 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Alkyl functionalized barbituric and thiobarbituric acid bithiophene derivatives for vacuum deposited n-channel OFETs†
Ying
Shu
a,
Annabel
Mikosch
a,
Kevin N.
Winzenberg
a,
Peter
Kemppinen
a,
Christopher D.
Easton
a,
Ante
Bilic
b,
Craig M.
Forsyth
c,
Christopher J.
Dunn
a,
Th. Birendra
Singh
a and
Gavin E.
Collis
*a
aCSIRO Materials Science and Engineering, Private Bag 10, Clayton South MDC, VIC 3169, Australia. E-mail: Gavin.Collis@csiro.au
bCSIRO Computational Informatics, Private Bag 33, Clayton South, VIC 3169, Australia
cSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
First published on 16th April 2014
Abstract
A family of barbituric and thiobarbituric acid end capped small molecule semiconductors were synthesized, characterized and shown to exhibit n-channel organic thin film transistor properties. By changing the N-alkyl substituent from methyl to ethyl, a dramatic increase in electron mobilities was observed with values nearing 0.3 cm2 V−1 s−1.
Research on organic semiconducting materials has progressed rapidly in recent years on account of the potential of these materials to be applied in low cost, large area and mechanically flexible devices, such as organic photovoltaics (OPVs), organic light emitting diodes (OLEDs) and organic field effect transistors (OFETs). In the field of OFETs many high performance p-type organic semiconductors have been reported and continue to be developed.1,2 In contrast, the discovery of n-type materials has been slow with only a handful of reported compounds showing high electron mobility.1,3–8 In order to realize the potential of organic semiconductors in complementary circuits and ambipolar transistors,9 it is essential that research continues into the discovery of new high electron mobility and air stable n-type materials.
Previously we have employed a structure-performance based approach in the design and discovery of lead p-type dibenzochrysene materials for OPVs10 and high mobility OFETs (>1 cm2 V−1 s−1).11,12 Recently, we used this strategy to develop a high performance non-fullerene n-type material based around the 1,3-dicarbonyl acceptor, indan-1,3-dione, for bulk heterojunction OPVs.13
Another similar 1,3-dicarbonyl acceptor, barbituric acid, has been developed primarily as a small molecule donor–acceptor (D–A) and acceptor–donor–acceptor (A–D–A) p-type materials for use with fullerenes in OPVs14,15 and dyes for p-type dye sensitized solar cells.16 To the best of our knowledge, the only reports of barbituric acid functionalized semiconductor materials for OFETs have shown hole mobilities ranging from 10−7 to 10−4 cm2 V−1 s−1.17,18
In line with our ongoing efforts to design and develop new p- and n-type materials we report in this communication the study of a series of simple end-capped barbituric and thiobarbituric acid-bithiophenes for electron transport in OFETs. These materials were easily synthesized, purified by vacuum sublimation and characterized by thin film studies. We observe that subtle variation of the alkyl substituent (methyl vs. ethyl) and the terminal heteroatom (O vs. S) have a dramatic impact on the electron mobility of the material in OFETs. We screened and identified a number of compounds with electron mobility values approaching 0.3 cm2 V−1 s−1.
