 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid phase synthesis of functionalised SAM-forming alkanethiol–oligoethyleneglycols†
James
Murray
ab,
Dominika
Nowak
bc,
Laurynas
Pukenas
bd,
Rizuan
Azhar
e,
Mathieu
Guillorit
a,
Christoph
Wälti
c,
Kevin
Critchley
bd,
Steven
Johnson
e and
Robin S.
Bon
*ab
aSchool of Chemistry, University of Leeds, LS2 9JT, UK. E-mail: r.bon@leeds.ac.uk
bAstbury Centre for Structural Molecular Biology, University of Leeds, UK
cSchool of Electronic and Electrical Engineering, University of Leeds, UK
dSchool of Physics and Astronomy, University of Leeds, UK
eDepartment of Electronics, University of York, Heslington York, YO10 5DD, UK
First published on 23rd April 2014
Abstract
We present an efficient solid phase synthesis methodology that provides easy access to a range of functionalised long-chain alkanethiol–oligoethyleneglycols that form well-defined self-assembled monolayers on gold and are compatible with pre- or post-assembly conjugation of (bio)molecules. We demonstrate the versatility of our synthetic route by synthesising LCAT–OEGs with a range of functional moieties, including peptides, electro-active redox groups, chemical handles for post-assembly conjugation of (bio)molecules, and demonstrate the application of our LCAT–OEG monolayers in immunosensing, where they show good biocompatibility with minimal biofouling.
Self-assembled monolayers (SAMs) of functionalised long-chain alkanethiolates (LCATs) on metals1 provide an excellent and versatile platform for the chemoselective immobilisation of (bio)molecules on planar and nanoparticle surfaces. Their well-defined thickness, structure, and dielectric properties2 make them ideally suited for applications where a high degree of control of the physical and chemical properties of surfaces is required. Functionalised LCATs also enable the creation of patterned surfaces3 critical for protein and DNA microarrays. SAMs used for protein biosensing applications usually require LCATs incorporating oligoethylene glycol (OEG) linkers to help reduce non-specific protein adsorption4 and functional groups that allow the further tuning of surface chemistry providing control over the immobilisation, density and orientation of capture molecules. Thus, easy access to a variety of functionalised LCAT–OEGs is essential for optimisation of, for example, immobilisation chemistry and OEG and alkanethiolate length. However, commercial availability of functionalised LCAT–OEGs is severely limited and synthetic routes often laborious. In this study, we present a versatile solid phase synthesis route that provides easy access to a range of functionalised LCAT–OEGs. Using a range of surface characterisation techniques including surface plasmon resonance (SPR) and electrochemical spectroscopy, we demonstrate that the resulting LCAT–OEGs form dense, well-defined mixed SAMs of high quality, which are compatible with on-surface bioorthogonal ligation reactions.
Customised SAMs presenting ligands and/or biomolecules can be prepared according to two general strategies (Fig. 1): (i) conjugation of (bio)molecules to LCAT–OEGs after SAM formation (post-assembly conjugation); (ii) total synthesis of functionalised LCAT–OEGs followed by SAM formation (pre-assembly conjugation). Post-assembly conjugation requires robust, chemoselective, and mild chemistry to ensure complete surface functionalisation without side reactions.2,5 For pre-assembly conjugation approaches, total synthesis of LCAT–OEGs terminated with, amongst others, biotin,6 peptides,6c,7 ferrocene,8 azides,9 aminooxy groups,9 and sugars10 has been demonstrated. Such functionalised LCAT–OEGs are usually synthesised in solution, which requires multiple chromatographic steps and suffers from poor overall yields. Solid phase chemistry presents an attractive alternative for synthesising LCAT–OEGs. The groups of Whitesides and Mrksich have demonstrated the attachment of a thiol-protected LCAT–OEG to resin-bound peptides,7b,11 while Albericio et al. have successfully used 2-chlorotrityl chloride resin to construct biotinylated6b and peptide-containing7a LCAT–OEGs. However, for applications that require a quick, iterative fine-tuning of SAM properties/functionalities, currently available syntheses are often not sufficient. To address this need, we developed a flexible solid phase synthesis that gives access to a range of LCAT–OEGs through late-stage functionalisation, while minimising intermediate purification.
