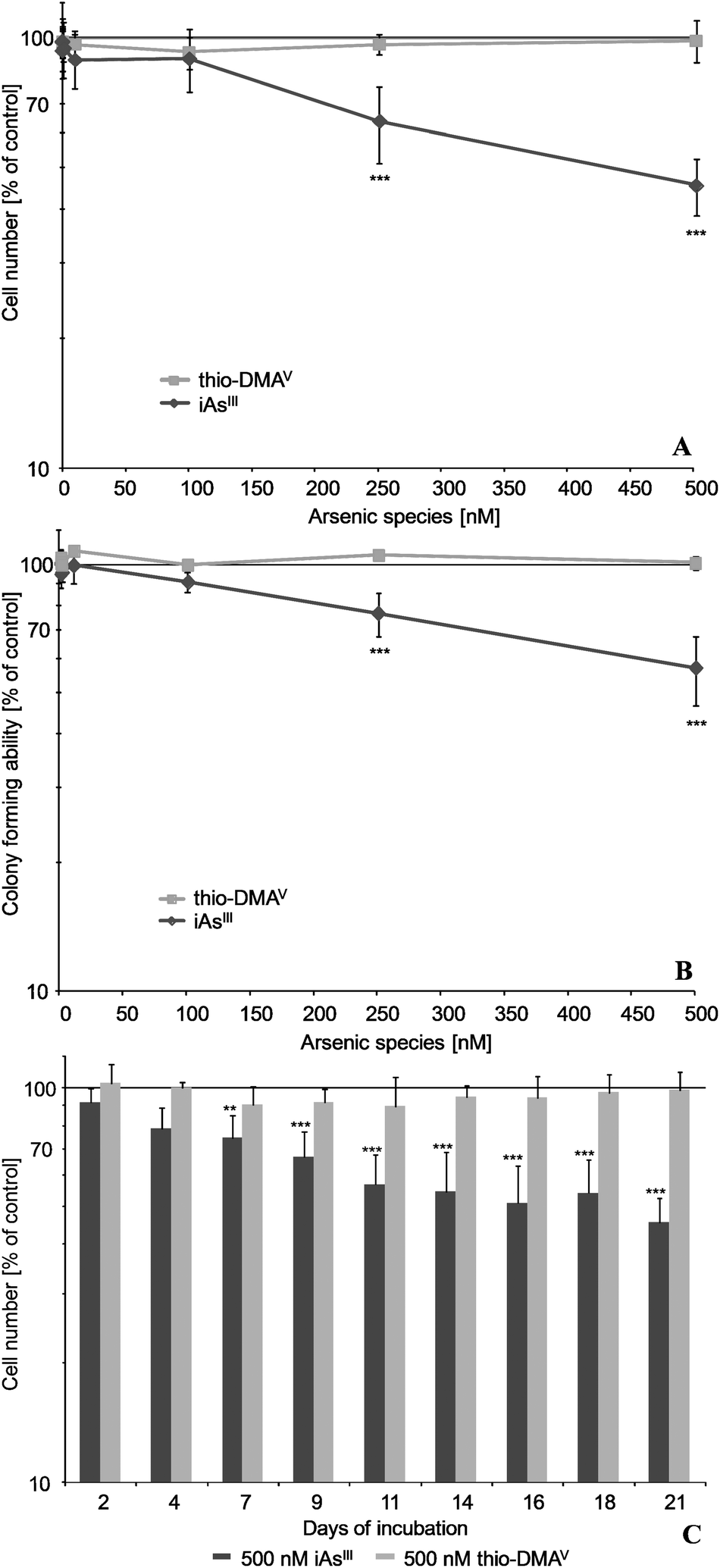 | ||
| Fig. 1 Cytotoxicity of iAsIII and its metabolite thio-DMAV in UROtsa cells after long-term incubation. Presented are effects on cell number (A, C) as well as colony forming ability (B) after 21 (A, B) and 2–21 days (C) of incubation, respectively. Shown are mean values of at least 3 independent determinations ±/+ SD. Colony forming ability of control cells was 80%. | ||
Cellular total glutathione
iAsIII and thio-DMAV showed different effects on the cellular total glutathione level (Fig. 2A and B). Thus, after 7, 14 and 21 days incubation, nanomolar iAsIII concentrations decreased the total glutathione level in UROtsa cells. Thio-DMAV lowered cellular total glutathione after 7 days incubation. Nevertheless, after 14–21 days incubation with thio-DMAV a strong increase in total cellular glutathione arose, pointing towards an adaption of the cellular system towards this arsenical.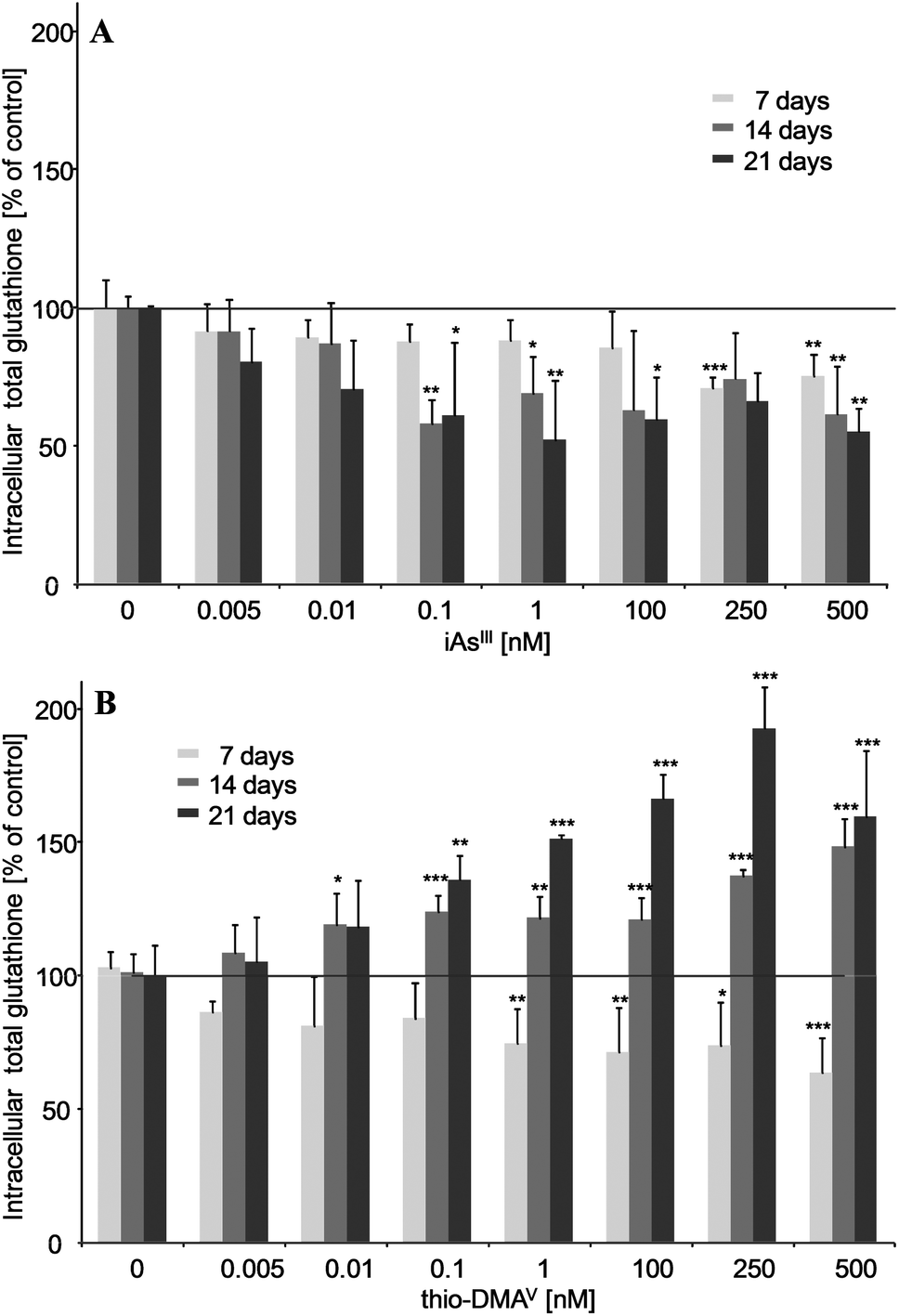 | ||
| Fig. 2 Intracellular total glutathione after 7, 14 and 21 days of incubation of iAsIII (A) or thio-DMAV (B) in UROtsa cells. Shown are mean values of at least 3 independent determinations +SD. | ||
The intracellular GSH/GSSG ratio was always monitored in parallel and was neither affected by iAsIII nor thio-DMAV (data not shown).
Micronuclei formation
Micronuclei formation was increased after long-term incubation with iAsIII (Fig. 3). A significant increase occurred already in case of the subcytotoxic concentration 100 nM iAsIII. In contrast, thio-DMAV did not induce micronuclei formation in the observed concentration-range. The number of bi- and multinucleated cells were not affected by iAsIII or thio-DMAV (data not shown).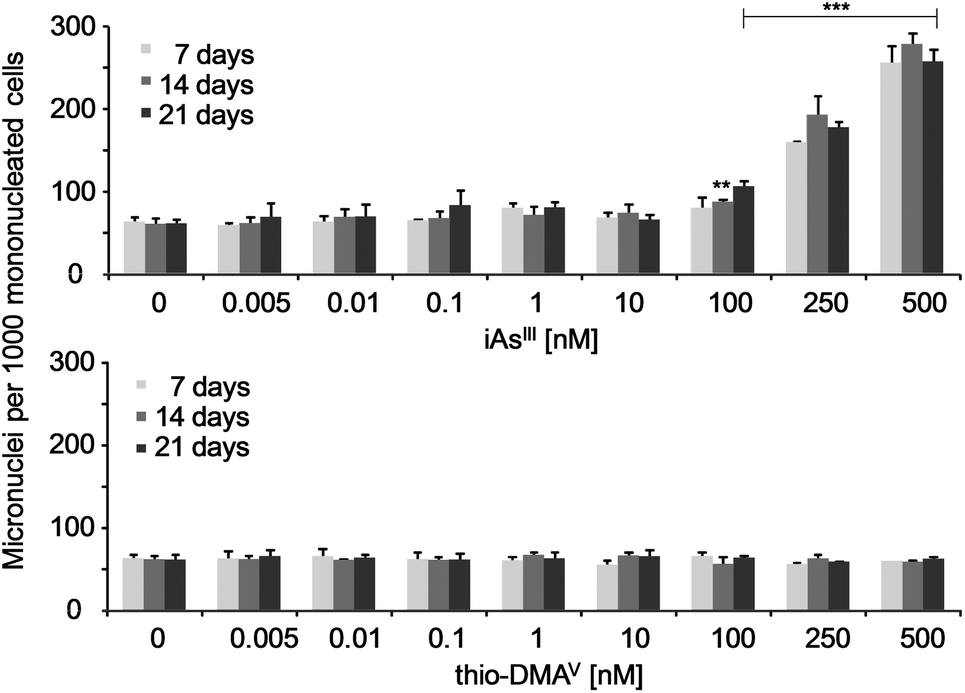 | ||
| Fig. 3 Micronuclei formation in UROtsa cells after 7, 14 and 21 days of incubation with iAsIII or thio-DMAV. Displayed are mean values of at least three independent determinations +SD. | ||
