
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduced roughness for improved mobility in benzodipyrrolidone-based, n-type OFETS†
J. W.
Rumer
*a,
S.
Rossbauer
b,
M.
Planells
a,
S. E.
Watkins
c,
T. D.
Anthopoulos
b and
I.
McCulloch
a
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK. E-mail: jwrumer@imperial.ac.uk
bDepartment of Physics and Centre for Plastic Electronics, Imperial College London, London SW7 2AZ, UK
cCSIRO Manufacturing Flagship, VIC 3169, Australia
First published on 4th September 2014
Abstract
A series of six phenyl-flanked benzodipyrrolidone-based copolymers are designed and synthesised: three exhibit backbone in-plane curvature or torsional twisting, while the other three remain planar and linear. While the former may appear less crystalline by X-ray diffraction, they afford a smoother thin-film surface topology and increased electron mobility in top-gate, bottom-contact organic field-effect transistors.
Introduction
Benzodipyrrolidones are promising building blocks for organic semiconductor materials, in part due to their innate stability and deep colour which lead to their early use as dye pigments.1 For example, similar bis-lactam-based conjugated materials such as diketopyrrolopyrrole,2 isoindigo3 and dihydropyrroloindoledione4,5 often exhibit high performance in organic field-effect transistors (OFETs)6–8 and solar cells.9,10 Such devices are particularly interesting due to their solution processed nature, facilitating printing of large-scale, flexible and light-weight displays.11 However, recent progress in achieving ultra-high hole mobilities12,13 and well balanced ambipolarity14,15 garners renewed interest in electron transporting (n-type) organic semiconductor materials.16 Moreover, as commercially viable performance targets are met,17 a new focus on material stability has emerged, particularly for air and water stable transistor materials.18–20Recently, a number of copolymers based on phenyl-flanked benzodipyrrolidone (BPP)21–25 and its analogues26,27 have been prepared, with charge carrier mobilities as high as 0.1 cm2 V−1 s−1 under optimal processing conditions.28 With appropriate copolymers, improved solid-state morphologies and high ionisation potentials may be realised, which can confer innate stability to electronic devices, with increased and preferential electron mobility. Indeed, such molecular design plays an important role in governing self-assembly in the solid-state, with resultant texture directly affecting OFET performance.29
To this end we report a series of six BPP copolymers for systematic comparison (Fig. 1). The synthesis, morphology, thermal, optical and electronic properties and computational models of the polymers are presented. Device measurements for organic field-effect transistors (OFETs) are reported, with analysis of the film morphology by atomic force microscopy (AFM). We demonstrate that charge carrier mobility correlates to film roughness, itself being related to the conjugated polymer backbone conformation and molecular architecture.
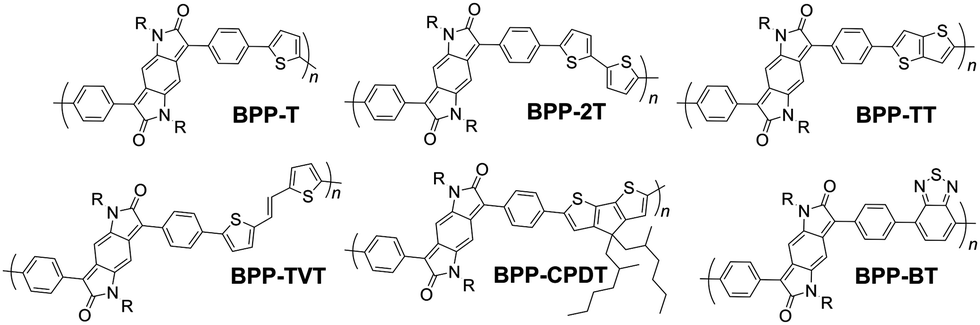 | ||
| Fig. 1 The series of six BPP copolymers; R = N-2-decyl-tetradecane. | ||
Results and discussion
Synthesis
The BPP polymers were prepared in five steps from p-phenylenediamine (Scheme 1). Condensation with 4-bromo-DL-mandelic acid followed by ring closure in concentrated sulphuric acid furnishes the tricyclic core, reduction by sodium hydroxide and potassium persulfate and subsequent alkylation then affording the dibrominated monomer as a dark brown powder solid in 35% overall yield (four steps). Polymerisation with either a bis-boronic ester or bis-stannylated comonomer was then effected by palladium catalysed cross-coupling (Suzuki or Stille, respectively) (Scheme S1†).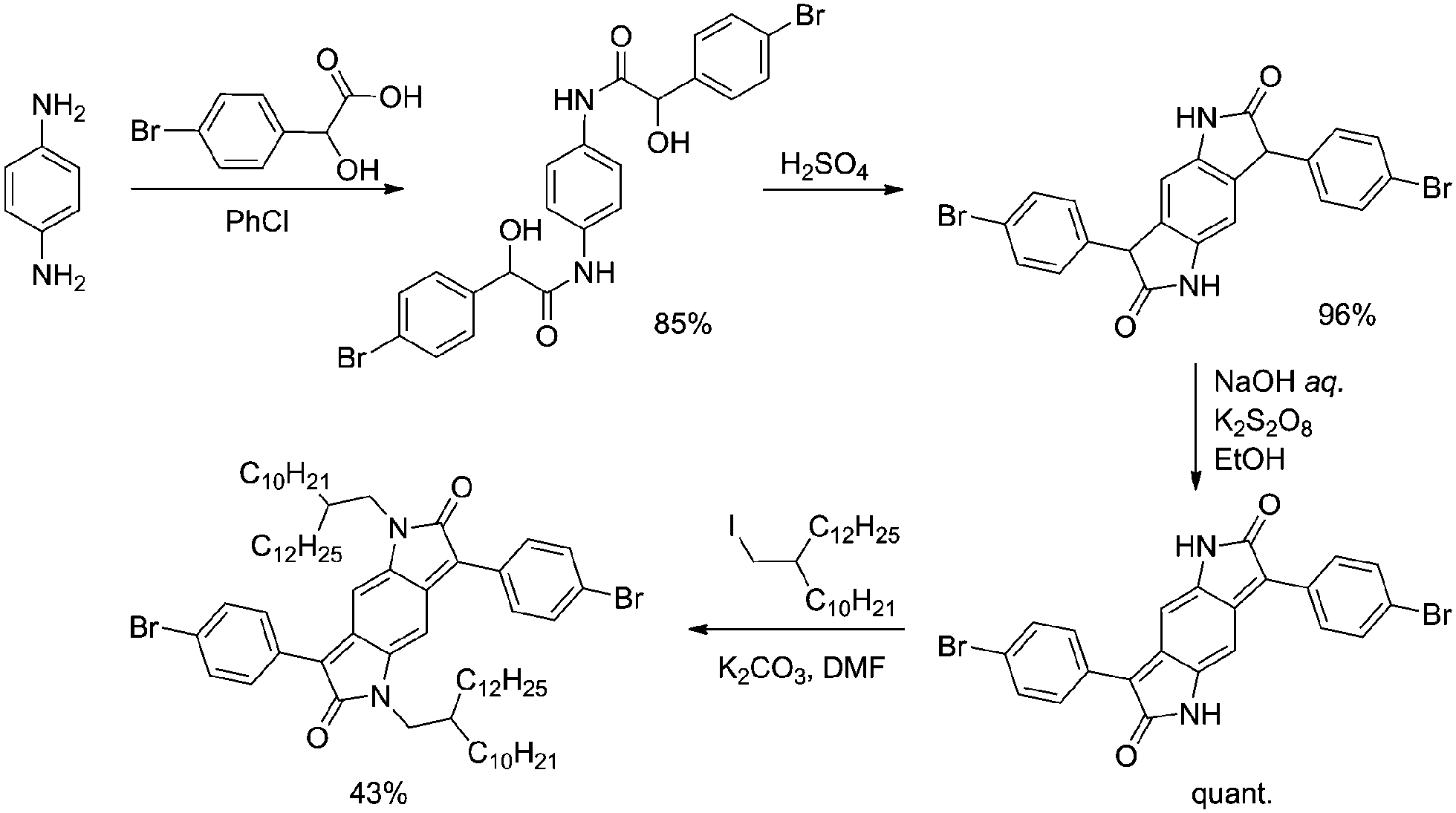 | ||
