A ruthenium(III) phosphonate complex on polyallylamine functionalized carbon nanotube multilayer films: self-assembly, direct electrochemistry, and electrocatalysis†
Received
14th October 2013
, Accepted 31st October 2013
First published on 1st November 2013
Abstract
On the basis of the electrostatic and donor–acceptor interactions between carboxylated multiwalled carbon nanotubes (MWCNT-COOH) and polyallylamine hydrochloride (PAH), PAH functionalized MWCNT multilayer films ({PAH/MWCNT-COOH}n) were readily formed on a glassy carbon (GC) electrode surface through a layer-by-layer (LBL) self-assembly method. Then, the PAH functionalized MWCNT multilayer films were used as a functional interface to effectively immobilize the ruthenium(III) ethylenediamine-tetramethylene phosphonate (EDTMP–RuIII) complex through the strong electrostatic and/or hydrogen bonding interactions between –NH2 and –PO3H2 groups. Fourier transform infrared spectroscopy (FT-IR), ultraviolet-visible spectroscopy (UV-vis), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), energy dispersive spectrum (EDS) mapping, and Raman measurements were used to characterize the self-assembly process, structure, composition and morphology of the EDTMP–RuIII/{PAH/MWCNT-COOH}n/GC electrode. In the EDTMP–RuIII/{PAH/MWCNT-COOH}n/GC electrode, the immobilized EDTMP–RuIII complex could directly exchange electrons with the substrate electrode and showed excellent electrocatalytic activity towards iodate reduction. Thus, the fabricated EDTMP–RuIII/{PAH/MWCNT-COOH}n/GC electrode could be used as an electrochemical sensor for iodate detection.
Introduction
The chemically designed inorganic–organic hybrid materials have attracted a considerable amount of attention because of the combination of properties with respect to the inorganic and organic components.1–4 Carbon nanotubes (CNTs), a star nanostructure in the carbon family, have attracted a great amount of interest in both academic and industrial sectors, owing to their extraordinary physical, chemical and mechanical properties such as high electrical conductivity, excellent chemical stability, large surface area, and good corrosion resistance.5–8 The integration of CNTs and polymers can impart CNTs with new interesting electrical, optical and chemical properties.9–18 In the methods of fabricating inorganic–organic hybrid material films, layer-by-layer (LBL) self-assembly has become a very promising technique due to its versatility, simplicity and robustness, which generally involves the alternative adsorption of charged polyelectrolytes and nanomaterials with opposite charge on various solid substrates.19–23 Until now, various CNT/polymer films with unique physical/chemical properties and precise control over film composition and thickness have been developed by LBL self-assembly techniques, which are widely applied in various fields, including chemical sensors,24,25 electrochemical sensors,26–29 and biological sensors,9,30 electrochemical capacitors,31,32 stimuli-responsive functional materials,33 Li-ion batteries,34 and so on. In particular, CNT/polymer ultrathin films have considerable potential applications in various electrochemical sensors because CNTs not only facilitate electron transfer but also serve as a good scaffold for the immobilization of catalytic materials.26–29
Iodine is a necessary micronutrient playing an important role in cell growth and brain function. A deficiency of iodine can result in a serious delay in neurologic development.35 In contrast, an excess of iodate can cause goiter, hypothyroidism and hyperthyroidism.36 Thus, a reliable, accurate, sensitive, rapid, and low-cost determination of iodate is important in a variety of areas such as foods,37 drugs,38 and industrial applications.39 Among various analytical techniques such as spectrophotometry,40,41 chromatography,42 capillary electrophoresis,43 and electrochemistry,44 the electrochemical sensing of iodate is especially attractive due to its simplicity, cost-effectiveness, fast response, high sensitivity, and ease of miniaturization. Until now, various metal nanoparticles (Pt45 and Au46), metal oxides (W,47 Mo,48 and Ru49), metal complexes (V,50 Os,51 and Fe52) and enzyme (catalase53) have been successfully used to fabricate the iodate electrochemical sensors. To the best of our knowledge, among the electrochemical methods for the determination of iodate, no Ru-complex based iodate electrochemical sensor has been reported.