Establishment of a HPLC-HRMS method to analyse nucleosides
The HRMS method was established using standard solutions of all DNA nucleosides which were separated by HPLC and identified by their extracted base peaks of DNA bases [M + H]+ from total ion scans: m/z dC 112.0501, medC 126.0657, dG 152.0565, T 127.0494, dA 136.0614 (±10 ppm), respectively. Identification was confirmed via the characteristic fragmentation spectra of nucleosides (Fig. 4). Strikingly, the five DNA bases were not ionised in similar manner. To include the differences in ionisation efficiency in the calculation of cellular methylation, before each run standards of methylated and non-methylated bases with known concentrations were analysed. The results of these measurements were used to define the relative ionisation efficiency of bases, which was then used for correction of the areas from measurements of samples for determination of the ratio (medC/(dC + medC)).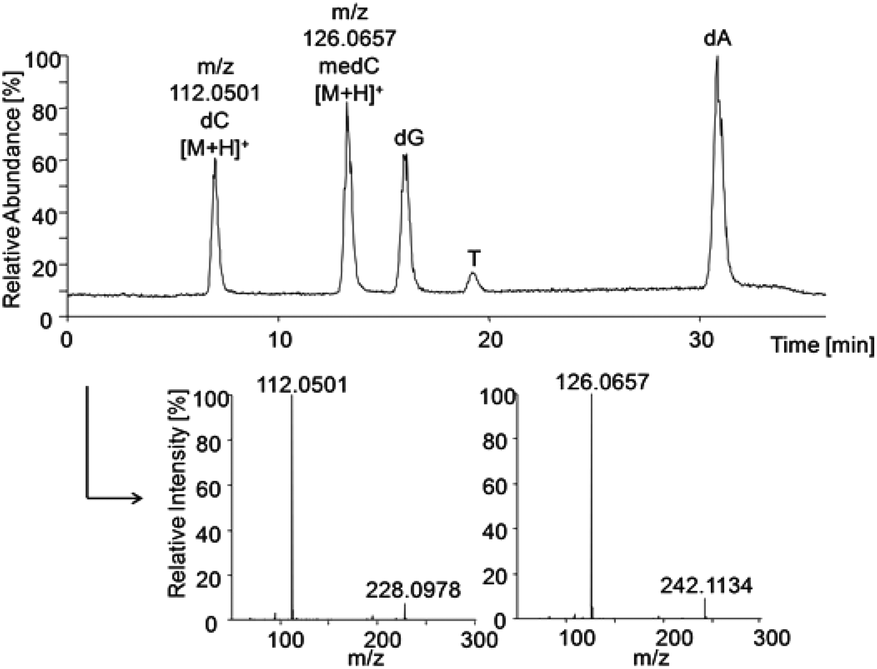 | ||
| Fig. 4 HPLC-HRMS total ion chromatogram of pooled standards (each 0.1 μM) and extracted mass spectra of dC and medC to identify the nucleosides via fragmentation to the corresponding bases. | ||
Applicability of the method was approved by quantification of DNA methylation in non-incubated control cells to 3.55% ± 0.69%, matching to postulated ranges in mammalian cells in culture.28–30
Global DNA methylation
5-Aza-dC induced effects on global DNA methylation were comparable when quantified by the immunofluorescence-based and the established HRMS-based test system (Fig. 5A and B). Thus, 24–120 h incubation with 1 μM 5-Aza-dC significantly decreased DNA methylation by around 50%.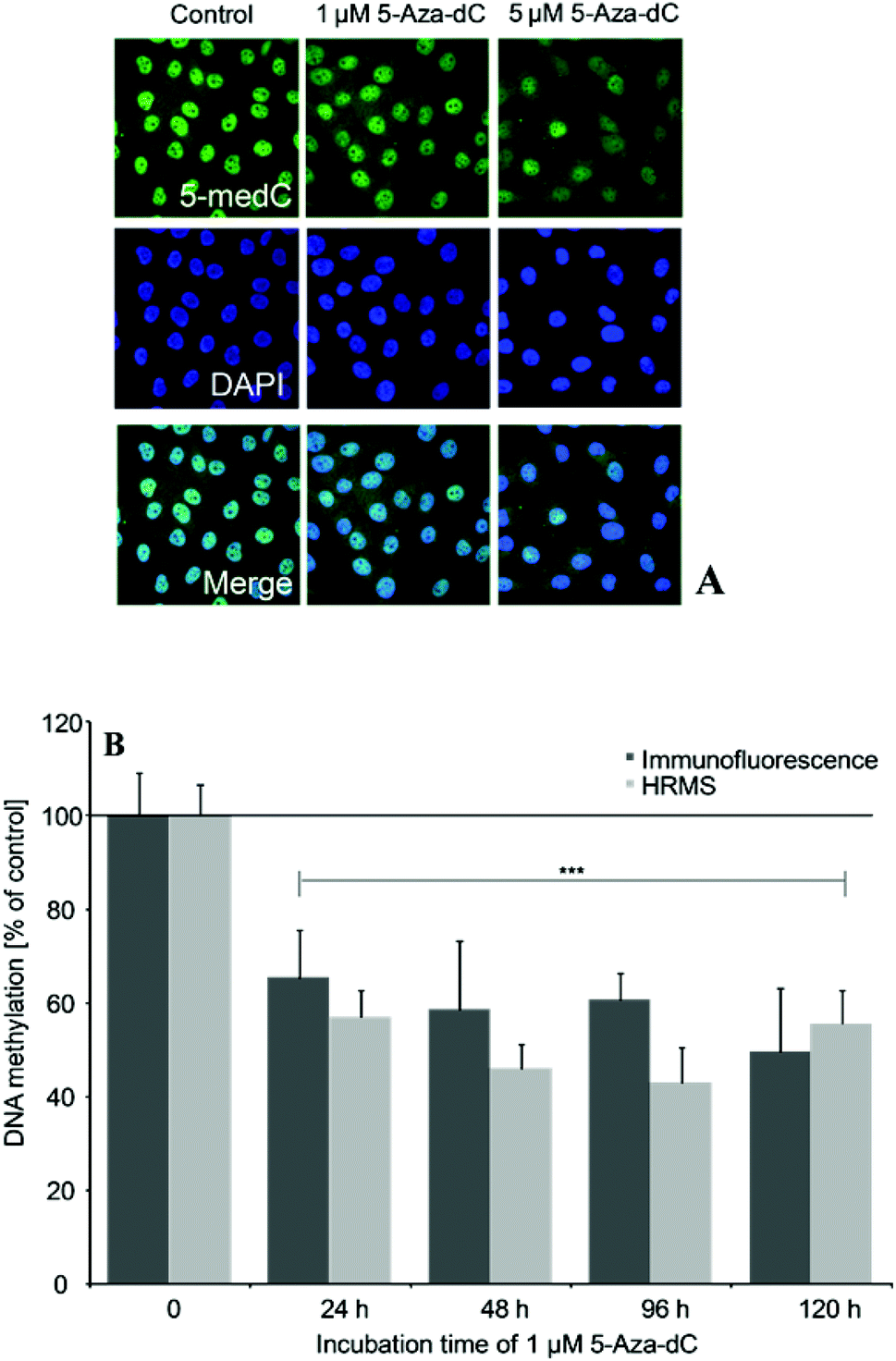 | ||