| Scheme 1 Synthesis of the dibrominated BPP monomer unit for use in Pd cat. cross-coupling polymerisations. | ||
In total, five alternating electron rich-poor, push–pull type BPP-based polymers were prepared (see Fig. 1 for structures), with the following electron-rich repeat units: thiophene (T), 2,2′-bithiophene (2T), thieno[3,2-b]thiophene (TT), (E)-1,2-bisthiophen-2-yl-ethene (TVT) and 4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT). In addition a copolymer was prepared with the electron-poor benzothiadiazole (BT) unit.
All BPP-based polymers bore the same N-alkyl side chain 2-decyltetradecane. The polymers were typically purified by precipitation from methanol followed by Soxhlet extraction using methanol, acetone, hexane, chloroform and, if necessary, chlorobenzene. The latter fraction(s) were then separately heated and vigorously stirred with aqueous sodium diethyldithiocarbamate to remove residual catalytic metal impurities. Lower molecular weight oligomers were readily removed in the acetone and hexane fractions, affording satisfactory molecular weights – as determined by gel permeation chromatography (GPC) – for all six polymers (see Table 1 for data and Fig. S1–S6† for GPC elution traces).
| Polymer | Soxhlet fraction | M n (kDa) | M w (kDa) | PDIa | T 5% wt loss (°C) | T melt (°C) | λ absmax (nm) | E optg (eV) | Energy levelsf (eV) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solution | Thin-film | EHOMO | ELUMO | ||||||||
| a Determined by GPC using polystyrene standards and PhCl as the eluent at 80 °C. b Determined by thermogravimetric analysis (TGA). c As observed by differential scanning calorimetry (DSC). d Solutions (dilute) in PhCl; thin-films drop-cast from 10 mg mL−1 PhCl solutions on glass substrates. e Determined from the absorption onset of the thin-film. f EHOMO found by AC2 (PESA) measurement; ELUMO = EHOMO + Eoptg. | |||||||||||
| BPP-T | CHCl3 | 25 | 25 | 1.64 | 386 | — | 602 | 622 | 1.66 | −5.68 | −4.02 |
| BPP-2T | PhCl | 17 | 42 | 2.47 | 393 | ∼230 | 602 | 624 | 1.58 | −5.63 | −4.05 |
| BPP-TT | CHCl3 | 15 | 51 | 3.41 | 337 | — | 613 | 631 | 1.61 | −5.68 | −4.07 |
| BPP-TVT | CHCl3 | 20 | 46 | 2.28 | 391 | — | 622 | 647 | 1.49 | −5.55 | −4.06 |
| BPP-CPDT | CHCl3 | 21 | 41 | 1.96 | 394 | — | 661 | 687 | 1.49 | −5.44 | −3.95 |
| BPP-BT | CHCl3 | 54 | 66 | 1.25 | 376 | — | 543 | 554 | 1.85 | −5.84 | −3.99 |
Thermal properties & morphology
Thermal properties and thin-film morphology were probed by thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and X-ray diffraction (XRD). The decomposition temperatures (5% weight loss) were all in the range 335–395 °C (Fig. S7†). However, no obvious transitions were observed by DSC (in the range −30 to 350 °C) (Fig. S8†), with the exception of BPP-2T, which exhibited a melt at ∼230 °C and corresponding crystallisation at ∼200 °C (Fig. S9†).From X-ray diffraction experiments (Fig. 2), the polymers were all determined to be crystalline as-cast, with an out-of-plane reflection peak at 4–5°, corresponding to the (100) peak diffraction. For BPP-2T and BPP-TT, where the (100) diffraction peak is more intense, the second order reflection peaks (200) were also visible at 9–10°. The low angle diffraction peaks are characteristic of lamellar-type crystallinity in π stacked conjugated polymers. In addition to the (100) and (200) diffraction peaks, the (010) peak associable with co-facial π-stacking between polymeric backbones is distinctly observable for BPP-TT at ∼20°.
