 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecord-high hyperpolarizabilities in conjugated polymers†
Stijn
Van Cleuvenbergen
a,
Inge
Asselberghs
ad,
Wouter
Vanormelingen
b,
Thierry
Verbiest
a,
Edith
Franz
a,
Koen
Clays
ac,
Mark G.
Kuzyk
c and
Guy
Koeckelberghs
*b
aLaboratory of Molecular Electronics and Photonics, Katholieke Universiteit Leuven, Celestijnenlaan 200 D, B-3001 Leuven, Belgium
bLaboratory of Polymer Synthesis, Katholieke Universiteit Leuven, Celestijnenlaan 200 F, B-3001 Leuven, Belgium. E-mail: guy.koeckelberghs@chem.kuleuven.be
cDepartment of Physics and Astronomy, Washington State University, Pullman, USA
dIMEC, Kapeldreef 15, 3001 Leuven, Belgium
First published on 24th April 2014
Abstract
Disubstituted poly(phenanthrene), a conjugated polymer, has been studied by hyper-Rayleigh scattering. Although this compound lacks the donor–acceptor motif that is typically associated with a strong second-order nonlinear optical response, the (intrinsic) hyperpolarizability ranks among the highest ever measured, breaking the longstanding apparent limit. The linear and nonlinear optical properties of the polymer depend strongly on the solvent conditions, affecting the macromolecular organization. An explanation for these unexpected results is postulated and is based on modulation of conjugation along the polymer backbone. As the molecular structure of the compound does not at all fit into the classical paradigms, our observations put these theories into perspective.
1. Introduction
For over two decades, efforts have focused on the design of molecules to improve their nonlinear optical (NLO) behavior for various applications.1–3 For second-order NLO applications, these molecules typically have a D–π–A motif, i.e. a conjugated π-system endcapped with electron donating (D) and electron accepting (A) groups. In order to maximize the hyperpolarizability (β), many different combinations of donors, acceptors and conjugated π-systems have been studied. Although increasingly stronger donors and acceptors do not necessarily lead to a higher hyperpolarizability, an optimized D–π–A motif is required for achieving optimal hyperpolarizabilities.4A very common technique to measure a molecule's hyperpolarizability is hyper-Rayleigh scattering (HRS). The hyperpolarizability is in fact a tensor composed of 27 components. The actually measured hyperpolarizability using HRS (βHRS) is usually composed of several tensor components. In general, the HRS response of a molecule depends on both its symmetry and its electronic transitions. The former parameter governs the number of independent non-zero tensor components βijk, while the latter property determines their actual value. As a consequence, if one wants to compare the general second-order NLO efficiency of different molecules, βHRS suffices.
If, however, a particular tensor component must be evaluated, the symmetry of the molecule must be known in order to calculate the magnitude of that particular component.
One must note that increasing the strength of donors and acceptors and the length of the conjugation path to optimize β, also affects the absorption features of a molecule. More in particular, their increase red-shifts the absorption band. Therefore, the more efficient molecules typically also show a higher λmax. For example, up until recently, the largest hyperpolarizabilities were measured for protected polyene or thiophene-ring containing chromophores like the benchmark compound FTC (Fig. 1, βHRS,0 = 263 × 10−30 esu).5,6