| Fig. 5 Global DNA hypomethylation induced by 5-Aza-dC as measured by 5-medC immunostaining (A, B) or HRMS (B), respectively. Effects were normalized to non-treated control cells; shown are mean values of at least 3 independent determinations +SD. | ||
After 21 days incubation, both iAsIII and thio-DMAV caused DNA hypomethylation in UROtsa cells (Fig. 6A and B). Thereby significant effects were measured applying both techniques already after incubation with picomolar concentrations of the arsenicals. DNA hypomethylation induced by the arsenicals was less intensive, when quantified via HRMS.
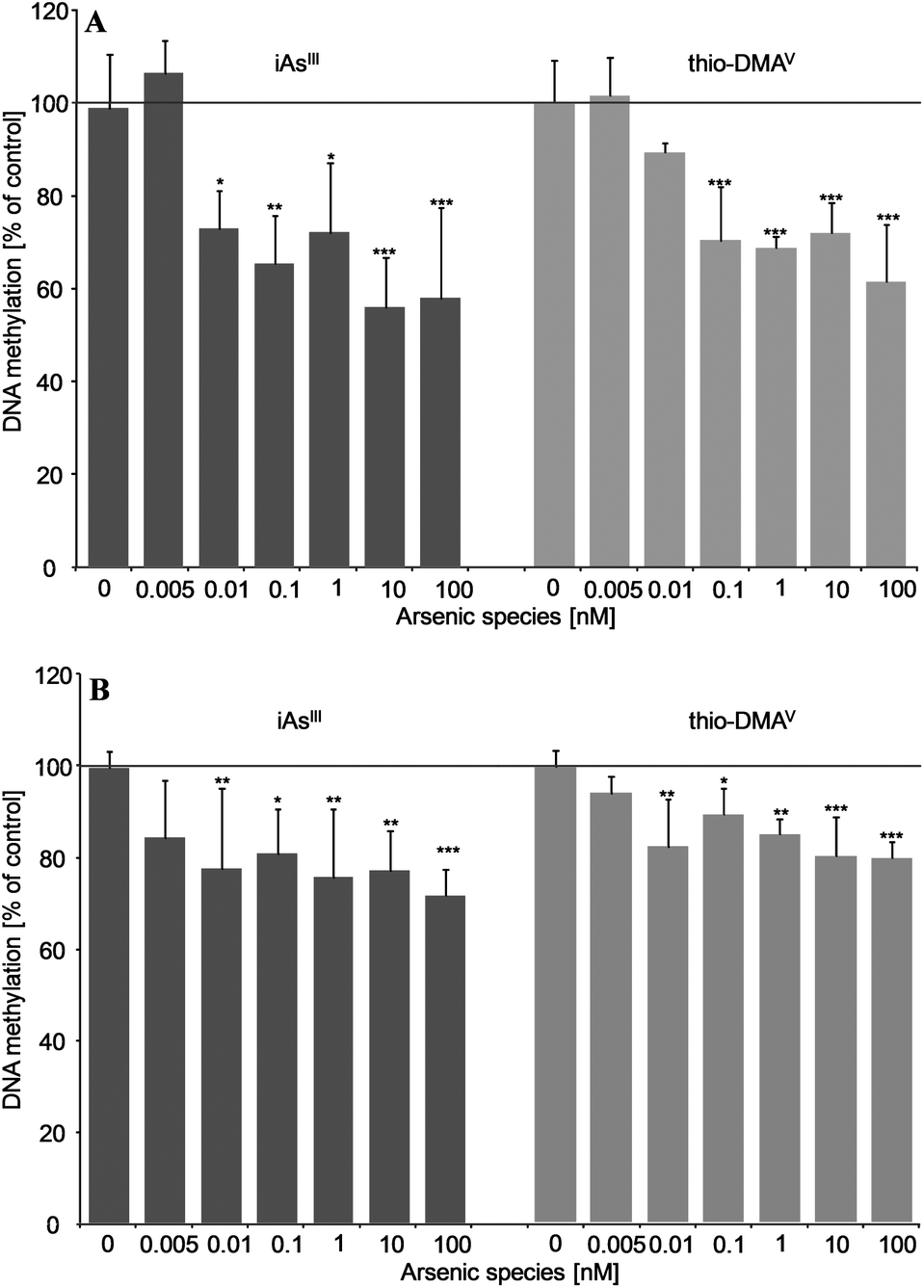 | ||
| Fig. 6 Effects on global DNA methylation in UROtsa after long-term incubation (21 days) with iAsIII or thio-DMAV as measured by 5-medC immunostaining (A) and HPLC-HRMS (B); shown are mean values of at least three independent determinations +SD. | ||
Discussion
This study compared for the first time the cytotoxic, genotoxic and epigenetic effects of a chronic long-term incubation of human urothelial (UROtsa) cells with pico- to nanomolar concentrations of iAsIII and its metabolite thio-DMAV.Recent studies in UROtsa cells have reported that after 24–48 hours incubation thio-DMAV exerted its cytotoxicity in a similar concentration range or at even lower concentrations as compared to iAsIII.20,31 Nevertheless, after 21 days incubation iAsIII is the more potent cytotoxic arsenical in these cells. Referring to the respective IC70 values and comparing 21 days with 24 h incubation (Table 1), iAsIII-induced effects on cell number and colony forming ability were evident at 18.6- and 16.3-fold lower concentrations, respectively.
| a Ebert et al. 2013.20 b Leffers et al. 2013.31 | |||
|---|---|---|---|
| iAsIII | |||
| 30% reduction in | 24 h | 48 h | 21 days |
| Cell number | 3900 nMa | 3600 nMb | 210 nM |
| Colony-forming ability | 5200 nMa | 2000 nMb | 320 nM |