 | ||
| Fig. 1 SAM functionalisation strategies. (a) SAM formation (in the example: a mixed SAM, with a hydroxy-terminated diluent, on a gold surface); (b) conjugation of LCAT 1 with reagent 2 (in which functional groups X and Y react to form linker X′Y′). | ||
OEGs are often linked to LCATs through an ether bond. However, in analogy to Albericio's approach,6b,7a we chose to use an amide linker, which has been shown to improve SAM stability through the formation of networks of hydrogen bonds.12 We first synthesised amino-OEGs 6 through the silver(I) oxide-mediated mono-tosylation of oligoethyleneglycols 4 (ref. 13) followed by tosyl displacement with sodium azide and Staudinger reduction (Scheme 1A).14 Next, 6a, 6b and 7 were coupled to 2-chlorotrityl resin-bound 11-mercaptoundecanoic acid (11-MUA) 9 (Scheme 1B). The terminal alcohol of 10c was tosylated and treated with sodium azide to afford resin-bound azide 12. Reduction of 12 under Staudinger conditions proved sluggish, but proceeded rapidly with tin thiophenolate15 to afford resin-bound amine 14.
 | ||
| Scheme 1 Synthesis of LCAT–OEGs: (A) preparation of amino-OEGs 6a and 6b. (B) Solid phase synthesis of functionalised LCAT–OEGs on 2-chlorotrityl chloride resin 8. (C) On-resin synthesis of peptide-labelled LCAT–OEGs 17 and methylene blue-labelled LCAT–OEG 18. Yields of 11a, 11b, 13, 15, and 16–18 were calculated after chromatography. Insets: Structures of 2-(2-aminoethoxy)ethanol 7 and methylene blue derivative 16. Abbreviations: Ac = acetyl; DCM = dichloromethane; DIC = diisopropylcarbodiimide; DIPEA = N,N-diisopropylethylamine; DMAP = 4-dimethylaminopyridine; DMF = N,N-dimethylformamide; Fmoc = 9-fluorenylmethyloxycarbonyl; HOBt = 1-hydroxybenzotriazole; 11-MUA = 11-mercaptoundecanic acid; SPPS = solid phase peptide synthesis; TES = triethylsilane; TFA = trifluoroacetic acid; Ts = 4-toluenesulfonyl. | ||
Using Fmoc-solid phase peptide synthesis, we demonstrated that 14 is an excellent starting point for the synthesis of LCAT–OEG–peptide conjugates such as 17 (for pre-assembly functionalisation strategies) or LCAT–OEG 18 incorporating the redox probe methylene blue (MB; Scheme 1C). MB was chosen for electrochemical analysis of SAMs (see below) because of its superior aqueous stability compared to ferrocene.16 MB-functionalised LCAT–OEG 18 was synthesised by acylating resin-bound amine 14 with carboxylate 16, which was prepared by adapting a procedure described by Pheeney et al. (Scheme S1.1†).17 Reactions were allowed to run until all resin-bound starting material was consumed, according to LC-MS analysis after micro-cleavage. The immobilised molecules can be cleaved from the solid support at any point using trifluoroacetic acid and triethylsilane (at least one equivalent to scavenge resin-bound trityl cations). After purification by chromatography (either SiO2 or C18), the pure LCAT–OEGs 11a, 11b, 13, 15, 17 and 18 were obtained in good total yields, based on the loading of 2-chlorotrityl chloride resin 8.‡