We have found that density functional theory (DFT) calculations serve as a rapid screening tool to estimate the frontier orbital energy levels of potential p- and n-type candidates.10,12,13 Based on the DFT calculation of the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) levels, compounds 7, 9, 11 and 13 were identified as potential candidates (Scheme 1). Here the average HOMO energy level is −6.46 eV, while the average LUMO energy level is −3.90 eV, sufficiently deep for electron transport and electron injection with common electrode materials (Table 1). Encouragingly, the HOMO and LUMO orbital plots reveal that the electron density of both are evenly delocalized throughout the conjugated π system of the barbituric acid end-capped system, as exemplified by compound 7 in Fig. 1. However, the presence of the terminal sulphur atom in the thiobarbituric group has a pronounced influence on the HOMO energy orbitals (see ESI, Table S1†). Previous studies on the frontier molecular orbitals of simple thiobarbituric acid derivatives has shown the presence of degenerate HOMO orbitals.19 The terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond is comprised of σ- and π-type molecular orbitals. As the CS bond length is longer than the CO bond (1.6 vs. 1.25 Å),20,21 there is a greater contribution from the σ-type HOMO molecular orbital that results in doubly degenerate HOMOs as shown in Table S1.† We find that the thiobarbituric derivatives are predicted to exhibit 0.2 to 0.4 eV smaller bandgap than their barbituric analogues.21
S bond is comprised of σ- and π-type molecular orbitals. As the CS bond length is longer than the CO bond (1.6 vs. 1.25 Å),20,21 there is a greater contribution from the σ-type HOMO molecular orbital that results in doubly degenerate HOMOs as shown in Table S1.† We find that the thiobarbituric derivatives are predicted to exhibit 0.2 to 0.4 eV smaller bandgap than their barbituric analogues.21
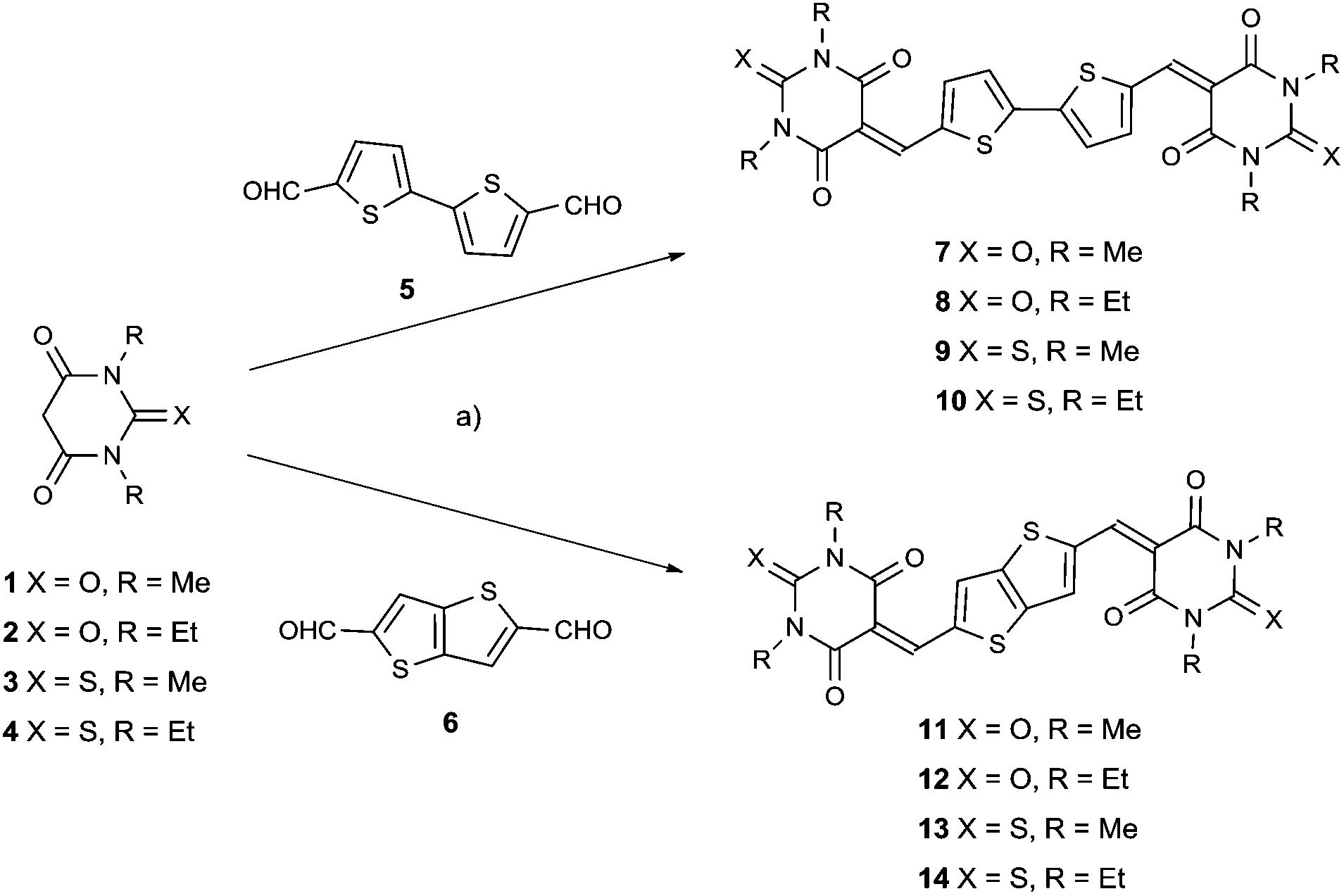 | ||
| Scheme 1 Synthetic routes to target compounds 7–14. Reaction conditions: (a) t-butanol, catalytic piperidine, reflux, 4–6 h. | ||
| Entry | Theoretical (DFT) | Experimental | |||||
|---|---|---|---|---|---|---|---|
| E HOMO (eV) | E LUMO (eV) | E g (eV) | IPa (eV) | EAb (eV) | UV/Vis λonset (nm) | E g (eV) | |