| thio-DMAV | |||
| 30% reduction in | 24 h | 48 h | 21 days |
| Cell number | 4300 nMa | 2300 nMb | >500 nM |
| Colony-forming ability | 5200 nMa | 1500 nMb | >500 nM |
The absence of a similar incubation-time dependent increase in the cytotoxic effects of thio-DMAV, might be due to an adaption of the cellular system towards thio-DMAV after chronic exposure. This assumption is supported by the glutathione data. Thus, after long-term incubation with thio-DMAV, total glutathione levels in UROtsa cells increased, whereas in case of iAsIII total glutathione levels were decreased. This suggests that thio-DMAV induces cellular GSH synthesis, which subsequently counteracts thio-DMAV induced cytotoxic effects. In contrast, iAsIII-induced decrease in glutathione is likely to partly cause its cytotoxicity.
Genotoxicity after long-term incubation with the arsenicals was studied on the chromosomal level by the use of the micronuclei assay.32,33 In case of iAsIII micronuclei formation was already significantly increased at a subcytotoxic concentration of 100 nM iAsIII. In contrast, likewise after short-term incubation,19,31 thio-DMAV did not exert genotoxicity (as measured by micronuclei formation) in the subcytotoxic concentration range.
To accurately quantify potential epigenetic effects induced by the arsenicals after long-term in vitro incubation we measured global DNA methylation by a further optimised immuno-fluorescence-based assay24 and in parallel by a newly established HPLC-HRMS-based test system. Performance of the test systems was compared applying cells incubated with the demethylating agent 5-Aza-2′-deoxycytidine (5-Aza-dC). This cytosine analog is well known to alter global DNA methylation by inhibiting DNA methyltransferases. After incorporation into DNA it forms an irreversible covalent complex with DNMT