Next, we analysed the quality of SAMs assembled from our functionalised LCAT–OEGs on sputtered gold surfaces. Using X-ray photoelectron spectroscopy (XPS), we found S 2p peaks of SAMs of 11a, 11b, and 13 to have binding energies consistent with the formation of thiolate bonds (Fig. S2.1†). The C 1s region consisted of three peaks for all SAMs, corresponding to the alkyl chain, the OEG chain, and carbonyls (Fig. 2A). The analysis of the N 1s region for SAMs of 13 was complicated due to rapid degradation of the azide group under XPS conditions (see the ESI†). The azide-terminated SAM 13 resulted in a surface that was of slightly lower surface energy than SAMs of 11a or 11b. This enabled the degree of mixing in the SAMs between diluents 11a or 11b and azide 13 to be estimated by contact angle analysis by applying the Cassie equation18 (Fig. 2B). The insulating properties of SAMs containing different ratios of 11a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 or 11a:17a were assessed by electrochemical impedance spectroscopy (Fig. S3.1 and S3.2†) The minimum phase angles of −88° to −83° for SAMs of 11a:15 and −88° to −87° for SAMs of 11a:17a, measured at 0.1 Hz, correspond to the formation of well-packed, insulating monolayers that are almost free of pinholes and collapsed sites effects. Furthermore, atomic force microscopy (AFM) of a SAM of 11a:17a (1:1) revealed uniform surfaces absent of macroscopic islands resulting from phase separation (Fig. S5.1†). Cyclic voltammetry (CV) measurements of mixed SAMs formed from different ratios of 11b:15 (Fig. S6.1–S6.3†) revealed SAM capacitance values of 4.4 × 10−6 F cm−1, which corresponds well with the capacitance of simple LCAT monolayers.19 Furthermore, the absence of redox peaks due to electron transfer to solution phase redox probe confirms the absence of pin holes within the LCAT–OEG monolayers.
:15 or 11a:17a were assessed by electrochemical impedance spectroscopy (Fig. S3.1 and S3.2†) The minimum phase angles of −88° to −83° for SAMs of 11a:15 and −88° to −87° for SAMs of 11a:17a, measured at 0.1 Hz, correspond to the formation of well-packed, insulating monolayers that are almost free of pinholes and collapsed sites effects. Furthermore, atomic force microscopy (AFM) of a SAM of 11a:17a (1:1) revealed uniform surfaces absent of macroscopic islands resulting from phase separation (Fig. S5.1†). Cyclic voltammetry (CV) measurements of mixed SAMs formed from different ratios of 11b:15 (Fig. S6.1–S6.3†) revealed SAM capacitance values of 4.4 × 10−6 F cm−1, which corresponds well with the capacitance of simple LCAT monolayers.19 Furthermore, the absence of redox peaks due to electron transfer to solution phase redox probe confirms the absence of pin holes within the LCAT–OEG monolayers.
 | ||