| a Ionisation potential (IP) was determined from UPS. b Electron affinity (EA) was estimated using IP and optical bandgap (Eg). c Optical bandgap (Eg) was determined from the UV/Vis absorption offset using the equation, Eg = 1240 (eV nm)/λonset. | |||||||
| 7 | −6.36 | −3.69 | 2.67 | −6.32 | −4.30 | 615 | 2.02 |
| 8 | −6.42 | −3.74 | 2.67 | −6.28 | −4.23 | 606 | 2.05 |
| 9 | −6.37 | −3.97 | 2.40 | −6.22 | −4.31 | 648 | 1.91 |
| 10 | −6.34 | −3.93 | 2.41 | −6.65 | −4.78 | 664 | 1.87 |
| 11 | −6.66 | −3.85 | 2.81 | −6.44 | −4.32 | 584 | 2.12 |
| 12 | −6.64 | −3.84 | 2.80 | −6.29 | −4.23 | 602 | 2.06 |
| 13 | −6.45 | −4.08 | 2.37 | −6.17 | −4.26 | 649 | 1.91 |
| 14 | −6.42 | −4.03 | 2.38 | −6.19 | −4.33 | 667 | 1.86 |
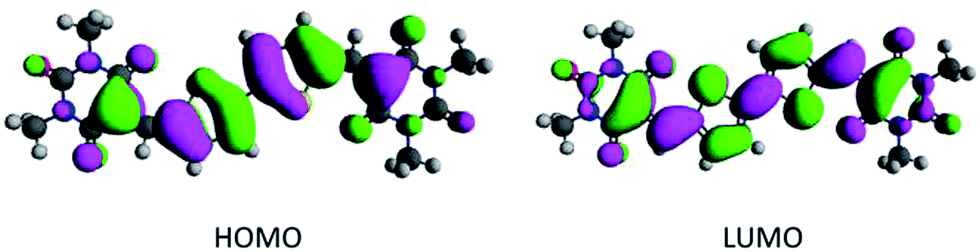 | ||
| Fig. 1 DFT HOMO and LUMO orbital energy plots of compound 7. | ||
Our previous work12,13 and the current studies (see ESI, Table S1†) have confirmed that the DFT calculations show little electronic perturbation caused by changing the alkyl groups, but previous experimental data suggests the impact on the physical properties can be dramatic for OFET materials.12 To investigate this point, an analogous series of N-ethyl functionalized compounds, 8, 10, 12 and 14, were studied for comparison (Scheme 1). Interestingly, the DFT calculations show some variation between analogues sharing the bithiophene or thieno[3,2-b]thiophene core unit (e.g. compounds 7 and 11) with a slight deepening of the HOMO and LUMO energy levels.
These eight target compounds were synthesized by base catalyzed Knoevenagel condensation of the bis-aldehyde 5 or 6, with the appropriate thio/barbituric acid derivatives, 1, 2, 3 or 4, as shown in Scheme 1. The products were extremely insoluble, therefore easily isolated and purified by vacuum sublimation. The chemical formula of these compounds was confirmed by elemental analysis and by high resolution mass spectrometry. Thermal gravimetric analysis and differential scanning calorimetry plots (ESI, Fig. S1–S16†) indicated that all compounds follow a similar trend with melting points >400 °C and excellent thermal stability. Analysis of compounds 7–14 by powder fourier transform infrared spectroscopy indicated typical stretching modes for CO, and aromatic CC around 1650 to 1720 and at 1550 cm−1 respectively (ESI†).