enzymes, primarily with the de novo methyltransferases DNMT3a and DNMT3b.34–36 The levels of 5-Aza-dC-induced DNA hypomethylation were comparable when quantified by immunostaining and HRMS and fit nicely to the levels published in literature.30 The immunofluorescence-based technique is faster and seems to be a suitable screening tool, assessing effects in a few hundred cells. The more time consuming and costlier HRMS method has the advantage of being more reproducible. This is most likely because the respective DNA-methylation levels represent mean levels of a large number of cells, which have been pooled for DNA isolation. Thus HRMS might be used to verify effects measured by immunofluorescence and produce valid data.
Applying both techniques we could show that picomolar concentrations of iAsIII and thio-DMAV cause global DNA hypomethylation in UROtsa cells. The respective intensity of DNA hypomethylation fits to observations in cancer studies: although the frequency of cancer-associated hypomethylation depends on grade, stage and individual tumor, a large loss of global methylation between 20–60% of 5-medC compared to healthy cells is postulated.37–39 In this case, most organisms are able to survive dramatic global DNA hypomethylation, which is believed to emerge early in carcinogenesis.40,41
Depletion in global DNA methylation after in vitro exposure to arsenite was recorded several times before.13,42–46 Nevertheless, effective arsenite concentrations in the present study are at least 8000 fold lower, than reported prior to this.13,42–47 Furthermore, thio-DMAV has never been shown to exert epigenetic effects before.
Several mechanisms might account for the epigenetic effect of the respective arsenicals. Both arsenicals might disturb DNA methyltransferases or cause a lack of the methylating substrate S-adenosyl-methionine (SAM).