 | ||
| Fig. 2 X-ray diffraction (XRD) diagrams of the BPP-based copolymers as thin-films drop-cast on Si substrates. | ||
Table 2 showed the interlayer d-spacing distance obtained from XRD experiments. Calculated lamellar stacking distances were ∼19 Å for all polymers, expect for BPP-TT (21 Å) and BPP-T (21 Å).
| Substrate | Polymer | Lamellar packing | |||||
|---|---|---|---|---|---|---|---|
| 2θ/° | Intensity | d-spacing/Å | 2θ/° | Intensity | d-spacing/Å | ||
| n = 1 | n = 2 | ||||||
| a Out-of-plane reflections observed by X-ray diffraction for drop-cast films (from 10 mg mL−1 PhCl solution) on Si substrates dried in air. | |||||||
| As cast | BPP-T | 4.11 | 2.1 × 103 | 21.5 | — | — | — |
| BPP-2T | 4.72 | 9.6 × 103 | 18.7 | 9.33 | 1.2 × 103 | 18.9 | |
| BPP-TT | 4.98 | 9.7 × 103 | 17.7 | 9.80 | 2.0 × 103 | 18.0 | |
| BPP-TVT | 4.72 | 2.3 × 103 | 18.7 | — | — | — | |
| BPP-CPDT | 4.68 | 2.0 × 103 | 18.9 | — | — | — | |
| BPP-BT | 4.75 | 1.3 × 103 | 18.6 | — | — | — | |
The shorter d-spacing distance of BPP-TT is consistent with the increased intensity of the π-stacking peak, indicating a high degree of crystallinity. In comparison to BPP-2T, which also exhibits a high intensity lamellar packing peak, the increased π-stacking in BPP-TT may be attributed to the fused nature of the TT comonomer unit improving coplanarity along the polymer backbone. Moreover, the phenyl-TT-phenyl linkage is strictly linear in comparison to the other five comonomers which all exhibit some degree of torsional or in-plane deviation. The larger lamellar d-spacing for BPP-T could be due to the decreased donor–acceptor spacing aspect ratio along the conjugated backbone hindering packing. However, the similar lamellar spacing for the other polymers indicates that the alkyl side-chains on the benzodipyrrolidone unit are the dominant steric factor in dictating lamellar packing interactions (d-spacing distance), as opposed to contributions from the comonomer.
Optical & electronic properties
The optical and electronic properties of the BPP-based copolymers were investigated by UV-Vis absorption spectroscopy (Fig. 3) and photo-electron spectroscopy in air (PESA) and are summarised in Table 1. The thin-film absorption maxima are all in the range 620–650 nm with the exception of BPP-BT which is blue-shifted to ∼550 nm and BPP-CPDT which is red shifted to ∼690 nm. This trend can be explained be the dual-band theory for semiconducting polymers.30 For the BPP-BT polymer both monomeric units are considered electron-deficient. For that reason, there is not a strong donor–acceptor character and the intermolecular charge transfer band (ICT) associated to the BPP-BT polymer is blue shifted compared to our series. Therefore, as we introduce stronger donor moieties (T, 2T, TT and TVT) the ICT band is red-shifted up to 650 nm. The far-reaching case is for BPP-CPDT, where the λmax is further red-shifted to 690 nm. In this case, CPDT is a stronger donor due to the planarization of the bithiophene unit.31 Consequentially, while the BPP-T, -2T and -TT polymers exhibited an effective band gap of ∼1.6 eV as determined from the thin-film absorption onset, this was increased to ∼1.9 eV for BPP-BT and decreased to ∼1.5 eV for BPP-TVT and BPP-CPDT polymers.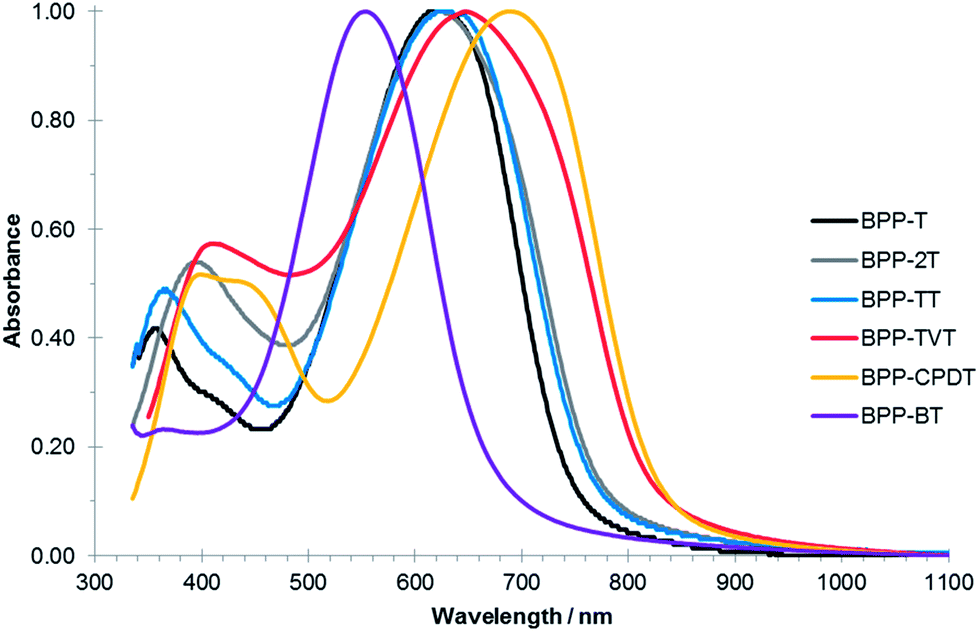 | ||
| Fig. 3 Normalised UV-Vis absorption spectra of the BPP-based copolymers in solid-state thin-films (spin-cast from 10 mg mL−1 chlorobenzene solutions on glass substrates). | ||
In addition, the thin-film absorption maxima are typically red-shifted by some 20–25 nm in comparison to the solutions (dilute in chloroform) (Fig. S10†), indicating that solid-state interactions likely improve ordering, suppressing torsional twisting and dihedral angles, which results in increased delocalisation and subsequent red-shifting. Such orbital overlap is necessary for charge transport between conjugated backbones, with stronger interactions likely increasing mobilities in field-effect transistor devices. However, this effect is limited for BPP-BT which exhibits only an 11 nm red-shift from solution to solid, possibly due to the torsional twisting due to the phenyl–phenyl link inhibiting π-stacking. Notably the polymers are free from aggregation shoulders in both their solution and thin-film absorption profiles, implying good solubility and solution processability, and that the observed properties are inherent to the molecular architecture and interactions.