The compounds 7–14 were further characterized by thin film studies of the materials evaporated onto glass substrates. Normalized thin film UV/Vis absorption spectra of the bithiophene and thieno[3,2-b]thiophene derivatives are shown in Fig. 2. In the bithiophene series (Fig. 2a) compounds 7–10 show similar key absorption maxima occurring at 466, 482, 484 and 498 nm respectively. However, the presence of an additional methylene group in 8 compared to the methyl-capped 7 has a dramatic influence on the shape of the absorption profile. The methyl compound 7 has a very broad absorption band from 360 nm through to 600 nm with absorption maxima occurring at 462 and 560 nm. In contrast, the ethyl derivative has a sharper absorption profile from 420 to 600 nm and well defined absorption maxima occurring at 485 and 566 nm. The same trend is also observed with methyl- and ethyl-thiobarbituric acid derivatives, compounds 9 and 10, respectively. The presence of the terminal sulphur atom in these compounds also results in a red shift in all absorption bands. In contrast, the thieno[3,2-b]thiophene derivatives 11–14 show very simple absorption profiles with absorption maxima occurring at 440, 445, 478, and 479 nm respectively (Fig. 2b). In this series we observe no significant difference between the methyl and ethyl derivatives, but a red shift in the absorption maxima is again observed for the terminal sulphur compounds 13 and 14.
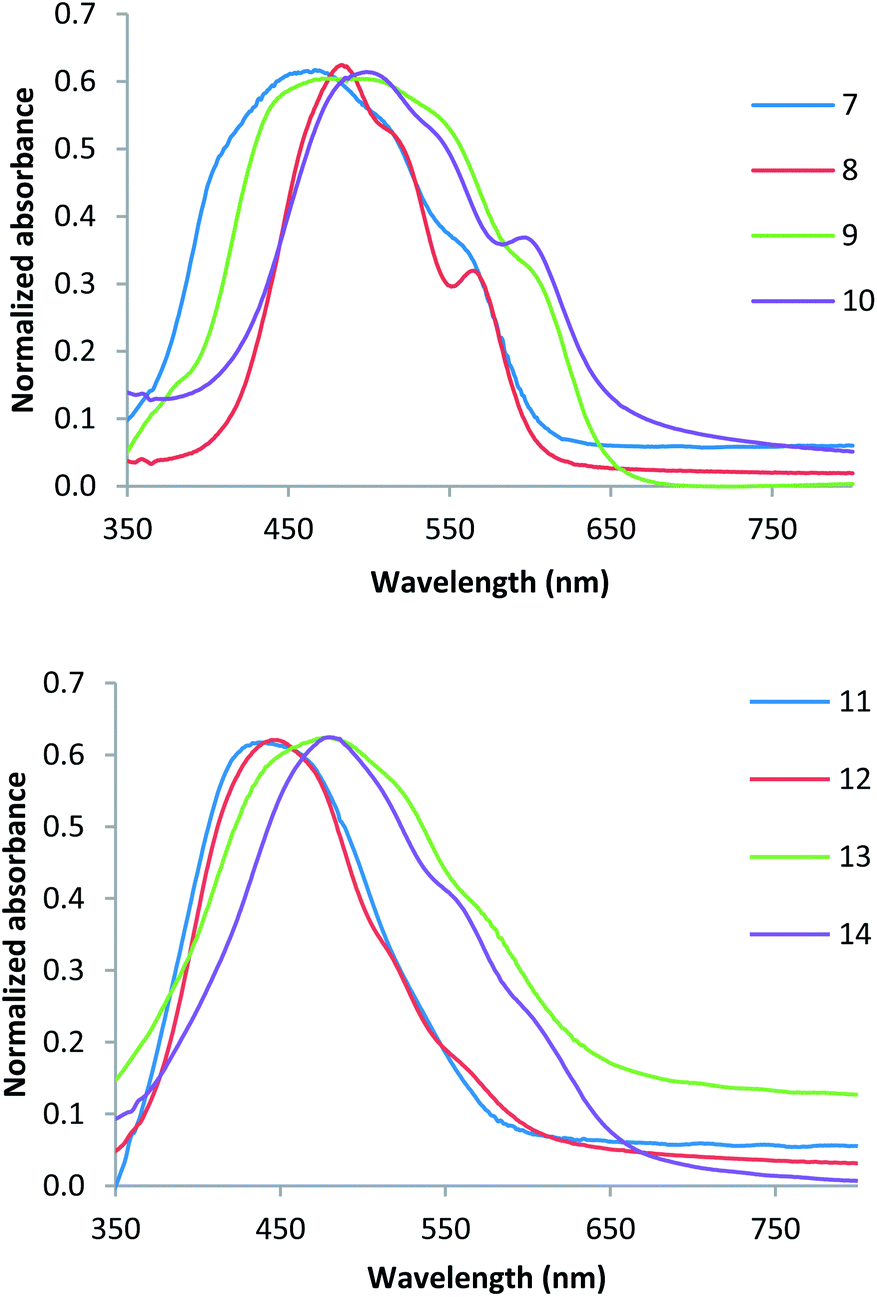 | ||
| Fig. 2 Evaporated thin film UV/Vis spectra of (a) bithiophene compounds 7–10 and (b) thieno[3,2-b]thiophene compounds 11–14. | ||
The electronic properties of compounds 7–14 were determined by analysis of the thin film samples. X-ray photoelectron spectroscopy (XPS) was used to confirm the elemental composition of the compounds deposited and curve fitting of the high resolution C 1s spectra indicated that the carbon-based functional groups were as expected from the chemical structures (ESI†). Ionisation potentials (IP) were calculated from ultraviolet photoelectron spectroscopy (UPS)22 measurements (Table S4 and ESI†). We observed that the calculated DFT EHOMO levels agree well with experimentally determined IP from by UPS. The determined electron affinities (EA) were found to be deeper by 0.3 to 0.5 eV compared to the calculated ELUMO.