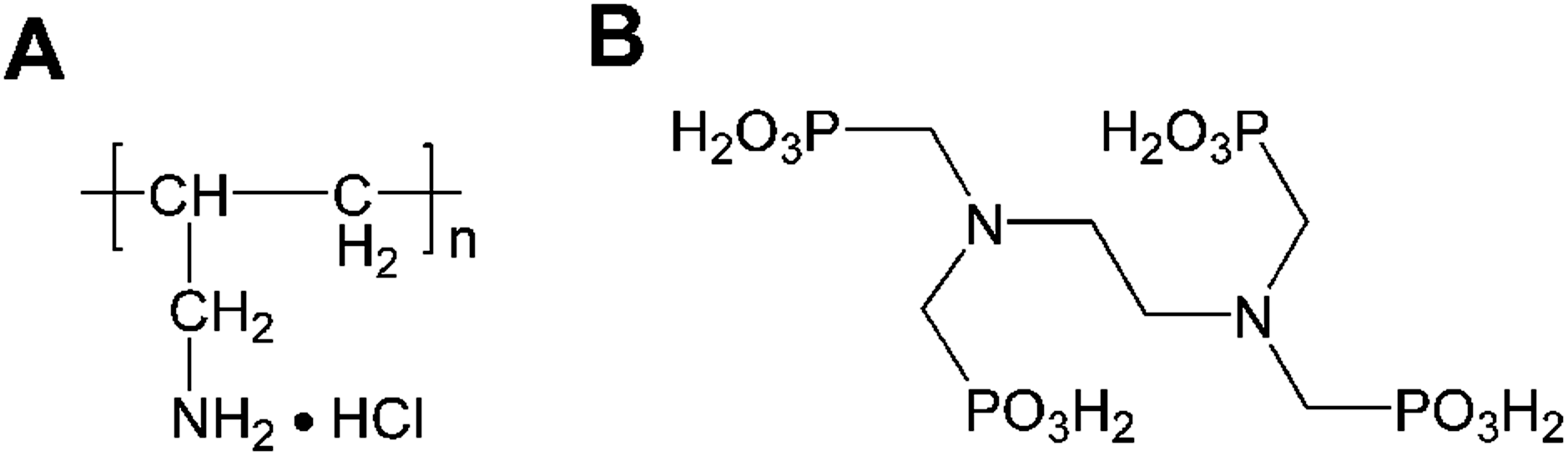
Polyallylamine hydrochloride (PAH, shown in Scheme 1), a positively charged polyelectrolyte with a large number of primary amine groups, has excellent coordination capability, good hydrophilic property, high chemical stability, and strong adhesion ability on a carbon material surface.54–57 In the present work, we demonstrate a simple strategy for fabricating PAH/CNT multilayer films through a LBL self-assembly method. Then, the corresponding PAH/CNT multilayer films were used to immobilize the ruthenium(III) ethylenediamine-tetramethylene phosphonate (EDTMP–RuIII) complex. Various physical characterizations were used to investigate the self-assembly process, composition, morphology and structure of the hybrid nanomaterial films. Furthermore, the direct electrochemistry and electrocatalysis of the modified electrode were investigated by using various electrochemical techniques, including cyclic voltammetry, multipotential step, and chronoamperometry.