The ionisation potentials (HOMO levels) were obtained by PESA and shown in Table 1. The BPP-BT polymer exhibits the deepest HOMO energy level (−5.84 eV) due to the inductive effect from both electron-poor BT and BPP units. The addition of electron-donating units (T, 2T and TT) raised the HOMO energy level by 0.2 eV to approximately −5.65 eV. Finally, stronger electron-donating TVT and CPDT units further raised the HOMO to −5.55 and −5.44 eV, respectively. The corresponding LUMO energy levels obtained by the addition of the optical band gaps to the HOMO values were in the (narrow) range of −3.95 to −4.07 eV, being suitably deep to permit electron injection in transistor devices. Furthermore, the deep HOMO energy levels (below −5.30 eV) could indicate good stability towards unintentional doping by atmospheric species such as oxygen and water.
Computational models
The optimized structures of the BPP-based copolymers and visualisations of the HOMO and LUMO molecular orbital distributions are shown in Fig. S11,† as calculated by density functional theory (DFT) using Gaussian 09 software at the B3LYP/6-31G* level of theory with N-methyl substitution, due to the weak influence of the alkyl side-chains on the electronic structure of the conjugated core. Each polymer is modelled as a tetramer, except BPP-2T and BPP-TVT, which are modelled as trimers due to the larger size of the systems.The dihedral angle between the carbon atoms of the lactam rings on the central benzodipyrrolidone (BP) unit and flanking phenyl rings is found to be ∼30° in all cases (Fig. 4 and Table 3; phenyl-BP C–C–C–C). Meanwhile the dihedral angle between the carbon atoms of the phenyl rings and the comonomer (phenyl-comonomer) is ∼20° in all cases except BPP-BT, where the torsional twist is much greater (34°), due to the increased steric repulsion in phenyl-phenyl couples compared to phenyl–thiophene.
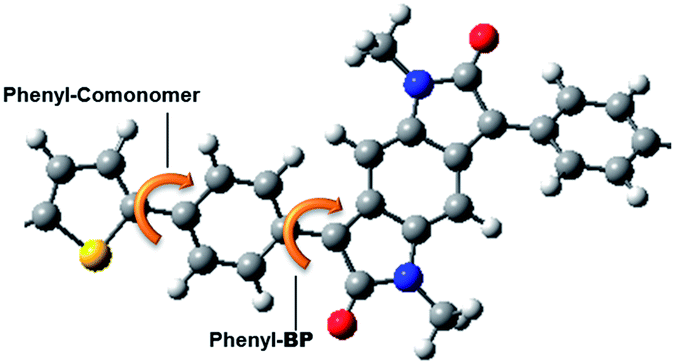 | ||
| Fig. 4 The phenyl-comonomer and phenyl-BP C–C–C–C dihedral angles are indicated for a repeat unit of BPP-T. | ||
The HOMO molecular orbital distribution is largely delocalised over the conjugated π-system for all the BPP-based polymers while the LUMO is mainly located on the BPP electron deficient unit (Fig. 5).
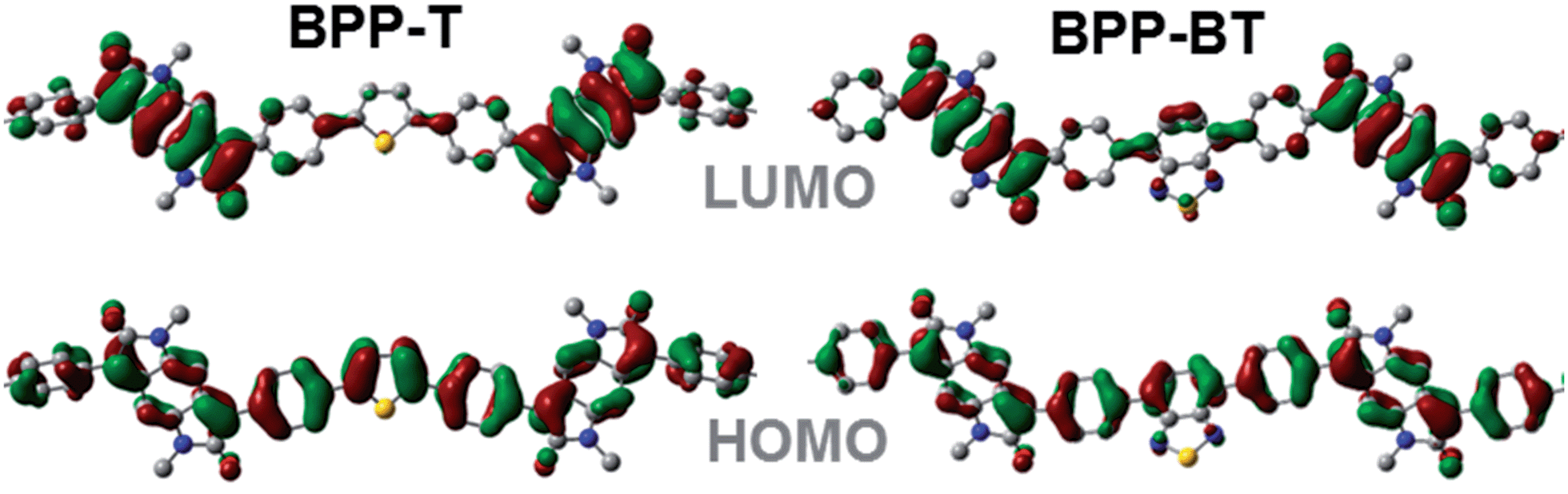 | ||
| Fig. 5 Segments of the computational models of the BPP-based copolymers BPP-T and BPP-BT, showing the energy minimised structures with N-methyl substitution and visualisation of the HOMO and LUMO energy distributions (hydrogen atoms are omitted from view for clarity). | ||
Notably the BPP and different comonomer units have the possibility for a range of conformations, orienting in various different directions, as exemplified for BPP-T in Fig. 6. BPP-T is able to theoretically adopt such a range of conformations as the thiophene comonomer does not afford a co-linear connection to its neighbouring BPP units. Instead, the thiophene introduces an in-plane kink angle to the conjugation pathway of the polymer backbone. The same is true of the CPDT comonomer. Allowing for perpendicular bonds to the neighbouring units, the six comonomers can be crudely modelled as trapeziums (T and CPDT) or rectangles (TVT, 2T, BP, TT), the latter of four units being co-linear (Fig. 7). The number of different theoretically possible conformations increases exponentially with the number of repeat units considered. However, even with just four repeat units, a simple but meaningful model can be considered. In all cases an overall linear conformation of the conjugated polymer backbone is possible. However, when the comonomer is non-co-linear and introduces a kink (T and CPDT) a curved shape can also evolve along the conjugation pathway in the plane of the backbone, which may hamper solid-state ordering.