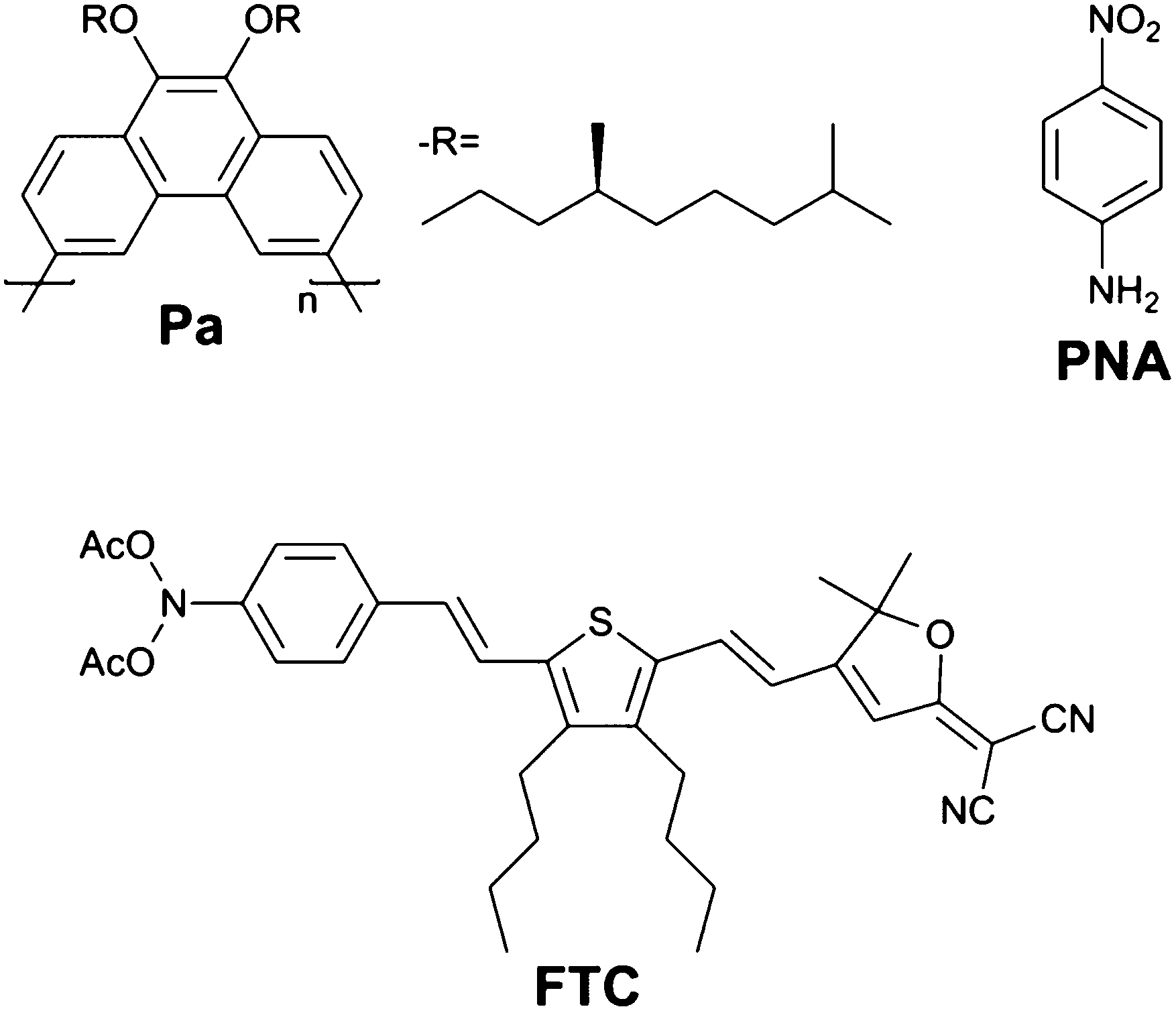 | ||
| Fig. 1 Structures of the studied conjugated polymer Pa, para-nitroaniline (PNA) and benchmark compound FTC.5,6 | ||
While these values are much higher than classical compounds such as para-nitroaniline (PNA, βHRS,0 = 10 × 10−30 esu),5,7 the λmax is also considerably redshifted: from 348 nm for PNA to 650 nm for FTC. This limits the window of wavelengths that can be used for applications.
In this manuscript, we report that Pa (disubstituted poly(phenanthrene), Fig. 1), a conjugated polymer which completely lacks the typical D–π–A structure and has a rather blue-shifted absorption, shows nevertheless a record-high hyperpolarizability. An explanation is postulated to account for this unexpected result.
2. Experimental
2.1 Materials
The polymer studied (Pa) is a poly(phenanthrene) with![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 7.3 kg mol−1, polydispersity = 2.5, degree of polymerization = 15. The molar mass has been measured by gel permeation chromatography (GPC) in THF towards poly(styrene) standards. Its synthesis has been described elewhere.8
n = 7.3 kg mol−1, polydispersity = 2.5, degree of polymerization = 15. The molar mass has been measured by gel permeation chromatography (GPC) in THF towards poly(styrene) standards. Its synthesis has been described elewhere.8
2.2 Methods
UV-vis and CD spectra have been recorded on a Perkin-Elmer Lambda-900 and a JASCO J810 spectrophotometer, respectively. HRS measurements have been performed at a wavelength of 800 nm. The set-up is described in full detail elsewhere.9 All samples are analyzed towards crystal violet in methanol (βHRS,800 nm = 208.6 × 10−30 esu), a standard reference compound in this wavelength range. The differences in solvent and molecular symmetry between the reference and samples are accounted for by the standard local-field correction factors at optical frequencies and the appropriate factors for the contributing tensor components, respectively. To account for resonance enhancement, a static dispersion-free hyperpolarizability value (β0) is obtained using a standard two-level dispersion term.10 Due to the presence of multi-photon fluorescence, the high-frequency demodulation technique has been applied to obtain fluorescence-free first hyperpolarizabilities.9,11 For all solvent conditions, the HRS signal without any fluorescence contribution could be detected at a modulation frequency of 880 MHz. Depolarization measurements have been carried out at this fluorescence-free modulation frequency.2.3 Theoretical calculations
To evaluate the efficiency of the second-order nonlinear optical response the polymer's intrinsic (size-independent) hyperpolarizability, βINT, is calculated as:with βzzz,0,MAX:
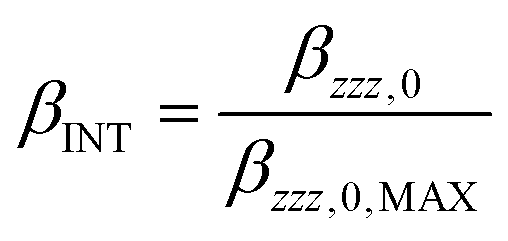
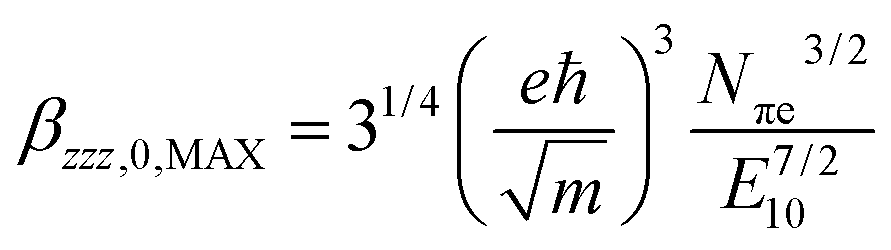
N πe is the effective number of polarizable electrons, m the electron mass and E10 the energy difference between the ground and first excited state, proportional to the reciprocal of λmax.12 For Pa the number of polarizable electrons is 210, or 14 electrons per monomeric unit.