| Fig. 2 Characterisation of LCAT–OEG SAMs. (A) XPS data of C 1s region for reference SAMs of 11a (dotted), 11b (dashed) and 13 (solid) normalised to the alkyl chain peak at 284.9 eV. Spectra reflect similar carbonyl (288.3 eV), but varying amount of OEG (286.9 eV) carbon. (B) A linear change in cosine of the contact angle with molar ratio of azide 13 in mixed SAMs was observed. Triangles facing down represent 11a:13; facing up – 11b:13 SAMs; advancing and receding angles are filled and open symbols, respectively. Dotted and solid lines represent the line of best fit for the Cassie equation for contact angles for 11a:13 and 11b:13 SAMs, respectively. (C) Typical cyclic voltammograms (left) for the 2-electron oxidation/reduction of SAM-bound methylene blue (right) at scan rates of 200 (black), 400 (red), 600 (blue) and 800 mV s−1 (green). The inset shows the linear relationships between current and scan rate. | ||
To demonstrate the potential suitability of MB-functionalised LCAT–OEGs for molecular electronics applications, we assessed the redox behaviour of surface-bound MB by CV. CV measurements on SAMs of 18 showed clear oxidation and reduction peaks associated with the MB moiety around −130 mV vs. Ag/AgCl (Fig. 2C). No redox peaks were observed in equivalent CV measurements performed on non-redox active LCAT–OEG SAMs (compounds 15 and 17a, Fig. S6.1–S6.3†). The peaks are reasonably symmetric with only a small peak separation (9 mV at a scan rate of 200 mV s−1), typical of the electrochemical behaviour of a fully reversible redox-active monolayer. Furthermore, the peak anodic and cathodic current was found to increase linearly with scan rate (see inset of Fig. 2C), again characteristic of a surface-tethered redox-active group. From the gradient of the straight line fit to peak current vs. scan rate we calculated a surface coverage, Γ = 8.8 × 1013 molecules per cm2. This is slightly lower than the theoretical density predicted for a perfect SAM formed from LCATs only (4 × 1014 molecules per cm2) and is likely due to the steric hindrance of the bulky OEG and MB groups that inhibit denser monolayer assembly. Finally, the FWHM of the oxidation and reduction peaks was found to be 37 mV (at 200 mV s−1). While this is lower than the theoretical ideal (45.3 mV for a 2 electron process) we note that deviations in the FWHM are not uncommon in densely packed redox-active monolayers due to electrostatic interactions between adjacent charged species.
Confident that our molecules formed well-defined SAMs, we demonstrated that they also allow (bio)molecule immobilisation by common post-assembly conjugation techniques: Copper-catalysed azide-alkyne cycloaddition (CuAAC) and bis(sulfosuccinimidyl) suberate (BS3)-mediated amide coupling.
First, we ‘clicked’ propargyl biotin onto a SAM of 11a:13 (1:1; see ESI†) and tested for the presence of biotin attached covalently to the surface using a colorimetric assay (Fig. 3A). Briefly, we introduced streptavidin-alkaline phosphatase fusion protein to the surface and used western blue as a stain. Only spots where biotin had been successfully linked to the surface were stained purple (Fig. 3A, S7.1 and S7.2†). Finally, we tested the suitability of SAMs containing amine 15 for immobilisation of the clinically relevant human chorionic gonadotropin antibody (anti-hCG).20 A SAM of 11a:15 (1:1) was formed and activated using the bis-succinimide crosslinker BS3 on a Biacore SPR chip (Scheme S8.1†). Injection of anti-hCG gave a much stronger signal in the channel pre-treated with BS3 than in the control channel (no BS3), indicating successful covalent immobilisation of anti-hCG (Fig. 3B). The lack of binding observed on the control channel suggests the LCAT–OEG monolayer is efficient at minimising non-specific adsorption.