DNA methyltransferases, which are urgently needed for maintenance (DNMT1) as well as de novo (DNMT3a/b) DNA methylation can be divided into three different families, while DNMT2 shows quite weak activity in vitro.48,49 Since activation and stimulation of DNMT3a and b enzyme activity have been shown to be regulated by mutual interaction, effects on one methyltransferase can have wide-ranging consequences also for the other methyltransferase.50 The arsenicals might disturb the activity of the respective DNA methyltransferases via both a direct and an indirect interaction; effects on gene expression are also likely to occur.16 Thus, a repression of DNMT1 and DNMT3a enzyme activity as well as a decrease in the respective gene expression levels has been shown in various human cells after incubation with either iAsIII (ref. 10 and 44) or arsenic trioxide.13,46,51,52 One possible example for an indirect disturbance of the activity of DNA methyltransferases could be a compromised ability of DNMTs to methylate cytosines through hindrance of oxidative lesions on DNA, such as 8-oxoguanine.53,54 In this context, arsenite has been shown before to decrease repair of 8-oxoguanine in vitro.55
Besides providing methyl groups for DNA methylation, SAM represents the major methyl donor for inorganic arsenic biomethylation in humans. But in the cellular system applied in our study, iAsIII is not metabolised to its methylated metabolites (data not shown). Thus, SAM will not be exhausted by arsenic metabolism, which has been reported before in another cellular system.43 Nevertheless, the arsenicals might decrease cellular SAM production. Thus, arsenic species could disturb methionine metabolism through changes in the intracellular redox state e.g. via glutathione depletion.43 GSH and SAM metabolism are connected by shared requirement for homocysteine (Hcy).56 Chronically attained glutathione depletion, as observed in the case of iAsIII in the present study, could reduce DNA methylation via reduced methyl donating SAM cycle components because of transsulfuration of Hcy on GSH. Hcy, the breakdown product of S-adenosyl-homocysteine (SAH) and precursor of GSH, seems to be very important due to its connectivity to both, the one carbon as well as transsulfuration pathway.43,57,58
In the present study thio-DMAV induced DNA hypomethylation in the same concentration range like iAsIII. This points once more towards the high toxic potential of this iAs metabolite.
Nevertheless, as indicated before,19,20,23,59,60 toxic modes of action seem to be different for these two arsenicals. In case of iAsIII, the induced epigenetic and genotoxic effects might be linked to each other. Thus, increased micronuclei formation might result from a disturbance of certain DNA binding proteins and changes in DNA confirmation.61 This junction has been observed in other studies with mice;62,63 if this theory can be transferred to humans has to be further elucidated. In this context, Ramirez et al. demonstrated that exogenous addition of SAM results in a prevention of aneuploidy in human cells treated with iAsIII.64 The authors discussed that SAM might have a protective effect on the ‘machinery of chromosome segregation’ related to a ‘restoration of cell methylation status’.
Conclusions
The fact that both arsenite and its metabolite thio-DMAV caused DNA hypomethylation at really low, exposure-relevant65–67 concentrations in human urothelial cells suggests that this epigenetic effect might contribute to inorganic arsenic induced carcinogenicity. This, for sure, has to be clarified in future studies. Nevertheless, likewise arsenite,26 thio-DMAV might be classified now as an in vitro epimutagen, an agent ‘whose exposure induces stable and inheritable changes to epigenetic state’. Further investigations, including gene expression studies as well as the quantification of cellular SAM levels, might be helpful to understand whether both arsenicals cause DNA hypomethylation via similar or different mechanisms. Moreover, a detailed investigation of other epigenetic endpoints is likely to provide further insight in the toxic mechanisms of the arsenicals in these low concentration ranges. Finally, this study clearly demonstrates the potential of long-term incubation in vitro studies, especially when searching for mechanisms behind diseases following chronic exposure towards environmental contaminants.The authors declare that there are no conflicts of interests.
Acknowledgements
The authors especially thank the master student Cornelia Hoppe (Food Chemistry, WWU Muenster, Germany) for her practical work in the laboratory. This work was supported by the Graduate School of Chemistry (WWU Muenster, Germany) and the DFG grant number SCHW903/4-1 and the European Union's Seventh Framework Programme (FP7/2007-2013) [grant numbers 267038 (NOTOX) to JW].References
- I. P. Pogribny and I. Rusny, in Epigenetic Alterations in Oncogenesis Advances in Experimental Medicine and Biology 754, ed. A. R. Karpf, Springer, Berlin, 2013, vol. 754, pp. 215–232 Search PubMed.
- D. Deobagkar, in Toxicology and Epigenetics, ed. S. C. Sahu, Wiley, Chichester, UK, 2012, pp. 25–50 Search PubMed.
- B. Sadikovic, K. Al-Romaih, J. A. Squire and M. Zielenska, Curr. Genomics, 2008, 9, 394–408 CrossRef CAS PubMed.
- J. Sandoval and M. Esteller, Curr. Opin. Genet. Dev., 2012, 22, 50–55 CrossRef CAS PubMed.
- IARC, IARC Monographs on the Evaluation of the Carcinogenic Risks to Humans, suppl. 7, Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs, Lyon, France, 1987 Search PubMed.
- IARC, A review of human carcinogens – Part C: Arsenic, metals, fibres and dusts, IARC Monographs, Lyon, France, 2012 Search PubMed.
- EFSA, Panel on Contaminants in the Food Chain (CONTAM), European Food Safety Authority (EFSA), Scientific Opinion on Arsenic in Food, EFSA J., 2009, 7(1–198), 1351 Search PubMed.
- A. Arita and M. Costa, Metallomics, 2009, 1, 222–228 RSC.
- J. F. Reichard and A. Puga, Epigenomics, 2010, 2, 87–104 CrossRef CAS PubMed.