3. Results and discussion
Pa adopts an unordered, coiled conformation in a good solvent such as CHCl3, in which chirality is not expressed, and folds into a helical conformation upon decreasing the solvent quality by addition of a nonsolvent, such as hexane or methanol.8 The transition to the helical conformation is accompanied by the occurrence of circular dichroism (CD), as depicted in Fig. 2. The conjugation length, however, is less affected by this transition and remains rather short, as is evidenced by its λmax, which is a measure of conjugation length (∼360 nm vs. 321 nm for the monomer).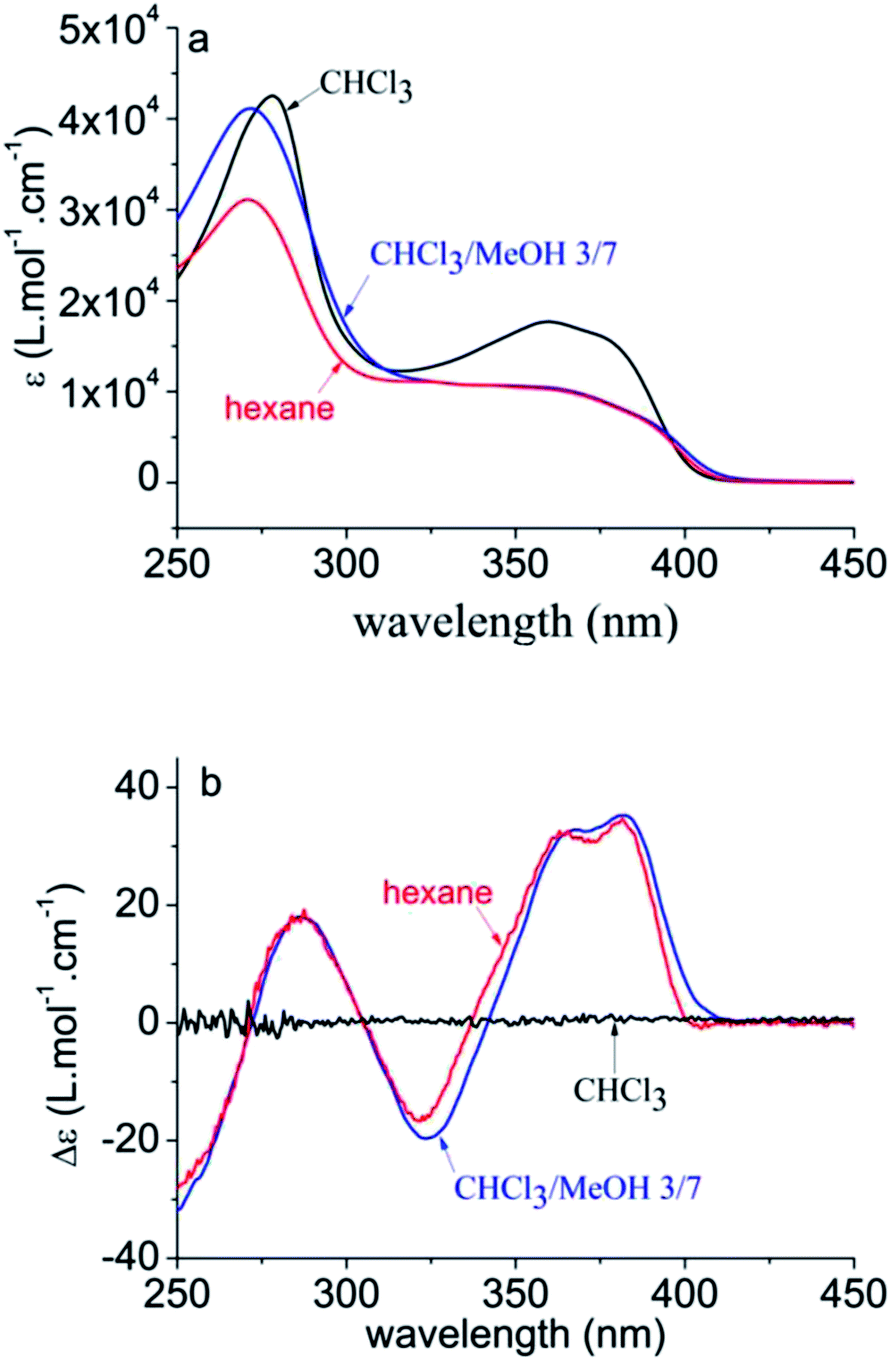 | ||
| Fig. 2 UV-vis (a) and CD (b) spectra of Pa in different solvent conditions, evidencing the transition from coiled to helical conformation for poor solvents. The broad HOMO–LUMO (π–π*) transition around 360 nm shows vibronic fine structure, as does the monomer (see ESI†). The used concentrations are 22 mg L−1 in CHCl3 and CHCl3–MeOH 3/7, and 27 mg L−1 in hexane. | ||
Pa shows a very large HRS response, which strongly depends on the solvent composition (Table 1). The measurements are performed at 800 nm with λmax similarly remote from the second-harmonic wavelength (i.e. 400 nm) in all conditions. The results are thus similarly affected by resonance enhancement or re-absorption. For both the samples and the reference, a series of 5 dilute concentrations has been measured. The linear increase of the second-harmonic signal with the sample concentration shows that the results are not influenced by aggregation of the polymer. βHRS,0 in CHCl3, in which the polymer adopts a disordered coil-state, amounts to 2300 × 10−30 esu, while in hexane or CHCl3–CH3OH mixture, in which a helix is formed, βHRS,0 amounts to 730 × 10−30 esu. The monomer itself, measured in chloroform, has a static hyperpolarizability of 9 × 10−30 esu, and therefore can on its own not account for the large NLO response of the polymer. If these values are compared with that of the classical D–π–A chromophore PNA (βHRS,0 = 10 × 10−30 esu), which has a similar λmax, it is clear that Pa is orders of magnitude stronger.7 Even the benchmark chromophore FTC (βHRS,0 = 263 × 10−30 esu), is outperformed by Pa.