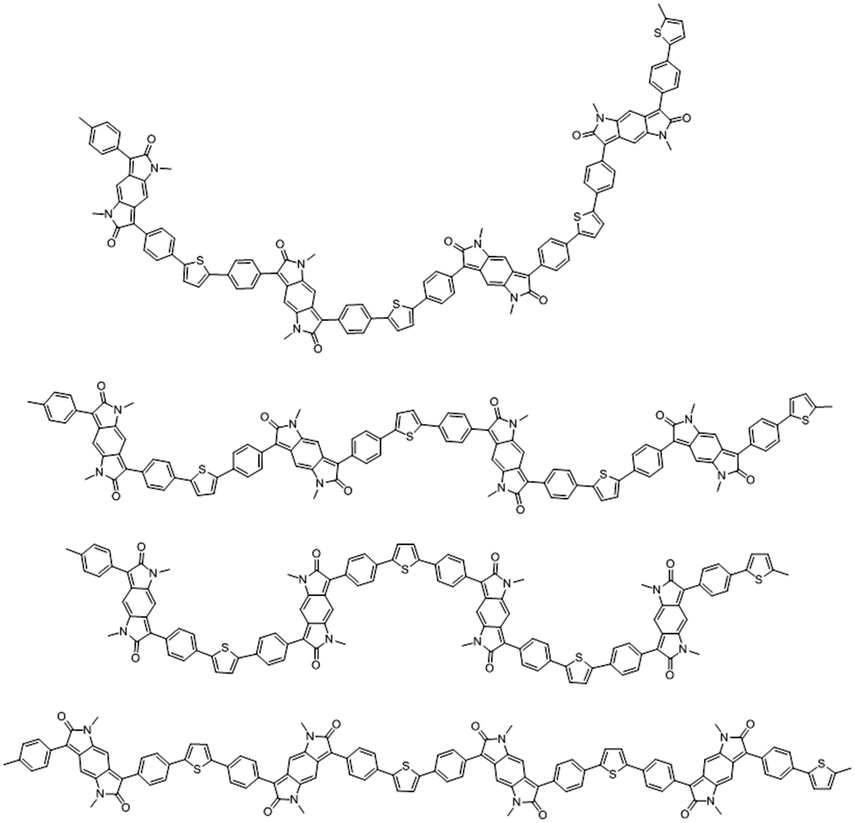 | ||
| Fig. 6 Some possible chain conformations for the BPP-T polymer shown over four repeat units to illustrate the various polymer chain shapes that can arise. | ||
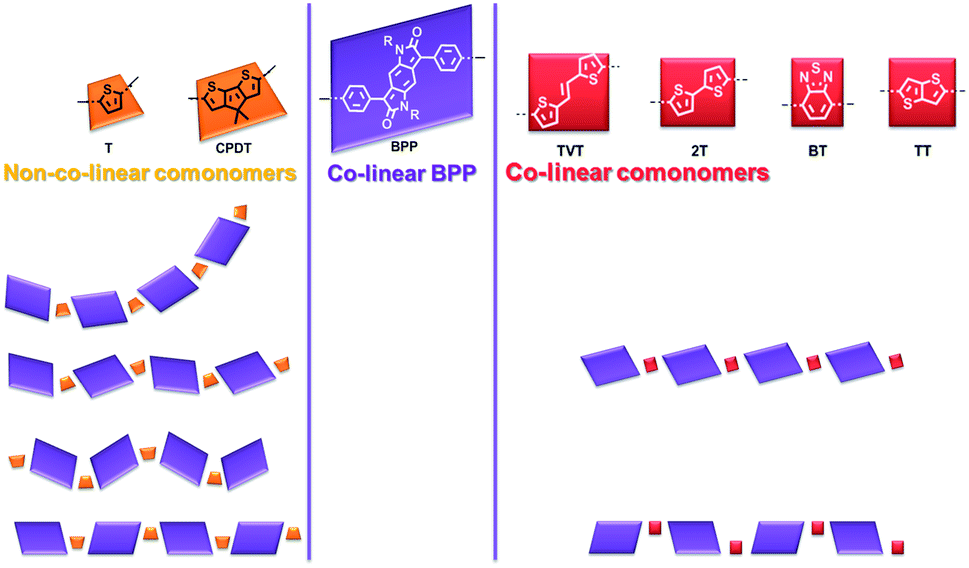 | ||
| Fig. 7 The BPP comonomers can be modelled as either trapeziums which introduce an in-plane kink angle to the conjugation pathway of the polymer backbone (non-co-linear), or rectangles which afford a co-linear join. The possible in-plane conformations of the BPP-based polymers over four repeat units are shown for the non-co-linear and co-linear comonomers. | ||
In all computational models the polymers have been modelled in the likely lowest energy state, which would allow the alkyl side-chains to point out into free space, reducing steric clashes. For BPP-T and BPP-CPDT this is an overall linear conformation with the benzodipyrrolidone units alternately ‘pointing’ in different directions, while for the other four polymers this is an overall linear conformation where the benzodipyrrolidone unit consistently orients in the same direction.