Compounds 7–14 were evaluated in bottom gate/top contact (BG/TC) OFET devices. The SiO2 surface was treated with octadecyltrichlorosilane (SiO2-ODTS)12 and the compounds were vacuum deposited onto this surface at a set substrate temperature (Tsub) of 25 °C. Device fabrication was completed by the deposition of top gold source and drain electrode through a shadow mask. All compounds tested exhibited typical electron transport behaviour. The OFET characteristics of all of the compounds show electron mobilities (μe) ranging from 0.009 to 0.26 cm2 V−1 s−1; the μe is highly dependent on the alkyl group substituent and terminal heteroatom (Table 2).
| Entry | μ (cm2 V−1 s−1) | I on/Ioff | V th (V) |
|---|---|---|---|
| a Tested in a glovebox. W/L ratio is 33, where W = 2000 μm and L = 60 μm. | |||
| 7 | 0.009 ± 0.002 | 103 | 38 |
| 8 | 0.26 ± 0.04 | 106 | 15 |
| 9 | 0.0040 ± 0.0008 | 103 | 10 |
| 10 | 0.093 ± 0.006 | 106 | 22 |
| 11 | 0.026 ± 0.008 | 106 | 34 |
| 12 | 0.20 ± 0.06 | 106 | 40 |
| 13 | 0.024 ± 0.008 | 104 | 19 |
| 14 | 0.066 ± 0.009 | 105 | 12 |
Of all the compounds tested, the highest μe of 0.26 cm2 V−1 s−1 was obtained from the ethyl bithiophene-barbiturate compound 8. Fig. 3 shows a transfer curve obtained by plotting drain current as a function of applied positive gate voltage for compound 8. The transistor device has an average turn-on threshold voltage of 15 V and an on/off ratio 106. Interestingly, in the thienothiophene series the analogous compound 12, with ethyl barbituric acceptor groups, also afforded a relatively high μe of 0.20 cm2 V−1 s−1. These results imply that the mobility is only slightly altered by the core aromatic unit (i.e. bithiophene vs. fused thienothiophene), but the most noticeable difference results from the functionality on the barbituric acceptor (i.e. the terminal heteroatom and alkyl group). All thiobarbituric acceptor compounds (i.e.9, 10, 13 and 14) performed moderately in devices, whereas the best devices were achieved employing barbituric acceptor groups with ethyl substituents (i.e.8 and 12). These results emphasise the important, yet subtle structural role functional groups have on device performance.