| Solvent | λ max (nm) | β HRS,800nm | β HRS,0 | β xyz,0 | β INT | |
|---|---|---|---|---|---|---|
| (×10 −30 esu) | ||||||
| Pa coil (polymer) | CHCl3 | 360 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
2300 | 3000 | ≥0.077 |
| Pa helix (polymer) | n-Hexane or CHCl3–CH3OH (3/7) | 360 | 4800 | 730 | 950 | ≥0.025 |
| Pa (monomer) | CHCl3 | 321 | 30 | 9 | N/A | <7 × 10−4 |
One might argue that the large response stems mainly from the substantial number of π-electrons. Indeed, it is evident that for an optimized system, the magnitude of the response increases with the number of polarizable electrons. Thus, to evaluate or compare the intrinsic efficiency of different compounds, the number of polarizable electrons must be accounted for. In first approximation, this is often done by scaling βHRS,0 towards the number of π-electrons (βHRS,0/Nπe). Even if the number of polarizable electrons is taken into account, Pa compares to the best traditional D–π–A chromophores.13–20 For example, βHRS,0/Nπe(FTC) = 10 × 10−30 esu, while βHRS,0/Nπe(Pa) = 11 × 10−30 esu. Note that in these calculations, Nπe of Pa is calculated based on the Mn obtained by GPC, which typically overestimates the molar mass of conjugated polymers.
Apart from βHRS or βHRS,0/Nπe, the NLO efficiency of molecules can also be compared more rigorously by their intrinsic (size-independent) hyperpolarizability (βINT), as defined by Kuzyk.22 This model uses Thomas-Kuhn sum rules to estimate a compound's maximum molecular first hyperpolarizability, βMAX, and defines βINT as the ratio of the experimentally determined static hyperpolarizability, β0 over the maximum molecular hyperpolarizability.
In 2004 a survey of the best chromophores up until then revealed that, despite the progress made in achieving ever higher hyperpolarizabilities, their intrinsic hyperpolarizabilities all fall a factor 10−3/2 short of the fundamental limit (i.e. when βINT is 1).23 Since it has been shown that this limit is not of a fundamental nature, it has been recognized as the apparent limit. Indeed, from 2007 on there have been reports of molecules breaching this limit.13,24–26
Since βMAX can be calculated for the zzz-component but not for the overall βHRS, βzzz,0 must be known. As already mentioned, this requires that the actual symmetry of the molecules is known. Applied to Pa, the symmetry of the particular random coil is unknown and in principle all tensor components can contribute. The C2 symmetry of a helix, on the other hand, restricts the number of independent, nonvanishing tensor components to four, βxyz, βyxx, βyzz, and βyyy.27,28 In order to determine the number of actually contributing tensor components, depolarization measurements were performed. The depolarization ratio, ρ, which is the ratio of the horizontal to vertical polarized second harmonic light intensity with respect to the vertical input polarization, amounts to 0.68 in all solvent conditions. In combination with the macromolecular topology, this reveals that only octupolar contributions27,28 contribute to the HRS-response and that therefore βxyz is the only relevant tensor component in all solvent conditions. As a consequence, βxyz,0, expressed per polymer, is calculated from all HRS measurements and amounts to 3000 × 10−30 esu in CHCl3 and 950 × 10−30 esu in hexane and CHCl3–methanol (3/7). βzzz,MAX amounts to 