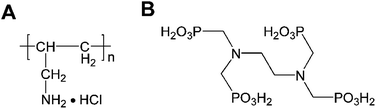 |
| | Scheme 1 The structures of (A) PAH and (B) EDTMP. | |
Experimental
Chemicals and reagents
The highly pure carboxylated multiwalled carbon nanotubes (MWCNT-COOH) (>99.9 wt% purity; outer diameter: 30–50 nm; surface area: >30 m2 g−1) used in this study were purchased from Chengdu Organic Chemicals Co. Ltd, Chinese Academy of Sciences (Chengdu, China). Polyallylamine hydrochloride (PAH, average molecular weight 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, shown in Scheme 1A) was supplied from Nitto Boseki Co., Ltd (Tokyo, Japan). Ruthenium(III) chloride (RuCl3) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Ethylenediamine-tetramethylene phosphonic acid (EDTMP, shown in Scheme 1B) was purchased from Shandong Taihe Water Treatment Co., Ltd (Shandong, China). Solution of pH 2.0 K2SO4–H2SO4 was prepared from 0.1 M K2SO4 by adjustment with 0.5 M H2SO4. Other reagents were of analytical reagent grade and used as received. All the aqueous solutions were prepared with Millipore water having a resistivity of 18.2 MΩ.
000, shown in Scheme 1A) was supplied from Nitto Boseki Co., Ltd (Tokyo, Japan). Ruthenium(III) chloride (RuCl3) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Ethylenediamine-tetramethylene phosphonic acid (EDTMP, shown in Scheme 1B) was purchased from Shandong Taihe Water Treatment Co., Ltd (Shandong, China). Solution of pH 2.0 K2SO4–H2SO4 was prepared from 0.1 M K2SO4 by adjustment with 0.5 M H2SO4. Other reagents were of analytical reagent grade and used as received. All the aqueous solutions were prepared with Millipore water having a resistivity of 18.2 MΩ.
LBL self-assembly of PAH/MWCNT-COOH multilayer films
A glassy carbon (GC) electrode (3 mm diameter) was used as the substrate electrode. The GC electrode was polished first with emery paper and then with aqueous slurries of fine alumina powders (0.3 and 0.05 μm) on a polishing cloth, and was finally cleaned with Millipore water and ethanol under an ultrasonic bath, each for 5 min. LBL assembly of PAH and MWCNT-COOH on the GC electrode was conducted by first immersing the GC substrate into positively charged PAH (0.5 wt%) aqueous solution for 30 min, and then alternatively dipping the PAH/GC electrode into 0.6 mg mL−1 aqueous dispersion of the MWCNT-COOH and 0.5 wt% PAH each for 30 min. After every layer deposition, the electrode was repeatedly immersed into Millipore water 2–3 times to remove the excess of assembling materials and then dried with nitrogen gas. The obtained electrode is referred to as {PAH/MWCNT-COOH}n/GC, (n denotes the number of PAH/MWCNT-COOH layers). For a typical demonstration, the GC substrate modified with eight layers of PAH and MWCNT-COOH (hereafter designated as {PAH/MWCNT-COOH}8/GC) was used for the electrochemical measurements and for the fabrication of the iodate electrochemical sensor.
Synthesis and immobilization of the EDTMP–RuIII complex
EDTMP–RuIII complex solution was obtained by adjusting the solution pH of the mixture of 5 mM EDTMP and 5 mM RuCl3 to 9.0 and mechanically stirred for 8 h. Immobilization of the EDTMP–RuIII complex on the {PAH/MWCNT-COOH}8/GC electrode was accomplished by immersing the {PAH/MWCNT-COOH}8/GC electrode in a 5 mM EDTMP–RuIII complex solution for 24 h at room temperature. Then, the electrode was rinsed with Millipore water to remove the non-firmly adsorbed EDTMP–RuIII complex and allowed to dry at ambient temperature. The fabricated electrode is denoted as the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode and stored in air when not in use.
Physical characterization
High-resolution X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Thermo VG Scientific ESCALAB 250 spectrometer with an Mg Kα radiator, and the vacuum in the analysis chamber was maintained at about 10−9 mbar or lower. The binding energy was calibrated by means of the C1s peak energy of 284.6 eV. Raman spectra of carbon materials were examined with a Labram HR 800 UV Raman spectrometer, equipped with a confocal microscope and an Ar ion laser (λ = 514.5 nm) to probe the sp2− (ordered) and sp3− (disordered) hybridized C peaks. Fourier transform infrared (FT-IR) measurements were carried out using a Nicolet 520 SXFTIR spectrometer. Ultraviolet-visible spectroscopy (UV-vis) measurements were performed at room temperature on a Shimadzu UV3600 spectrophotometer. Scanning electron microscopy (SEM) images were obtained using a Hitachi S4800 scanning electron microscope.