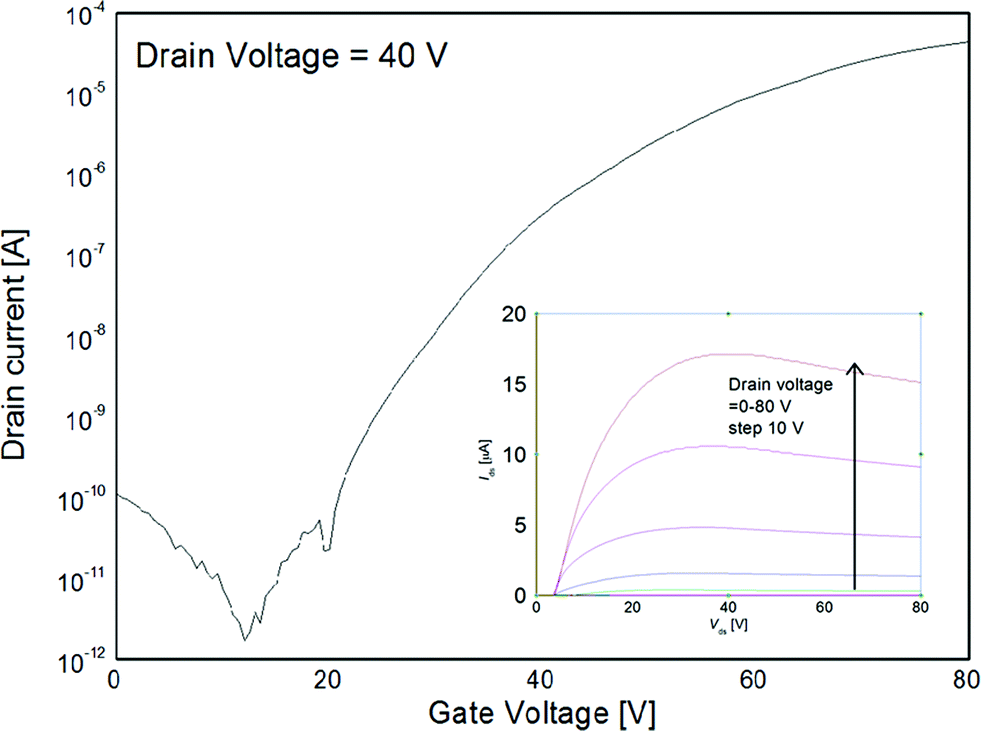 | ||
| Fig. 3 N-channel OFET characteristics with output curve shown as insert with compound 8 as active layer. | ||
Of all the compounds studied, only 7 and 8 gave, by thermal sublimation, crystals suitable for single crystal X-ray crystallography studies. While molecules of 8 are essentially planar (Fig. 4b), molecules of 7 exhibit a slight curvature of the end barbituric groups (Fig. 4a). The methyl-capped 7 exhibits face-to-face π stacking with an interstack distance of 3.59 Å (Fig. 4a). At this π–π distance, neighboring molecules are out of C–C van der Waals radius of each other. However, the ethyl-compound 8 packs in a series of 1-D slipped stacks (Fig. 4d). Within each stack, the interstack distance was found to be considerably shorter at 3.39 Å, favouring charge transport. Additionally with ethyl-capped 8, each barbituric end group is spatially arranged in the solid state to minimize terminal carbonyl to carbonyl electrostatic repulsion with other barbituric end groups (Fig. 4d). We hypothesize that ethyl substitution provides adequate insulation as a buffer between carbonyl to carbonyl electrostatic repulsion over methyl substitution.
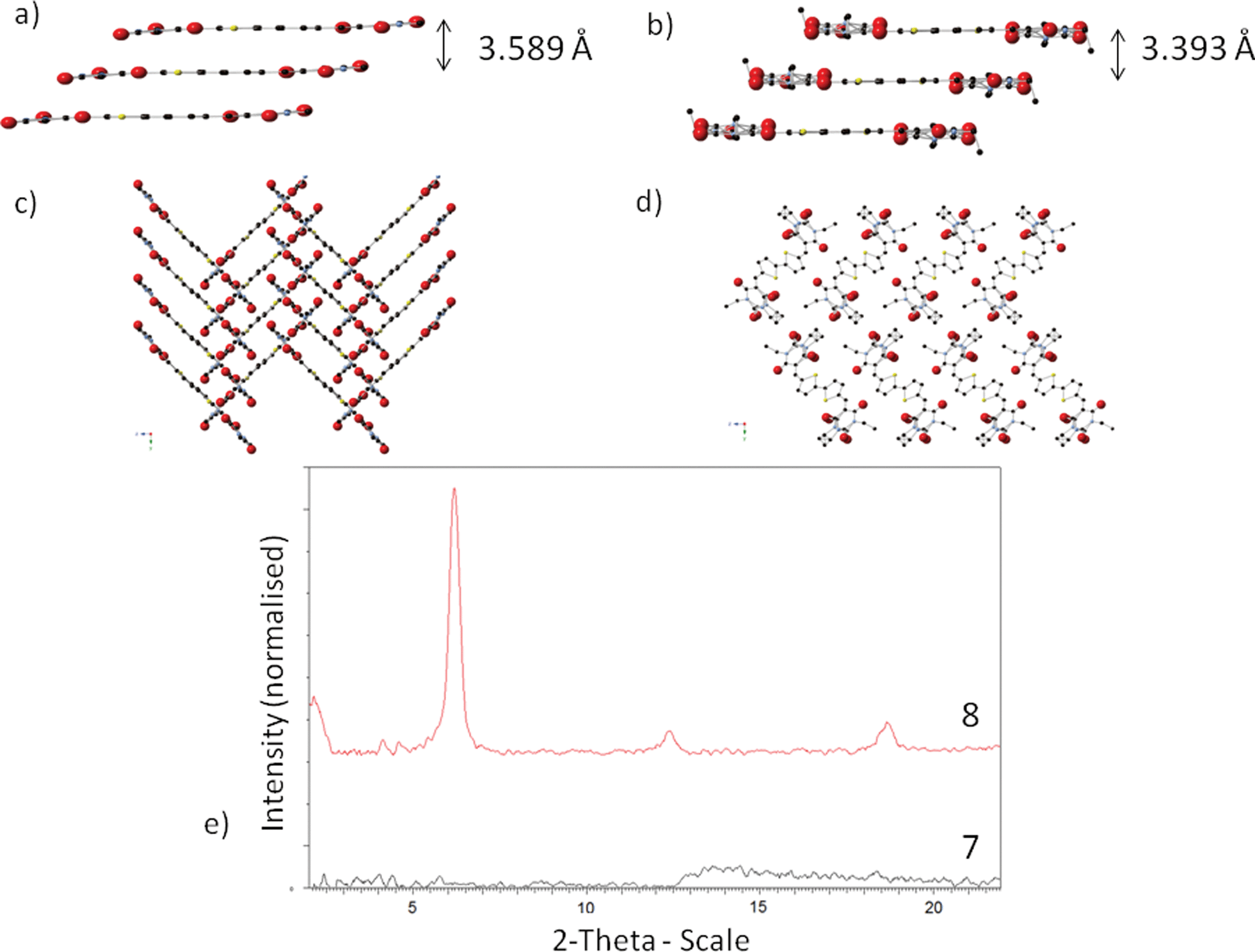 | ||
| Fig. 4 Cross-section view of compound (a) 7 and (b) 8. Representative crystal packing of (c) 7 and (d) 8 along the a-axis. (e) Thin film XRD of compounds 7 and 8. | ||
Films of 7–14 appeared smooth and continuous under optical microscopy. Film surface microstructures were studied via scanning electron microscopy (SEM). In general the films all appeared to have crystalline grains on the order of 0.1 micron or smaller (ESI, Fig. S20†). We