43000 × 10−30 esu in all solvent conditions. Since βxyz,MAX ≤ βzzz,MAX29 it can be concluded that βINT ≥ 0.077 (coil) and βINT ≥ 0.025 (helix), revealing record-high βINT and breaking the apparent limit in the coiled conformation. Note that the assumptions that have been made in order to calculate βINT each underestimate βINT. Indeed, the first assumption is βxyz,MAX ≤ βzzz,MAX. Second, Nπe− is estimated from Mn measured by GPC. This technique typically overestimates the molar mass of conjugated polymers and, consequently, βMAX will be overestimated as well. As a consequence, the actual βINT is likely to be even higher than the one tabulated. We must finally mention that, while classical chromophores are typically rigid molecules with a fixed conformation (and hyperpolarizability), this is not at all the case for Pa. In contrast, the polymer molecules can adopt many different conformations, each with a different hyperpolarizability. Since an averaged hyperpolarizability is measured, this implies that some molecules display a hyperpolarizability which is even higher than the one calculated. Indeed, Méreau et al. have demonstrated that the hyperpolarizability in a dynamic structure significantly depends on the actual conformation.30
It is clear that Pa shows a record-high hyperpolarizability, regardless of the criterion used. This opens of course the question what the origin of this phenomenon is. A clue to this question can be found in the evolution of the hyperpolarizability and the UV-vis and CD spectra upon decreasing the solvent quality. For this purpose, the following parameters were defined and plotted (Fig. 3c and d):
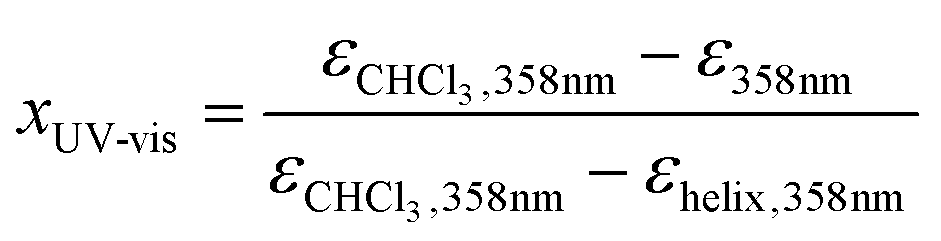
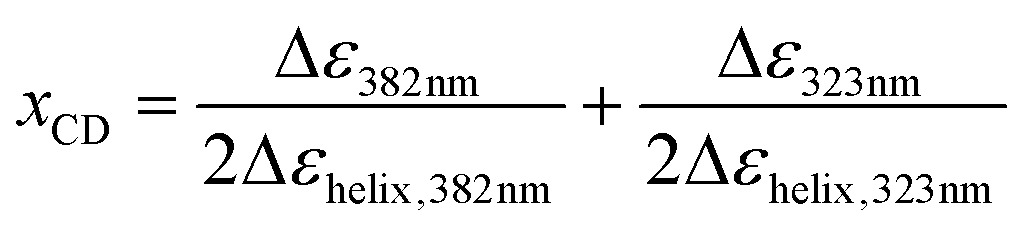
in which the “helix” conditions refer to CHCl3–methanol (3/7) or hexane. These parameters reflect the stepwise transformation from the coiled (x = 1) to the helical conformation (x = 0). The evolution of the CD and UV-vis spectra depends on the amount of (one-handed) helices and therefore probes the coil-helix transition and the change in conjugation related with it.
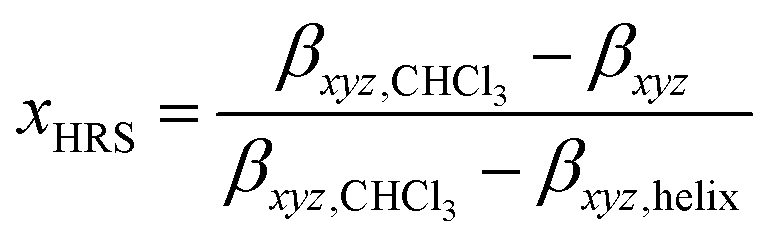
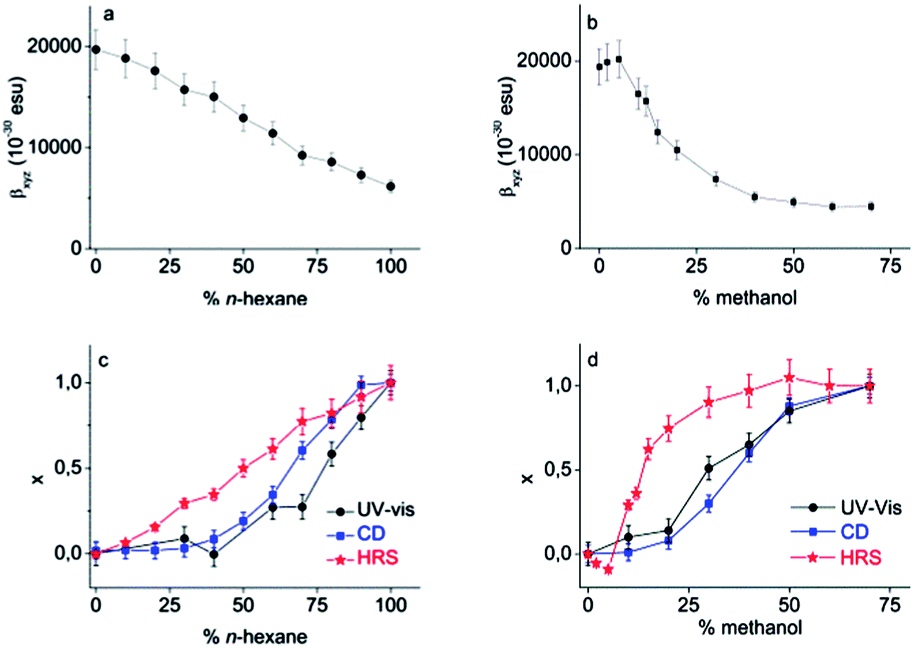 | ||
| Fig. 3 β HRS in chloroform mixtures upon addition of (a) n-hexane and (b) methanol. Relative evolution of the coil → helix transition probed by UV-vis and CD spectroscopy and HRS in chloroform upon addition of (c) n-hexane and (d) methanol (c = 92 mg L−1). | ||
In all solvent conditions, also in the intermediate mixtures, ρ is unaffected. Therefore, βxyz remains the sole important contribution to the HRS response in all conditions. As a consequence, the evolution of the HRS intensity in different solvent conditions cannot be ascribed to a symmetry change, which might affect the number of nonzero-components, but must originate from a different magnitude of βxyz. The magnitude of the hyperpolarizability is affected by the transition dipole moment of the HOMO–LUMO transition, which is proportional to the strength of the peak in the extinction spectrum. Although the HOMO–LUMO band becomes weaker upon addition of nonsolvent, it is apparent that this change in absorption does not coincide with the measured βxyz values (Fig. 3). This implies that the change in transition dipole moment cannot solely explain the evolution of the hyperpolarizability in different solvent conditions.