Electrochemical measurements
All electrochemical experiments were performed by using a CHI 660B electrochemical analyzer (CH Instruments, Shanghai Chenhua Co.) in a conventional three-electrode electrochemical cell. A Pt plate auxiliary electrode and a saturated calomel reference electrode (SCE) were used. All potentials in this study were reported with respect to the SCE. Prior to the electrochemical measurements, N2 was bubbled through the solution for 10 min to remove the dissolved O2. During experiments, a continuous N2 flow was maintained over the solution. All the electrochemical measurements were carried out at 25 ± 1 °C in a water-jacketed glass vessel connected to a circulating water bath.
Results and discussion
LbL self-assembly of PAH/MWCNT-COOH multilayer films
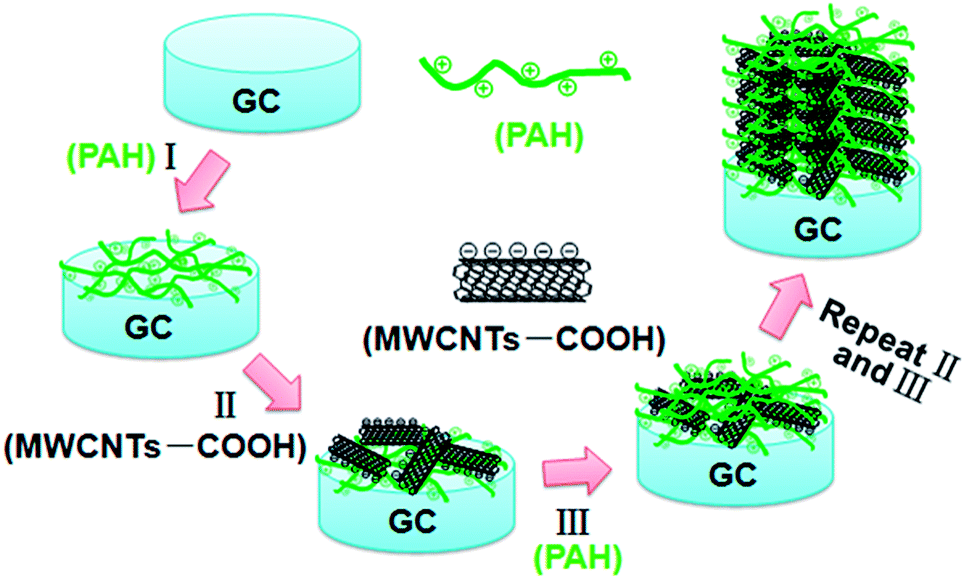
The strategy for the synthesis of PAH/MWCNT-COOH multilayer films (i.e., {PAH/MWCNT-COOH}n, n denotes the number of PAH/MWCNT-COOH layers) is described in detail in the Experimental section. The schematic diagram of the stepwise self-assembling procedure of the {PAH/MWCNT-COOH}n/GC electrode is shown in Scheme 2.