could establish no discernable pattern between the rather unremarkable surface features of all the films and their corresponding device performance. Instead, we rely on out of plane film X-ray diffraction (XRD) to provide an insight into how the film microstructure correlates with OFET device performance.
To study the crystallinity of compounds 7–14 in devices, XRD patterns were recorded on films prepared via thermal evaporation onto SiO2–ODTS substrates. For N-methyl compounds 7, 9, 11 and 13, no diffraction peaks were observed, suggesting that these films are amorphous (ESI, Fig. S21†). In contrast, all of the N-ethyl compounds 8, 10, 12 and 14 exhibit a primary diffraction peak at 2θ = ∼6.2°, corresponding to a d-spacing of 14.3 Å. Thin film XRD data of 8 exhibits distinct out of plane diffraction peaks at 2θ = 6.2°, 12.4° and 18.7°.
Subtle differences in the XRD patterns were observed between bithiophene and thienothiophene analogues such as between compound 8 and compound 12, or between compound 10 and compound 14. However, for N-ethyl thiobarbituric end-capped compounds 10 and 14, there are two additional small angle XRD peaks below 5°, indicating the possibility of multiple crystalline orientations within the film that is absent for their barbituric end capped analogues 8 and 12. Although some of these compounds show features that indicate highly crystalline films, the lower mobility of some compounds, especially in thiobarbituric derivatives, may be a reflection of the crystal packing motifs and/or electronic factors (see earlier section on UV/Vis thin film data analysis and DFT calculations, ESI Table S1†) that do not favour suitable charge transfer.