While in solution the polymer may be able, with sufficient energy, to transition between different shapes, the conformation will be kinetically frozen on casting into the solid-state. With appropriate casting conditions, such as high boiling point solvents or additives, the polymer may be able to settle into a thermodynamically preferred low energy orientation that affords ordered solid-state packing, as indicated by X-ray diffraction.
Organic field-effect transistor (OFET) device performance
Organic field-effect transistors (OFETs) were prepared with the BPP-based polymers. A top-gate, bottom-contact device architecture (see Fig. S13†) was used, fabricated by spin-coating polymer solution (5 mg mL−1 in hot [75 °C] ortho-dichlorobenzene) and drying under a nitrogen atmosphere at 100 °C. In preliminary testing annealing at 200 °C lead to worse performance (as seen for similar BPP-based polymers),22 as did bottom-gate, bottom-contact devices, exhibiting large hysteresis and poor performance. The top-gate devices consisted of Cytop dielectric and Al/Au electrodes on glass substrates. Notably these devices only exhibited electron transport for all six BPP-based polymers, with moderate threshold voltages (30–65 V) and satisfactory Ion/off ratios all >103 (Table 4). The output and transfer curves are shown in Fig. 8, S14 and S15.†| Polymer | Vth (V) | μe (cm2 V−1 s−1) | I on/Ioff | RMS roughness (nm) |
|---|---|---|---|---|
| a Top-gate, bottom-contact architecture devices annealed at 100 °C. Mobility values are accurate to one significant figure. | ||||
| BPP-T | 60 ± 5 | 0.007 | 4 ± 1 × 103 | 0.54 |
| BPP-2T | 49 ± 3 | 0.001 | 2 ± 1 × 103 | 1.37 |
| BPP-TT | 34 ± 1 | 0.001 | 1 ± 0.5 × 103 | 1.63 |
| BPP-TVT | 59 ± 7 | 0.002 | 1 ± 0.6 × 103 | 1.82 |
| BPP-CPDT | 47 ± 1 | 0.01 | 4 ± 2 × 103 | 0.40 |
| BPP-BT | 43 ± 1 | 0.01 | 8 ± 3 × 103 | 0.68 |
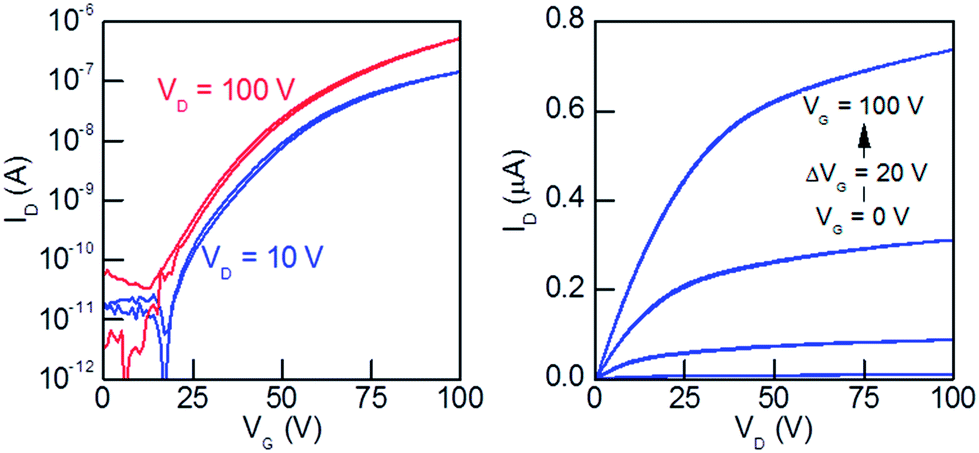 | ||
| Fig. 8 The BPP-BT copolymer OFET device characteristics: (left) the output curves and (right) the transfer curves; (top-gate, bottom-contact architecture; annealed at 100 °C). Of the three higher electron mobility polymers BPP-BT also exhibited the highest Ion/off ratio and lowest threshold voltage. | ||
The BPP-based polymer mobilities are of the magnitude ×10−2 cm2 V−1 s−1 (BPP-2T, -TT and -TVT) or ×10−3 cm2 V−1 s−1 (BPP-T, -CPDT and -BT), independent of threshold voltage or Ion/off ratio. To further investigate this trend the OFET device morphology was probed by atomic force microscopy (AFM) imaging of polymer films prepared in an identical fashion (Fig. S16†).32 There is a clear correlation to root mean square (RMS) roughness of the films, with the lower mobility polymers exhibiting a roughness at least double that of the higher mobility polymers; for example BPP-CPDT has the lowest RMS roughness at 0.40 nm with a corresponding highest mobility of 0.01 cm2 V−1 s−1 whilst BPP-TVT exhibits the highest RMS roughness at 1.82 nm, accompanied by a lower mobility of 0.002 cm2 V−1 s−1 (Fig. 9). However in all cases the roughness is well below what is considered acceptable for OFET devices (up to 25 nm),8,18 indicating that the trend is attributable to the polymer structure, solid-state interactions and morphology, as opposed to poor processing or fabrication technique.