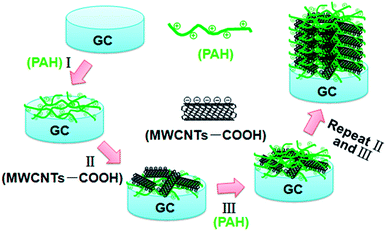 |
| | Scheme 2 Schematic illustration for preparation of the {PAH/MWCNT-COOH}n/GC electrode. | |
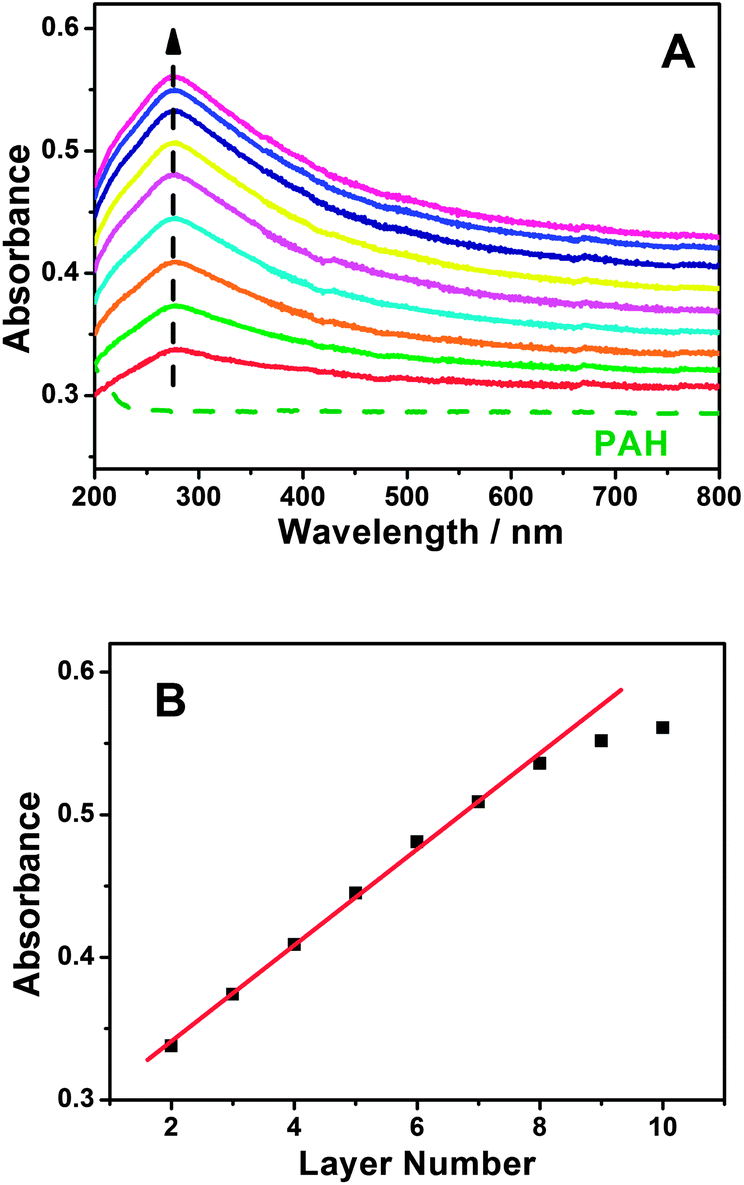
A GC electrode is used as the optimization substrate because PAH can irreversibly adsorb onto the carbon material surface through the donor–acceptor interaction.57–60 Meanwhile, PAH is positively charged whereas MWCNT-COOH is negatively charged in water solution, which results in strong electrostatic interactions between PAH and MWCNT-COOH. The evidence for the interactions between PAH and MWCNT-COOH was provided by XPS. After incubating the MWCNT-COOH in PAH solution for 30 min at room temperature, the N1s peaks (400.5 eV) are clearly observed for the PAH/MWCNT-COOH hybrids (Fig. S1, ESI†). Thus, the positively charged PAH molecules can bind easily the negatively charged MWCNT-COOH to the PAH/GC electrode surface and vice versa, which definitely allows LBL self-assembly between PAH and MWCNT-COOH. The self-assembly process of {PAH/MWCNT-COOH}n films was characterized by UV-vis. The spectrum of PAH is featureless whereas MWCNT-COOH shows characteristic absorption bands at ca. 277 nm (Fig. S2, ESI†). Thus, the remarkable absorption peak at 277 nm on {PAH/MWCNT-COOH}n films originates from the absorption peak of MWCNT-COOH (Fig. 1A). The clear increase in the absorbance with the assembling step is indicative of the film deposition on the substrate (Fig. 1A), and the linearity between the absorbance with the layer number of the PAH/MWCNT-COOH (Fig. 1B) further suggests that almost the same amount of MWCNT-COOH is loaded in each assembling step. As the assembly layer number increases to 9, the absorbance of {PAH/MWCNT-COOH}n films deviates from the straight line.