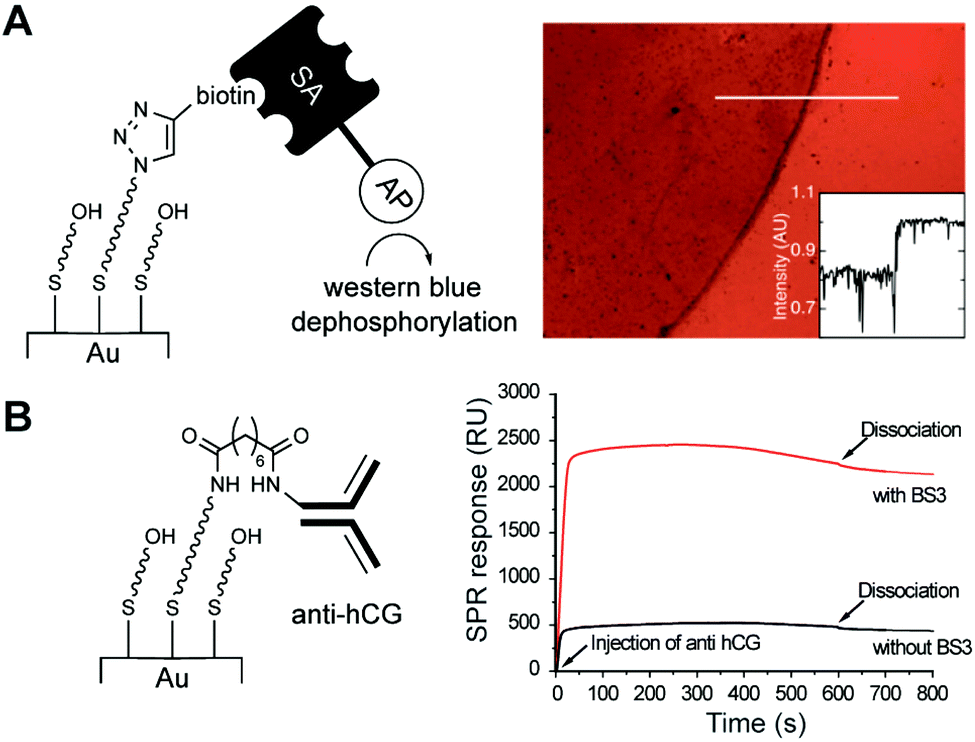 | ||
| Fig. 3 Post-assembly conjugation: (A) biotin immobilised through CuAAC on SAMs of 11a:13 (1:1; left) was detected colorimetrically, as purple spots (right), upon incubation of the gold slide with streptavidin-alkaline phosphatase (SA-AP) and western blue. (B) Immobilisation of anti-hCG on a BS3-activated amine SAM (11a:15, 1:1; left) was detected by SPR (right). | ||
In conclusion, we have established a robust and versatile solid phase synthesis of LCAT–OEGs functionalised with alcohols, azides, amines, peptides, and the redox probe methylene blue, thereby significantly expanding the range of LCAT–OEGs that are easily accessible through solid phase approaches such as those reported by Albericio.6b,7a All LCAT–OEGs could be isolated in good yields as pure materials, and were used to form dense and well-defined, high quality mixed SAMs absent of macroscopic island formation (AFM data) and pinholes (EIS and CV data). In addition, the ratios of functionalised LCAT–OEGs and diluents on the surface were directly proportional to the ratios of these molecules in the applied solutions. Finally, we demonstrated that in addition to total synthesis of functional LCAT–OEGs for pre-assembly conjugations, our solid phase methodology also allows the use of bioorthogonal post-assembly conjugation techniques.
Acknowledgements
This research was funded by the Biomedical and Health Research Centre of the University of Leeds (RSB and SJ), a Henry Ellison PhD Studentship (JM), EPSRC Grants EP/J010731/1 (RSB, DN and SJ), EP/J01513X/1 (KC), and EP/I014039/1 (LP), the Centre for Chronic Diseases and Disorders (C2D2; RA), the University of York (SJ), and WELMEC, a Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC, under grant number WT 088908/Z/09/Z (DN and CW). We thank Dr. Simon White for assistance with SPR measurements and Prof. Stephen Evans for assistance with interpretation of XPS data.Notes and references
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- (a) P. Jonkheijm, D. Weinrich, H. Schröder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS PubMed; (b) I. Zaccari, B. G. Catchpole, S. X. Laurenson, A. G. Davies and C. Wälti, Langmuir, 2014, 30, 1321–1326 CrossRef CAS PubMed.
- X. Zhou, F. Boey, F. Huo, L. Huang and H. Zhang, Small, 2011, 7, 2273–2289 CrossRef CAS PubMed.
- K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164–1167 CrossRef CAS.
- (a) J. Li, P. S. Thiara and M. Mrksich, Langmuir, 2007, 23, 11826–11835 CrossRef CAS PubMed; (b) D. Samanta and A. Sarkar, Chem. Soc. Rev., 2011, 40, 2567–2592 RSC; (c) N. Laurent, R. Haddoub, J. Voglmeir, S. C. C. Wong, S. J. Gaskell and S. L. Flitsch, ChemBioChem, 2008, 9, 2592–2596 CrossRef CAS PubMed.