- J. C. States, A. Barchowsky, I. L. Cartwright, J. F. Reichard, B. W. Futscher and R. C. Lantz, Environ. Health Perspect., 2011, 119, 1356–1363 CrossRef CAS PubMed.
- M. F. Hughes, B. D. Beck, Y. Chen, A. S. Lewis and D. J. Thomas, Toxicol. Sci., 2011, 123, 305–332 CrossRef CAS PubMed.
- H. H. Wang, M. M. Wu, M. W. Chan, Y. S. Pu, C. J. Chen and T. C. Lee, Arch. Toxicol., 2014 Search PubMed , Feb 24 Epub ahead of print.
- J. F. Reichard, M. Schnekenburger and A. Puga, Biochem. Biophys. Res. Commun., 2007, 352, 188–192 CrossRef CAS PubMed.
- S. B. Baylin and J. G. Herman, Trends Genet, 2000, 16, 168–174 CrossRef CAS.
- R. Martinez-Zamudio and H. C. Ha, Epigenetics, 2011, 6, 820–827 CrossRef CAS.
- X. Ren, C. M. McHale, C. F. Skibola, A. H. Smith, M. T. Smith and L. Zhang, Environ. Health Perspect., 2011, 119, 11–19 CrossRef CAS PubMed.
- C. J. Marsit, M. R. Karagas, H. Danaee, M. Liu, A. Andrew, A. Schned, H. H. Nelson and K. T. Kelsey, Carcinogenesis, 2006, 27, 112–116 CrossRef CAS PubMed.
- C. J. Marsit, M. R. Karagas, A. Schned and K. T. Kelsey, Annal. N. Y. Acad. Sci., 2006, 1076, 810–821 CrossRef CAS PubMed.
- M. Bartel, F. Ebert, L. Leffers, U. Karst and T. Schwerdtle, J. Toxicol., 2011, 2011, 373141 Search PubMed.
- F. Ebert, L. Leffers, T. Weber, S. Berndt, A. Mangerich, S. Beneke, A. Burkle and T. Schwerdtle, J. Trace Elem. Med. Biol., 2014, 28(2), 138–146 CAS.
- L. Leffers, M. Unterberg, M. Bartel, C. Hoppe, I. Pieper, J. Stertmann, F. Ebert, H. U. Humpf and T. Schwerdtle, Toxicology, 2013, 305, 109–119 CrossRef CAS PubMed.
- H. Naranmandura, M. W. Carew, S. Xu, J. Lee, E. M. Leslie, M. Weinfeld and X. C. Le, Chem. Res. Toxicol., 2011, 24, 1586–1596 CrossRef CAS PubMed.
- H. Naranmandura, Y. Ogra, K. Iwata, J. Lee, K. T. Suzuki, M. Weinfeld and X. C. Le, Toxicol. Appl. Pharmacol., 2009, 238, 133–140 CrossRef CAS PubMed.
- M. Wossidlo, J. Arand, V. Sebastiano, K. Lepikhov, M. Boiani, R. Reinhardt, H. Scholer and J. Walter, EMBO J., 2010, 29, 1877–1888 CrossRef CAS PubMed.
- K. E. Eblin, M. E. Bowen, D. W. Cromey, T. G. Bredfeldt, E. A. Mash, S. S. Lau and A. J. Gandolfi, Toxicol. Appl. Pharmacol., 2006, 217, 7–14 CrossRef CAS PubMed.
- B. W. Futcher, in Epigenetic Alterations in Oncogenesis Advances in Experimental Medicine and Biology 754, ed. A. R. Karpf, Springer, Berlin, 2013, vol. 754, pp. 179–194 Search PubMed.
- J. Sandhu, B. Kaur, C. Armstrong, C. J. Talbot, W. P. Steward, P. B. Farmer and R. Singh, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1957–1961 CrossRef CAS PubMed.
- L. Song, S. R. James, L. Kazim and A. R. Karpf, Anal. Chem., 2005, 77, 504–510 CrossRef CAS PubMed.
- X. Wang, Y. Suo, R. Yin, H. Shen and H. Wang, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1647–1652 CrossRef CAS PubMed.
- M. F. Paz, M. F. Fraga, S. Avila, M. Guo, M. Pollan, J. G. Herman and M. Esteller, Cancer Res., 2003, 63, 1114–1121 CAS.
- L. Leffers, F. Ebert, M. S. Taleshi, K. A. Francesconi and T. Schwerdtle, Mol. Nutr. Food Res., 2013, 57, 1270–1282 CAS.
- M. Fenech, Mutat. Res., 2000, 455, 81–95 CrossRef CAS.
- M. Fenech, M. Kirsch-Volders, A. T. Natarajan, J. Surralles, J. W. Crott, J. Parry, H. Norppa, D. A. Eastmond, J. D. Tucker and P. Thomas, Mutagenesis, 2011, 26, 125–132 CrossRef CAS PubMed.
- L. Schermelleh, F. Spada, H. P. Easwaran, K. Zolghadr, J. B. Margot, M. C. Cardoso and H. Leonhardt, Nat. Methods, 2005, 2, 751–756 CrossRef CAS PubMed.
- L. Schermelleh, F. Spada and H. Leonhardt, Curr. Protoc. Cell Biol., 2008 Search PubMed , ch. 22, unit 22.12.
- M. Oka, A. M. Meacham, T. Hamazaki, N. Rodic, L. J. Chang and N. Terada, Oncogene, 2005, 24, 3091–3099 CrossRef CAS PubMed.
- A. Portela and M. Esteller, Nat. Biotechnol., 2010, 28, 1057–1068 CrossRef CAS PubMed.
- L. Wild and J. M. Flanagan, Biochim. Biophys. Acta, 2010, 1806, 50–57 CAS.