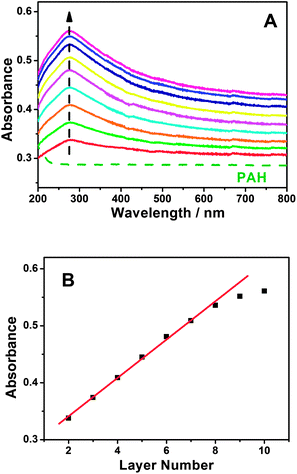 |
| | Fig. 1 (A) UV-vis spectra of {PAH/MWCNT-COOH}n films assembled on a quartz slide. The dashed curve represents the UV-vis spectrum for the PAH-modified quartz slide. (B) Plot of the absorbance at 277 nm versus the layer numbers. | |
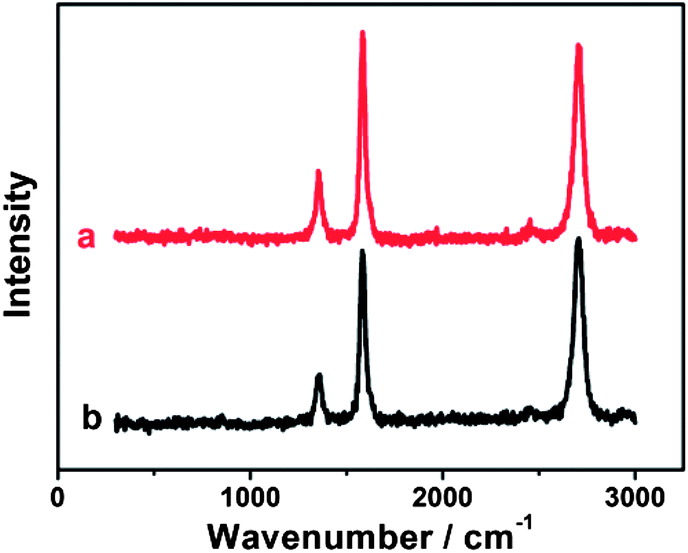
Compared with chemical modification of CNTs,61 the noncovalent modification of CNTs generally preserves the electronic structure of pristine CNTs.62 Raman spectroscopy is the most useful tool for examining the changes in the electronic structure of CNTs. The initial MWCNT-COOH exhibits three characteristic peaks at 1356.2, 1583.2 cm−1 and 2703.7 cm−1 (Fig. 2a), corresponding to the D, G, and 2D bands, respectively. Such three bands are also strongly observed for {PAH/MWCNT-COOH}8 films (Fig. 2b). The intensity ratio of the D band to the G band of CNTs, denoted as ID/IG, directly indicates the structural change of CNTs after hybridization. The ID/IG ratios of the pristine MWCNT-COOH and {PAH/MWCNT-COOH}8 are 0.30 and 0.34, respectively. The result indicates that MWCNT-COOH in {PAH/MWCNT-COOH}8 films almost completely preserves the electronic integrity of initial MWCNT-COOH, which facilitates the electron transfer in the electrochemical reactions.