- (a) C. Booth, R. J. Bushby, Y. Cheng, S. D. Evans, Q. Liu and H. Zhang, Tetrahedron, 2001, 57, 9859–9866 CrossRef CAS; (b) E. Prats-Alfonso, F. García-Martín, N. Bayo, L. J. Cruz, M. Pla-Roca, J. Samitier, A. Errachid and F. Albericio, Tetrahedron, 2006, 62, 6876–6881 CrossRef CAS PubMed; (c) R. Derda, D. J. Wherritt and L. L. Kiessling, Langmuir, 2007, 23, 11164–11167 CrossRef CAS PubMed.
- (a) E. Prats-Alfonso, S. Oberhansl, A. Lagunas, E. Martínez, J. Samitier and F. Albericio, Eur. J. Org. Chem., 2013, 7, 1233–1239 CrossRef; (b) B. T. Houseman and M. Mrksich, J. Org. Chem., 1998, 63, 7552–7555 CrossRef CAS.
- P. A. Bertin, M. J. Ahrens, K. Bhavsar, D. Georganopoulou, M. Wunder, G. F. Blackburn and T. J. Meade, Org. Lett., 2010, 12, 3372–3375 CrossRef CAS PubMed.
- Z. P. Tolstyka, W. Richardson, E. Bat, C. J. Stevens, D. P. Parra, J. K. Dozier, M. D. Distefano, B. Dunn and H. D. Maynard, ChemBioChem, 2013, 14, 2464 CrossRef CAS PubMed.
- M. Kleinert, N. Röckendorf and T. K. Lindhorst, Eur. J. Org. Chem., 2004, 18, 3931–3940 CrossRef.
- D. Ryan, B. A. Parviz, V. Linder, V. Semetey, S. K. Sia, J. Su, M. Mrksich and G. M. Whitesides, Langmuir, 2004, 20, 9080–9088 CrossRef CAS PubMed.
- (a) S.-W. Tam-Chang, H. A. Biebuyck, G. M. Whitesides, N. Jeon and R. G. Nuzzo, Langmuir, 1995, 11, 4371–4382 CrossRef CAS; (b) S. Svedhem, S. C. T. Svensson and B. Liedberg, Langmuir, 1999, 15, 3390–3394 CrossRef.
- A. Bouzide and G. Sauve, Org. Lett., 2002, 4, 8781–8783 Search PubMed.
- S. Svedhem, C. A. Hollander, J. Shi, P. Konradsson, B. Liedberg and S. C. Svensson, J. Org. Chem., 2001, 66, 4494–4503 CrossRef CAS PubMed.
- M. Bartra, P. Romea, F. Urpi and J. Vilarrasa, Tetrahedron, 1990, 46, 587–594 CrossRef CAS.
- D. Kang, X. Zuo, R. Yang, F. Xia, K. W. Plaxco and R. J. White, Anal. Chem., 2009, 81, 9109–9113 CrossRef CAS PubMed.
- C. G. Pheeney and J. K. Barton, Langmuir, 2012, 28, 7063–7070 CrossRef CAS PubMed.
- A. B. D. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546–551 RSC.
- M. S. Góes, H. Rahman, J. Ryall, J. J. Davis and P. R. Bueno, Langmuir, 2012, 28, 9689–9699 CrossRef PubMed.
- L. A. Cole, Clin. Chem., 1997, 43, 2233–2243 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of compounds, XPS, EIS, AFM, contact angle measurements, CV, colorimetry and SPR. See DOI: 10.1039/c4tb00573b |
| ‡ We routinely perform the synthesis of 11a, 11b, 13, 15, 17 and 18 on a 0.1–0.6 mmol scale per SPE tube, which provides plenty material for optimisation studies with SAMs. Instead of using chromatography, LCAT–OEGs incorporating basic amines can also be purified by precipitation (as their TFA salts) into mixtures of diethyl ether and hexane. |
| This journal is © The Royal Society of Chemistry 2014 |