For certain chiral systems, magnetic dipole interactions have to be included to accurately describe the NLO response. Indeed, strong delocalization of electrons in helical structures can induce magnetization, contributing to the hyperpolarizability of the polymer. This contribution is generally small, although measurements in the solid phase demonstrate that in some cases the magnitude of magnetic dipole components cannot be neglected.31,32 However, if the same experiment is repeated with a poly(phenanthrene) analogue substituted with achiral n-octyl groups, the CD response vanishes and similar results are obtained.33 Moreover, the fact that the HRS curve does not coincide with the CD and UV-vis curves, demonstrates that the HRS response is not determined by the ratio of coil/helix. The increase of the HRS signal at low methanol content is exemplary in this respect. In these conditions, no helices are yet formed and the conjugation length does not change (as shown by the CD and UV-vis spectra), but the HRS response changes. We conclude that neither helix formation, nor intrinsic chirality is required for a large second-order NLO response.
Other possible phenomena that can contribute to the measured second-harmonic response of Pa relate to short-range molecular interactions within the polymer chain, e.g. between different parts showing strong conjugation. Firstly, statistically dependent positional and orientational correlations between different sections along the polymer chain can give rise to coherent HRS.34,35 The total second-harmonic amplitude is then found as the coherent sum over these correlated sections. A second effect can be linked with the temporally and spatially fluctuating molecular field F, which becomes important in regions of near-range ordening.36–39 The low-frequency (∼dc) molecular field then gives rise to electric-field induced second-harmonic generation (EFISHG), in which case the contribution of the second hyperpolarizability γijkl should be considered and βijk becomes proportional to γijklF. Since this is in essence a third-order NLO process, this phenomenon also occurs for centrosymmetric molecules. However, in the limited amount of experimental studies on the subject this effect is found to be non-negligible but rather weak.36–38,40 Moreover, it is important to note that molecular correlations and interactions not only change the intensity, but also the polarization dependence, i.e. the depolarization ratio ρ, of HRS light.35–37 This effect has been exploited in several studies to investigate molecular correlations between chromophores or to determine the contribution of the local molecular field F.36–38,41–44 For Pa however, the depolarization ratio remains constant for different solvent conditions while the intensity changes drastically, implying that short-range interactions at least cannot explain the evolution of βxyz.
Importantly, addition of nonsolvent does influence the torsion angle θ between the consecutive phenanthrene units. Indeed, this induces a shrinking of the (coiled) polymer chains upon which a helical conformation is formed. Both processes – the shrinking of the coils and the formation of a helix – alter θ and, consequently, the conjugation.
We hypothesize that this extraordinary high hyperpolarizability relates to the theoretical findings of Kuzyk and coworkers. Their extensive numerical calculations point out that optimal hyperpolarizabilities are reached by effectively varying the potential energy landscape of the π-conjugation.45–47 It must be mentioned that a complete breaking of the conjugation (θ = 90°) is naturally also detrimental. Optimal hyperpolarizabilities are expected in molecules with strong, but varying conjugation. The evolution of the hyperpolarizability in different solvent conditions can then be explained by a change in θ, modulating conjugation and resulting in possibly extremely efficient chromophores. These calculations also suggest that enhanced hyperpolarizabilities, alongside optimized variation of the potential energy landscape, require the presence of two dominant electronic transitions.47 This seems to be in line with the absorption spectrum of Pa with one transition around 275 nm and one near 360 nm. These spectral features are in fact common to all conjugated polymers and are ascribed to absorption of the monomer unit for the short wavelength band, and a π–π* transition of the delocalized conjugated system along the polymer backbone for the HOMO–LUMO band. Interestingly, substantial hyperpolarizabilities were recently also measured in poly(3-alkylthiophene)s.48
Again, there have been some reports in recent years of molecules breaking the apparent limit. It must be noted that these molecules also obey the principle of potential energy modulation, either by combining groups with different effective conjugation13 or by tuning the angle between the electron donating moiety and the π-conjugated bridge.25 Importantly, these reports concern D–π–A molecules, while Pa does not show this typical D–π–A motif at all. Our results clearly deny the necessity of an electron donating and withdrawing moiety, i.e. the typical D–π–A structure, for achieving a high hyperpolarizability. As mentioned, a recent study has demonstrated that poly(3-alkylthiophene)s, despite the limited donor strength of the alkyl substituents, display a large second-order NLO response, be it substantially lower than Pa.48 Also, several reports showed that an octupolar symmetry by itself, even in absence of a D–π–A motif, can result in a substantial second-order NLO response.49–51 Indeed, such a D–π–A motif is one, but not the only way to provide the (i) required noncentrosymmetry and (ii) the conjugation modulation. The magnitude of the hyperpolarizability in Pa puts the importance of a D–π–A structure with C2v symmetry into perspective with respect to other symmetry-breaking paradigms, such as torsion between conjugated planes, resulting in D2 symmetry.52 Importantly, while D–π–A molecules typically concern planar molecules in order to maximize conjugation, efficient potential energy fluctuations are not favored in such flat molecules.