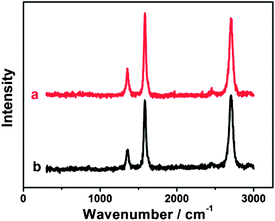 |
| | Fig. 2 Raman spectra of (a) pristine MWCNT-COOH and (b) {PAH/MWCNT-COOH}8 films. | |
Interaction between EDTMP and RuCl3
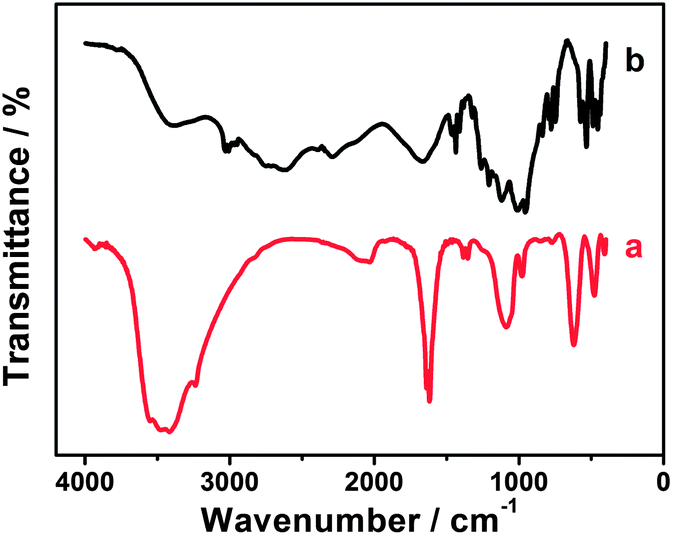
It is well known that EDTMP molecules possess a high coordinating capability and can form chelates with various metal ions such as EIII, TbIII, MnII, FeII, CoII, NiII, CdII, CuII, ZnII and PdII.63–68 Thus, we presume that EDTMP may interact with RuCl3 to generate the EDTMP–RuIII complex under appropriate conditions. When the pH of the mixture solution of EDTMP and RuCl3 is adjusted to 9.0, a black solution is obtained (Fig. 3a). In a controlled experiment, a lot of black Ru(OH)3 flocculent precipitate is generated immediately in a few seconds once adjusting the pH value of single-component RuCl3 solution to 9.0 (Fig. 3b). These above experimental phenomena confirm that EDTMP can interact with RuCl3 to generate the EDTMP–RuIII complex via coordination interactions under basic conditions. The interactions between EDTMP and RuCl3 can be further confirmed by the FT-IR measurement. After addition of ethanol to the EDTMP–RuIII complex solution, a black EDTMP–RuIII complex precipitate appears. The collected EDTMP–RuIII complex sample shows clear characteristic peaks of phosphonic acid groups located at 950–1300 cm−1 (Fig. 4a),69 which is consistent with those of bulk EDTMP (Fig. 4b). Thus, the FT-IR measurements also confirm that EDTMP can interact with RuCl3.
 |
| | Fig. 3 Photographs of a (a) mixture of EDTMP and RuCl3 solution and (b) single-component RuCl3 solution after adjusting the solution pH to 9.0. | |