- M. Ehrlich and M. Lacey, in Epigenetic Alterations in Oncogenesis Advances in Experimental Medicine and Biology 754, ed. A. R. Karpf, Springer, Berlin, 2013, vol. 754, pp. 31–56 Search PubMed.
- M. Ehrlich, Oncogene, 2002, 21, 5400–5413 CrossRef CAS PubMed.
- A. G. Rivenbark and W. B. Coleman, in Toxicology and Epigenetics, ed. S. C. Sahu, Wiley, Chichester, UK, 2012, pp. 317–338 Search PubMed.
- C. Q. Zhao, M. R. Young, B. A. Diwan, T. P. Coogan and M. P. Waalkes, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10907–10912 CrossRef CAS.
- J. F. Coppin, W. Qu and M. P. Waalkes, J. Biol. Chem., 2008, 283, 19342–19350 CrossRef CAS PubMed.
- T. J. Jensen, P. Novak, K. E. Eblin, A. J. Gandolfi and B. W. Futscher, Carcinogenesis, 2008, 29, 1500–1508 CrossRef CAS PubMed.
- T. J. Jensen, P. Novak, S. M. Wnek, A. J. Gandolfi and B. W. Futscher, Toxicol. Appl. Pharmacol., 2009, 241, 221–229 CrossRef CAS PubMed.
- L. Benbrahim-Tallaa, R. A. Waterland, M. Styblo, W. E. Achanzar, M. M. Webber and M. P. Waalkes, Toxicol. Appl. Pharmacol., 2005, 206, 288–298 CrossRef CAS PubMed.
- M. J. Mass and L. Wang, Mutat. Res., 1997, 386, 263–277 CAS.
- R. Jaenisch and A. Bird, Nat. Genet., 2003, 33(Suppl), 245–254 CrossRef CAS PubMed.
- R. J. Klose and A. P. Bird, Trends Biochem. Sci., 2006, 31, 89–97 CrossRef CAS PubMed.
- J. Y. Li, M. T. Pu, R. Hirasawa, B. Z. Li, Y. N. Huang, R. Zeng, N. H. Jing, T. Chen, E. Li, H. Sasaki and G. L. Xu, Mol. Cell Biol., 2007, 27, 8748–8759 CrossRef CAS PubMed.
- H. Y. Fu, J. Z. Shen, Y. Wu, S. F. Shen, H. R. Zhou and L. P. Fan, Oncol. Rep., 2010, 24, 335–343 CrossRef CAS.
- X. Cui, T. Wakai, Y. Shirai, N. Yokoyama, K. Hatakeyama and S. Hirano, Hum. Pathol., 2006, 37, 298–311 CrossRef CAS PubMed.
- S. A. Weitzman, P. W. Turk, D. H. Milkowski and K. Kozlowski, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1261–1264 CrossRef CAS.
- V. Valinluck and L. C. Sowers, Cancer Res., 2007, 67, 946–950 CrossRef CAS PubMed.
- F. Ebert, A. Weiss, M. Bultemeyer, I. Hamann, A. Hartwig and T. Schwerdtle, Mutat. Res., 2011, 715, 32–41 CrossRef CAS PubMed.
- K. A. Bailey, M. C. Wu, W. O. Ward, L. Smeester, J. E. Rager, G. Garcia-Vargas, L. M. Del Razo, Z. Drobna, M. Styblo and R. C. Fry, J. Biochem. Mol. Toxicol., 2013, 27, 106–115 CrossRef CAS PubMed.
- E. Mosharov, M. R. Cranford and R. Banerjee, Biochemistry, 2000, 39, 13005–13011 CrossRef CAS PubMed.
- D. H. Lee, D. R. Jacobs Jr. and M. Porta, Environ. Health Perspect., 2009, 117, 1799–1802 CrossRef CAS PubMed.
- H. Naranmandura, K. Ibata and K. T. Suzuki, Chem. Res. Toxicol., 2007, 20, 1120–1125 CrossRef CAS PubMed.
- T. Ochi, K. Kita, T. Suzuki, A. Rumpler, W. Goessler and K. A. Francesconi, Toxicol. Appl. Pharmacol., 2008, 228, 59–67 CrossRef CAS PubMed.
- H. Stopper, I. Eckert, P. Wagener and W. A. Schulz, Recent Results Cancer Res., 1997, 143, 183–193 CAS.
- A. Eden, F. Gaudet, A. Waghmare and R. Jaenisch, Science, 2003, 300, 455 CrossRef CAS PubMed.
- F. Gaudet, J. G. Hodgson, A. Eden, L. Jackson-Grusby, J. Dausman, J. W. Gray, H. Leonhardt and R. Jaenisch, Science, 2003, 300, 489–492 CrossRef CAS PubMed.
- T. Ramirez, H. Stopper, R. Hock and L. A. Herrera, Mutat. Res., 2007, 617, 16–22 CrossRef CAS PubMed.
- WHO, Arsenic and arsenic compounds, World Health Organization, Geneva, 2001 Search PubMed.
- R. Raml, A. Rumpler, W. Goessler, M. Vahter, L. Li, T. Ochi and K. A. Francesconi, Toxicol. Appl. Pharmacol., 2007, 222, 374–380 CrossRef CAS PubMed.
- J. Scheer, S. Findenig, W. Goessler, K. A. Francesconi, B. Howard, J. G. Umans, J. Pollak, M. Tellez-Plaza, E. K. Silbergeld, E. Guallar and A. Navas-Acien, Anal. Methods, 2012, 4, 406–413 RSC.
| This journal is © The Royal Society of Chemistry 2014 |