4. Conclusions
In conclusion, we have demonstrated that Pa, a conjugated polymer that lacks the push–pull (D–π–A) substitution pattern typically associated with strong NLO compounds, unexpectedly shows an extremely high hyperpolarizability. Even after scaling the NLO response to the number of polarizable electrons, by calculating the (size-independent) intrinsic hyperpolarizability, Pa ranks among the best chromophores ever measured. Indeed, it is one of the few compounds breaking the longstanding apparent limit, and is to the best of our knowledge the first compound that achieves this without relying on the traditional D–π–A design.The macromolecular conformation is found to be strongly dependent on the solvent conditions, evolving from an unordered coiled to a helical conformation upon addition of nonsolvent, as evidenced by CD and UV-vis spectroscopy. We found the largest βINT for the unordered coiled conformation, evidencing that chirality is not required for obtaining a large NLO response in this system. While the hyperpolarizabilty is dependent on the solvent conditions, it does not correlate clearly with the ratio helix/coil found in different solutions. Rather, depolarization measurements point out that the NLO response is governed by octupolar contributions for both the helical and the coiled state. In line with recent theoretical and experimental findings, we postulate that the variation in second-order NLO intensity upon addition of nonsolvent is related to modulation of conjugation along the polymer backbone, by influencing the torsion angle between consecutive phenanthrene units, and resulting in extremely efficient chromophores.
These unexpected results can lead to new insights for the design of efficient second-order NLO materials and put the common molecular design paradigms into perspective. In particular, the use of 3D macromolecules that allow for an efficient modulation of conjugation is an interesting path to explore. Additionally, the octupolar character of the second-order NLO response in Pa can be particularly attractive for the development of polarization-insensitive devices, while the limited conjugation length ensures transparency in the visible region. Further research will focus on the exact influence of the conformation of conjugated polymers on their hyperpolarizability and on determining the properties of the solid state. In this respect, it is important to note that aggregation in the solid state undoubtedly affects the conformation and hence the hyperpolarizability of the polymer. The fact that the actual hyperpolarizability of the polymer is, with the assumptions made, underestimated and the fact that an averaged hyperpolarizability of a broad scale of different conformations with different hyperpolarizability is measured, leaves the possibility to further increase the hyperpolarizability of conjugated polymers.
Acknowledgements
We acknowledge Dr Javier Pérez-Moreno for the fruitful discussions. We are grateful to the University of Leuven (GOA) and the Fund for Scientific Research (FWO-Vlaanderen) for financial support. MGK thanks the National Science Foundation (EECS-1128076).Notes and references
- P. N. Prasad and D. J. Williams, Introduction to Nonlinear Optical Properties of Molecules and Polymers, 1991 Search PubMed.
- T. Verbiest, S. Houbrechts, M. Kauranen and K. Clays, J. Mater. Chem., 1997, 7, 2175–2189 RSC.
- L. R. Dalton, W. H. Steier, B. H. Robinson, C. Zhang, A. Ren, S. Garner, A. Chen, T. Londergan, L. Irwin, B. Carlson, L. Fifield, G. Phelan, C. Kincaid, A. Jen, S. California and L. Angeles, J. Mater. Chem., 1999, 9, 1905–1920 RSC.
- S. R. Marder, B. Kippelen, A. K.-Y. Jen and N. Peyghambarian, ChemInform, 2010, 28, 845–851 Search PubMed.
- PNA = p-nitroaniline; FTC = 2-dicyanomethylen-3-cyano-4-[2-[trans-[4-(N,N-diacetoxyethylamino)]phenylene-3,4-dibutylthienyl-5]ethynylene]-5,5-dimethyl-2,5-dihydrofuran.
- Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119–122 CrossRef CAS PubMed.
- P. Kaatz and D. Shelton, J. Chem. Phys., 1996, 105, 3918–3929 CrossRef CAS.
- W. Vanormelingen, A. Smeets, E. Franz, I. Asselberghs, K. Clays, T. Verbiest and G. Koeckelberghs, Macromolecules, 2009, 42, 4282–4287 CrossRef CAS.
- G. Olbrechts, R. Strobbe, K. Clays and A. Persoons, Rev. Sci. Instrum., 1998, 69, 2233–2241 CrossRef CAS.
- J. Oudar and D. Chemla, J. Chem. Phys., 1977, 66, 2664–2668 CrossRef CAS.
- G. Olbrechts, K. Wostyn, K. Clays and A. Persoons, Opt. Lett., 1999, 24, 403–405 CrossRef CAS PubMed.
- M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC.
- J. Pérez-Moreno, Y. Zhao, K. Clays and M. G. Kuzyk, Opt. Lett., 2007, 32, 59–61 CrossRef.
- J.-M. Raimundo, P. Blanchard, N. Gallego-Planas, N. Mercier, I. Ledoux-Rak, R. Hierle and J. Roncali, J. Org. Chem., 2002, 67, 205–218 CrossRef CAS PubMed.
- J. Luo, Y.-J. Cheng, T.-D. Kim, S. Hau, S.-H. Jang, Z. Shi, X.-H. Zhou and A. K.-Y. Jen, Org. Lett., 2006, 8, 1387–1390 CrossRef CAS PubMed.
- B. H. Robinson, L. R. Dalton, A. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorova, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. K. Y. Jen, Chem. Phys., 1999, 245, 35–50 CrossRef CAS.
- Y. Liao, B. E. Eichinger, K. A. Firestone, M. Haller, J. Luo, W. Kaminsky, J. B. Benedict, P. J. Reid, A. K.-Y. Jen, L. R. Dalton and B. H. Robinson, J. Am. Chem. Soc., 2005, 127, 2758–2766 CrossRef CAS PubMed.
- P. Y. Moh, P. Cubillas, M. W. Anderson and M. P. Attfield, J. Am. Chem. Soc., 2011, 133, 13304–13307 CrossRef CAS PubMed.