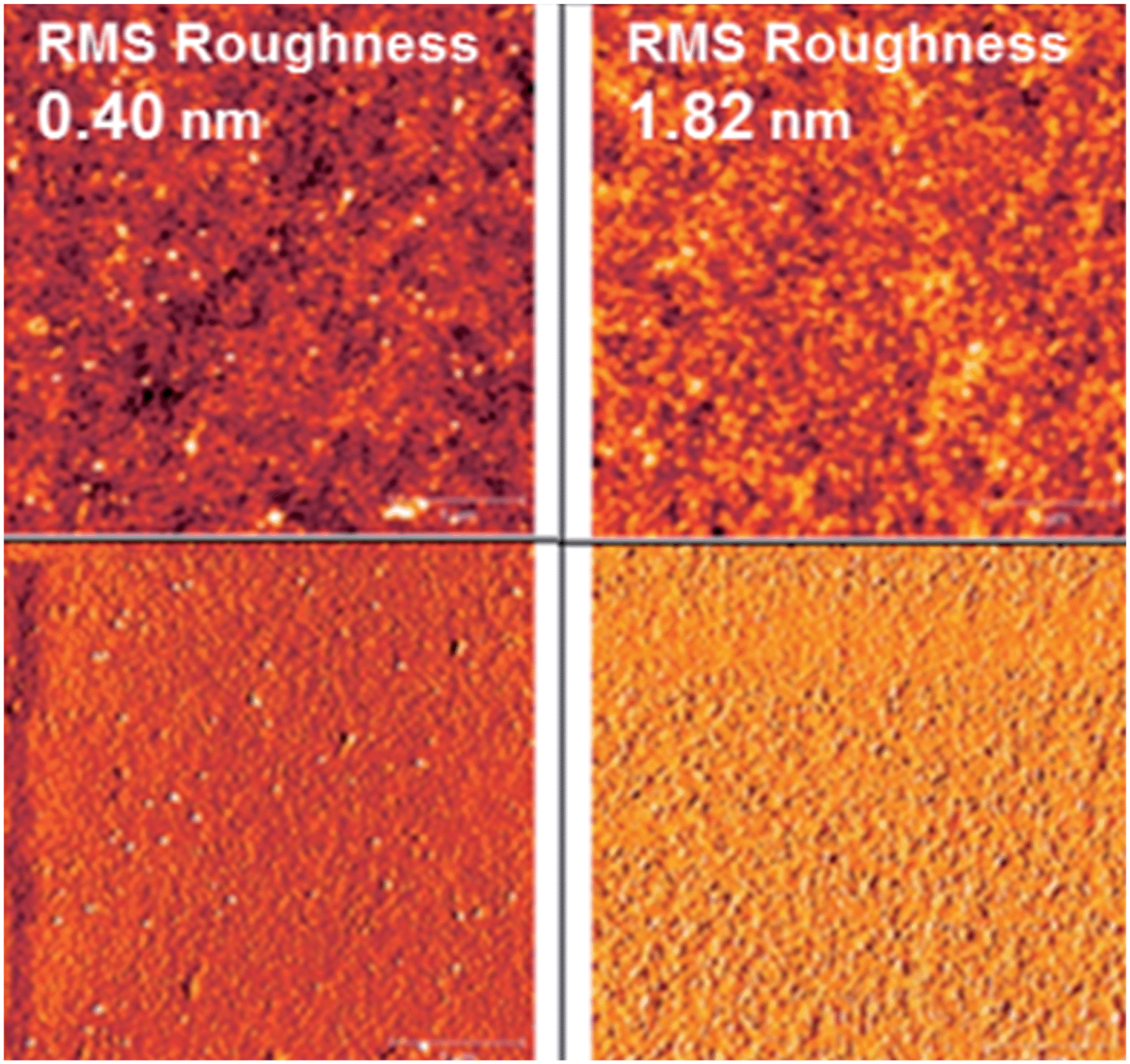 | ||
| Fig. 9 Atomic force microscopy (AFM) images of (left) the BPP-CPDT and (right) BPP-TVT thin-films to show the highest contrast in film roughness; (top) topography and (bottom) phase images, both 4 × 4 μm. | ||
Interestingly the mobility and roughness is apparently related to the crystallinity and potential ability of the polymer to adopt different conformations, energetically accessible in hot polymer solutions, which may then be kinetically frozen on casting the solid-state film. Both BPP-T and BPP-CPDT allow for curved or kinked polymer chain shapes (Fig. 7) whilst BPP-BT has a greater degree of torsional twisting along the backbone (Fig. 4 and Table 3). In addition, BPP-2T exhibits the highest degree of crystallinity with an observable melt by DSC and the most intense low angle XRD peak.
This indicates that while crystallinity in conjugated polymer systems can be beneficial for charge transport,33 π-stacking is much more important than lamellar packing, as expected from the nature of charge hopping between conjugated backbones via delocalised orbitals and their overlap.34 Polymers that could theoretically adopt a greater range of conjugated backbone shapes, achieved through either out-of-plane twisting or non-co-linearity with the conjugation pathway in this case, are able to achieve smoother topologies than planar and co-linear highly crystalline polymers. While high crystallinity may result in grain boundary defects, the ability of the polymer to coexist in amorphous regions, results in a reduced surface roughness, likely due to denser packing, giving improved charge carrier mobilities.35
Conclusions
In summary a series of six phenyl-flanked benzodipyrrolidone (BPP)-based copolymers were designed and synthesised, all bearing N-2-decyl-tetradecyl alkyl side-chains for high solubility in common organic solvents and solution processing. The polymers show good thermal stability, as-cast crystallinity, medium optical band gaps and deep HOMO and LUMO energy levels. Conjugated backbone planarity depends on the comonomer (BPP-BT exhibiting greater out-of-plane twisting), with all polymers exhibiting only electron conduction in top-gate, bottom-contact organic field-effect transistor (OFET) devices.Charge carrier mobility was observed to be dependent on polymer film surface roughness. Polymers that could theoretically exhibit a greater range of backbone shapes (e.g. curved or twisted: BPP-T, -CPDT and -BT), afford smoother films (RMS roughness' <1 nm) and exhibit mobilities an order of magnitude greater than their strictly linear counterparts (BPP-2T, -TT and -TVT; RMS roughness >1 nm).