In summary, a series of N-methylated and N-ethylated barbituric and thiobarbituric acid end capped bithophenes were synthesized, fully characterized and used for fabricating n-channel OFETs. The use of a structure-performance based approach in the design and discovery of a lead n-channel OFET semiconductor enabled the discovery of compound 8, an N-ethylated barbituric acid end capped bithophene, which exhibited an average charge carrier mobility of 0.26 cm2 V−1 s−1. We found that the heteroatom of the end capping groups had a larger influence on OFET performance than the central bithiophene or thienothiophene π bridge. We also found that subtle molecular perturbations resulting from changing from N-methylated to N-ethylated end groups had huge impact on OFET performance, which was consistent with the results from single X-ray crystal and thin-film XRD data.
Acknowledgements
This work was supported by the Flexible Electronics Theme and is part of CSIRO Future Manufacturing Flagship. We acknowledge support from CSIRO for an OCE Post-Doctoral Fellowship (YS) and OCE Julius Career Awards (AB and GEC). We thank Professor Andrew Holmes and Dr Mark Bown for helpful discussions.References
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- T. Mori, J. Phys.: Condens. Matter, 2008, 20, 184010 CrossRef.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- Y. Wen and Y. Liu, Adv. Mater., 2010, 22, 1–15 CrossRef.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- B. J. Jung, N. J. Tremblay, M.-L. Yeh and H. E. Katz, Chem. Mater., 2011, 23, 568–582 CrossRef CAS.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- K. N. Winzenberg, P. Kemppinen, G. Fanchini, M. Bown, G. E. Collis, C. M. Forsyth, K. Hegedus, T. B. Singh and S. E. Watkins, Chem. Mater., 2009, 21, 5701–5703 CrossRef CAS.
- K. B. Burke, Y. Shu, P. Kemppinen, B. Singh, M. Bown, I. Liaw, R. M. Williamson, L. Thomsen, P. Dastoor, W. Belcher, C. Forsyth, K. N. Winzenberg and G. E. Collis, Cryst. Growth Des., 2012, 12, 725–731 CAS.
- Y. Shu, G. E. Collis, C. J. Dunn, P. Kemppinen, K. N. Winzenberg, R. M. Williamson, A. Bilic, T. B. Singh, M. Bown, C. R. McNeill and L. Thomsen, J. Mater. Chem. C, 2013, 1, 6299–6307 RSC.
- K. N. Winzenberg, P. Kemppinen, F. H. Scholes, G. E. Collis, Y. Shu, T. B. Singh, A. Bilic, C. M. Forsyth and S. E. Watkins, Chem. Commun., 2013, 49, 6307–6309 RSC.
- D. Demeter, T. Rousseau, P. Leriche, T. Cauchy, R. Po and J. Roncali, Adv. Funct. Mater., 2011, 21, 4379–4387 CrossRef CAS.
- G. He, Z. Li, X. Wan, Y. Liu, J. Zhou, G. Long, M. Zhang and Y. Chen, J. Mater. Chem., 2012, 22, 9173–9180 RSC.
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Lin Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Licheng Sun, J. Phys. Chem. C, 2010, 114, 4738–4748 CAS.
- R. B. K. S. Siram, K. Tandy, M. Horecha, P. Formanek, M. Stamm, S. Gevorgyan, F. C. Krebs, A. Kiriy, P. Meredith, P. L. Burn, E. B. Namdas and S. Patil, J. Phys. Chem. C, 2011, 115, 14369–14376 CAS.
- R. B. K. S. Siram, M. Stephen, F. Ali and S. Patil, J. Phys. Chem. C, 2013, 117, 9129–9136 CAS.
- S. Millefiori and A. Millefiori, J. Heterocycl. Chem., 1989, 26, 639–644 CrossRef CAS.
- K. B. Wiberg and Y. Wang, ARKIVOC, 2011, 45–56 CAS.
- K. B. Wiberg, W. F. Bailey and G. A. Petersson, J. Phys. Chem. A, 2011, 115, 12624–12630 CrossRef CAS PubMed.
- J. H. Seo and T.-Q. Nguyen, J. Am. Chem. Soc., 2008, 130, 10042–10043 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The DFT calculations, synthetic procedures, DSC/TGA spectra, crystallographic information, XPS, UPS and SEM data. CCDC 964324 and 964325. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4tc00002a |
| This journal is © The Royal Society of Chemistry 2014 |