- C. Cai, I. Liakatas, M. Wong, M. Bo, C. Bosshard, P. Gu, S. Concilio, N. Tirelli and U. W. Suter, Org. Lett., 1999, 1, 1847–1849 CrossRef CAS.
- M. G. Kuzyk, Characterization Techniques and Tabulations for Organic Nonlinear Optical Materials, Taylor & Francis, 1998 Search PubMed.
- T. Verbiest, K. Clays and V. Rodriguez, Second-order Nonlinear Optical Characterization Techniques: An Introduction, 2009 Search PubMed.
- M. G. Kuzyk, J. Mater. Chem., 2009, 19, 7444 RSC.
- K. Tripathy, J. P. Moreno, M. G. Kuzyk, B. J. Coe, K. Clays and A. M. Kelley, J. Chem. Phys., 2004, 121, 7932–7945 CrossRef CAS PubMed.
- J. Pérez-Moreno, Y. Zhao, K. Clays, M. G. Kuzyk, Y. Shen, L. Qiu, J. Hao and K. Guo, J. Am. Chem. Soc., 2009, 131, 5084–5093 CrossRef PubMed.
- H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S.-T. Ho, E. C. Brown, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 3267–3286 CrossRef CAS PubMed.
- H. Kang, A. Facchetti, P. Zhu, H. Jiang, Y. Yang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S. Ho and T. J. Marks, Angew. Chem., Int. Ed., 2005, 44, 7922–7925 CrossRef CAS PubMed.
- J. Zyss, J. Chem. Phys., 1993, 98, 6583 CrossRef CAS.
- P. D. Maker, Phys. Rev. A, 1970, 1, 923–951 CrossRef CAS.
- M. G. Kuzyk, IEEE J. Sel. Top. Quantum Electron., 2001, 7, 774–780 CrossRef CAS.
- R. Méreau, F. Castet, E. Botek and B. Champagne, J. Phys. Chem. A, 2009, 113, 6552–6554 CrossRef PubMed.
- M. Kauranen, T. Verbiest, E. W. Meijer, E. E. Havinga, M. N. Teerenstra, A. J. Schouten, R. J. M. Nolte and A. Persoons, Adv. Mater., 1995, 7, 641 CrossRef CAS.
- T. Verbiest, S. Van Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS PubMed.
- β xyz,0 amounts to 1800 × 10−30 esu in pure ChCl3, and to 760 × 10−30 esu in CHCl3–MeOH (5/5).
- S. Kielich, Acta Phys. Pol., A, 1974, 45, 231–251 Search PubMed.
- D. P. Shelton, J. Chem. Phys., 2013, 138, 154502 CrossRef PubMed.
- S. Kielich, J. Lalanne and F. Martin, Phys. Rev. Lett., 1971, 26, 1295–1298 CrossRef CAS.
- S. Kielich, J. Raman Spectrosc., 1973, 1, 119–139 CrossRef CAS.
- J. Lalanne, F. Martin and S. Kielich, Chem. Phys. Lett., 1975, 30, 73–76 CrossRef CAS.
- S. N. Yaliraki and R. J. Silbey, J. Chem. Phys., 1999, 111, 1561 CrossRef CAS.
- D. P. Shelton, J. Chem. Phys., 2009, 130, 114501 CrossRef PubMed.
- J. Chen and K. Y. Wong, J. Chem. Phys., 2005, 122, 174505 CrossRef CAS PubMed.
- Y. C. Chan and K. Y. Wong, J. Chem. Phys., 2012, 136, 174514 CrossRef CAS PubMed.
- D. P. Shelton, J. Chem. Phys., 2013, 138, 054502 CrossRef PubMed.
- D. P. Shelton, J. Chem. Phys., 2010, 133, 234507 CrossRef PubMed.
- J. Zhou, M. G. Kuzyk and D. S. Watkins, Opt. Lett., 2006, 31, 2891–2893 CrossRef PubMed.
- S. Shafei, M. C. Kuzyk and M. G. Kuzyk, J. Opt. Soc. Am. B, 2010, 27, 1849 CrossRef CAS.
- J. Zhou, U. Szafruga, D. Watkins and M. Kuzyk, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76, 053831 CrossRef.
- S. Deckers, S. Vandendriessche, D. Cornelis, F. Monnaie, G. Koeckelberghs, I. Asselberghs, T. Verbiest and M. A. van der Veen, Chem. Commun., 2014, 50, 2741–2743 RSC.
- S. Van Cleuvenbergen, I. Asselberghs, E. M. García-Frutos, B. Gómez-Lor, K. Clays and J. Pérez-Moreno, J. Phys. Chem. C, 2012, 116, 12312–12321 CAS.
- R. Jua, M. Ramos, J. L. Segura, S. Van Cleuvenbergen, K. Clays, T. Goodson, J. T. Lo and J. Casado, J. Phys. Chem. C, 2013, 117, 626–632 Search PubMed.
- M. Quintiliani, J. Pérez-Moreno, I. Asselberghs, P. Vázquez, K. Clays and T. Torres, J. Phys. Chem. B, 2010, 114, 6309–6315 CrossRef CAS PubMed.
- T. V. Duncan, K. Song, S.-T. Hung, I. Miloradovic, A. Nayak, A. Persoons, T. Verbiest, M. J. Therien and K. Clays, Angew. Chem., Int. Ed., 2008, 47, 2978–2981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4tc00616j |
| This journal is © The Royal Society of Chemistry 2014 |