Acknowledgements
This work was carried out with funding from: EC FP7 Project X10D; the Centre for Plastic Electronics at Imperial College London, Doctoral Training Centre EP/G037515/1; EPSRC EP/I002936 and NSF CHE 1026664. The authors declare no competing financial interest.Notes and references
- C. W. Greenhalgh, J. L. Carey and D. F. Newton, Dyes Pigm., 1980, 1, 103 CrossRef CAS.
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2013, 26, 664 CrossRef.
- J. W. Rumer, S.-Y. Dai, M. Levick, Y. Kim, M.-B. Madec, R. S. Ashraf, Z. Huang, S. Rossbauer, B. Schroeder, L. Biniek, S. E. Watkins, T. D. Anthopoulos, R. A. J. Janssen, J. R. Durrant, D. J. Procter and I. McCulloch, J. Mater. Chem. C, 2013, 1, 2711 RSC.
- J. W. Rumer, S.-Y. Dai, M. Levick, L. Biniek, D. J. Procter and I. McCulloch, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1285 CrossRef CAS.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647 CrossRef CAS.
- M. J. Robb, S.-Y. Ku, F. G. Brunetti and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1263 CrossRef CAS.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636 CrossRef CAS PubMed.
- J. W. Rumer, B. C. Schroeder, C. B. Nielsen, R. S. Ashraf, D. Beatrup, H. Bronstein, S. J. Cryer, J. E. Donaghey, S. Holliday, M. Hurhangee, D. I. James, S. Lim, I. Meager, W. Zhang and I. McCulloch, Thin Solid Films, 2013, 560, 82 CrossRef PubMed.
- L. Dou, J. Gao, E. Richard, J. You, C.-C. Chen, K. C. Cha, Y. He, G. Li and Y. Yang, J. Am. Chem. Soc., 2012, 134, 10071 CrossRef CAS PubMed.
- H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158 CrossRef CAS PubMed.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. B. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Nat. Commun., 2014, 5, 3005 Search PubMed.
- C. Luo, A. K. K. Kyaw, L. A. Perez, S. Patel, M. Wang, B. Grimm, G. C. Bazan, E. J. Kramer and A. J. Heeger, Nano Lett., 2014, 14, 2764 CrossRef CAS PubMed.
- A. R. Mohebbi, J. Yuen, J. Fan, C. Munoz, M. F. Wang, R. S. Shirazi, J. Seifter and F. Wudl, Adv. Mater., 2011, 23, 4644 CrossRef CAS PubMed.
- J. W. Rumer, M. Levick, S.-Y. Dai, S. Rossbauer, Z. Huang, L. Biniek, T. D. Anthopoulos, J. R. Durrant, D. J. Procter and I. McCulloch, Chem. Commun., 2013, 49, 4465 RSC.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319 CrossRef CAS PubMed.
- O. Knopfmacher, M. L. Hammock, A. L. Appleton, G. Schwartz, J. Mei, T. Lei, J. Pei and Z. Bao, Nat. Commun., 2014, 5, 2954 Search PubMed.
- M. Yun, A. Sharma, C. Fuentes-Hernandez, D. K. Hwang, A. Dindar, S. Singh, S. Choi and B. Kippelen, ACS Appl. Mater. Interfaces, 2014, 6, 1616 CAS.
- Y. Ie, M. Ueta, M. Nitani, N. Tohnai, M. Miyata, H. Tada and Y. Aso, Chem. Mater., 2012, 24, 3285 CrossRef CAS.
- W. Cui, J. Yuen and F. Wudl, Macromolecules, 2011, 44, 7869 CrossRef CAS.
- K. C. Lee, W.-T. Park, Y.-Y. Noh and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 4872 CAS.
- W. Yue, X. Huang, J. Yuan, W. Ma, F. C. Krebs and D. Yu, J. Mater. Chem. A, 2013, 1, 10116 CAS.
- W. Hong, C. Guo, Y. Li, Y. Zheng, C. Huang, S. Lu and A. Facchetti, J. Mater. Chem., 2012, 22, 22282 RSC.
- P. Deng, L. Liu, S. Ren, H. Li and Q. Zhang, Chem. Commun., 2012, 48, 6960 RSC.
- W. Hong, B. Sun, C. Guo, J. Yuen, Y. Li, S. Lu, C. Huang and A. Facchetti, Chem. Commun., 2013, 49, 484 RSC.
- W. Hong, C. Guo, B. Sun, Z. Yan, C. Huang, Y. Hu, Y. Zheng, A. Facchetti and Y. Li, J. Mater. Chem. C, 2013, 1, 5624 RSC.
- G. W. P. van Pruissen, E. A. Pidko, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. C, 2014, 2, 731 RSC.
- J. W. Ward, R. Li, A. Obaid, M. M. Payne, D.-M. Smilgies, J. E. Anthony, A. Amassian and O. D. Jurchescu, Adv. Funct. Mater., 2014, 24, 5052 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396 CrossRef CAS PubMed.
- S. Barlow, S. A. Odom, K. Lancaster, Y. A. Getmanenko, R. Mason, V. Coropceanu, J.-L. Brédas and S. R. Marder, J. Phys. Chem. B, 2010, 114, 14397 CrossRef CAS PubMed.
- Y. Wen, Y. Liu, Y. Guo, G. Yu and W. Hu, Chem. Rev., 2011, 111, 3358 CrossRef CAS PubMed.
- A. Bagui and S. Sundar Kumar Iyer, Org. Electron., 2014, 15, 1387 CrossRef CAS PubMed.
- C. Deibel and V. Dyakonov, Rep. Prog. Phys., 2010, 73, 096401 CrossRef.
- T. Qin, W. Zajaczkowski, W. Pisula, M. Baumgarten, M. Chen, M. Gao, G. Wilson, C. D. Easton, K. Müllen and S. E. Watkins, J. Am. Chem. Soc., 2014, 136, 6049 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc01790k |
| This journal is © The Royal Society of Chemistry 2014 |