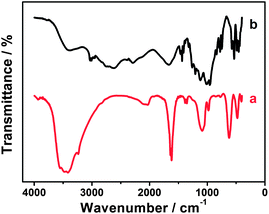 |
| | Fig. 4 FT-IR spectra of the (a) EDTMP–RuIII complex and (b) bulk EDTMP. | |
Characterization of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode
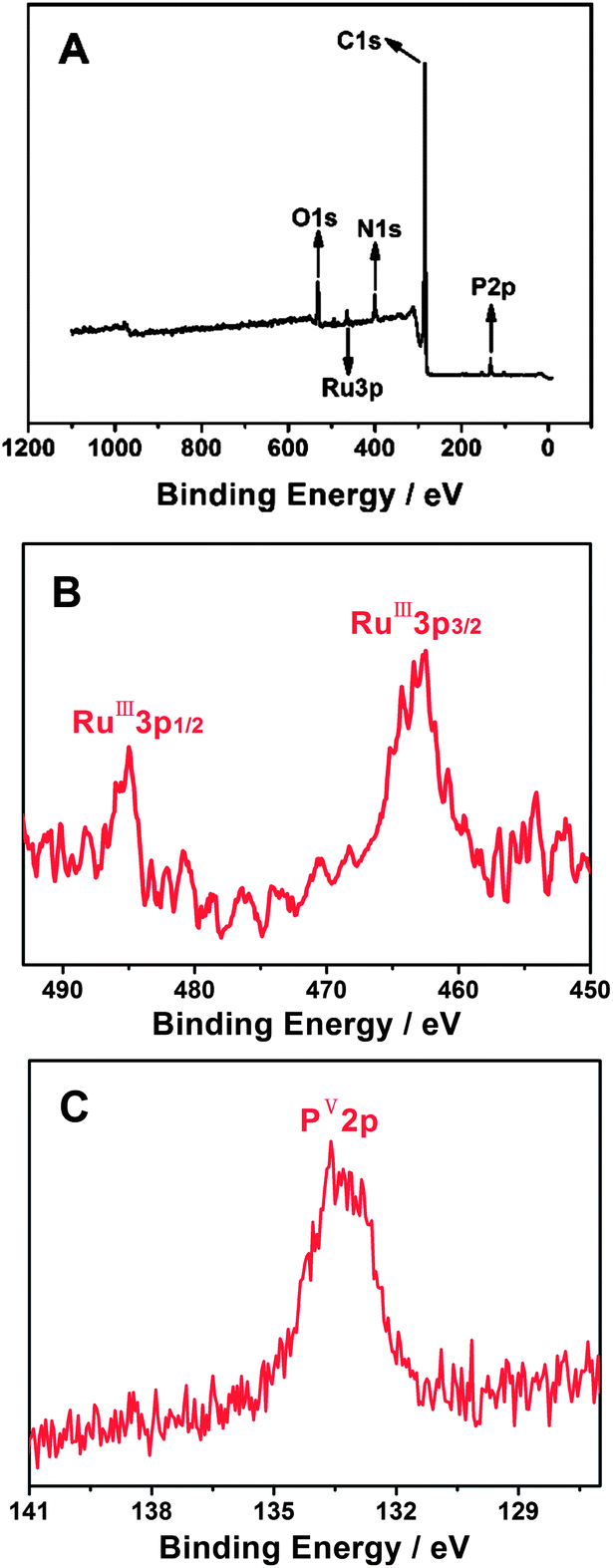
Similar to the structures of various EDTMP–metal complexes,63–65 the EDTMP–RuIII complex contains numerous uncoordinated –P–OH groups due to the particular polydentate coordination property of the EDTMP ligand. The uncoordinated –P–OH groups impart the EDTMP–RuIII complex with excellent self-assembly capacity on the PAH functionalized CNT surface, owing to the strong electrostatic and/or hydrogen bonding interactions between –P–OH groups and –NH2 groups.70–72 XPS was used to monitor the characteristic elements of the EDTMP–RuIII complex and to confirm the self-assembly of the EDTMP–RuIII complex on the {PAH/MWCNT-COOH}8/GC electrode. After immersing the {PAH/MWCNT-COOH}8/GC electrode into EDTMP–RuIII complex solution for 24 h, the appearance of RuIII3p3/2 (462.7 eV), RuIII3p1/2 (485.0 eV) and PV2p (133.6 eV) peaks confirms the successful immobilization of EDTMP–RuIII on the {PAH/MWCNT-COOH}8/GC electrode (Fig. 5), which originates from electrostatic and/or hydrogen bonding interactions between –P–OH groups in the EDTMP–RuIII complex and –NH2 groups in PAH/MWCNT-COOH hybrids.
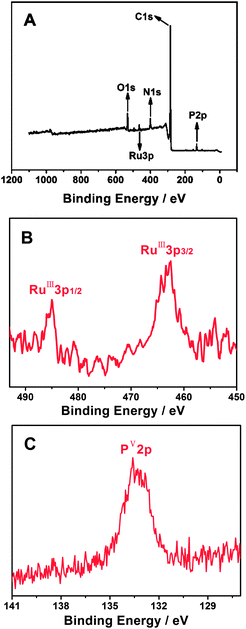 |
| | Fig. 5 XPS spectra of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC: survey scan (A) and narrow scan for Ru (B) and P (C) elements. | |
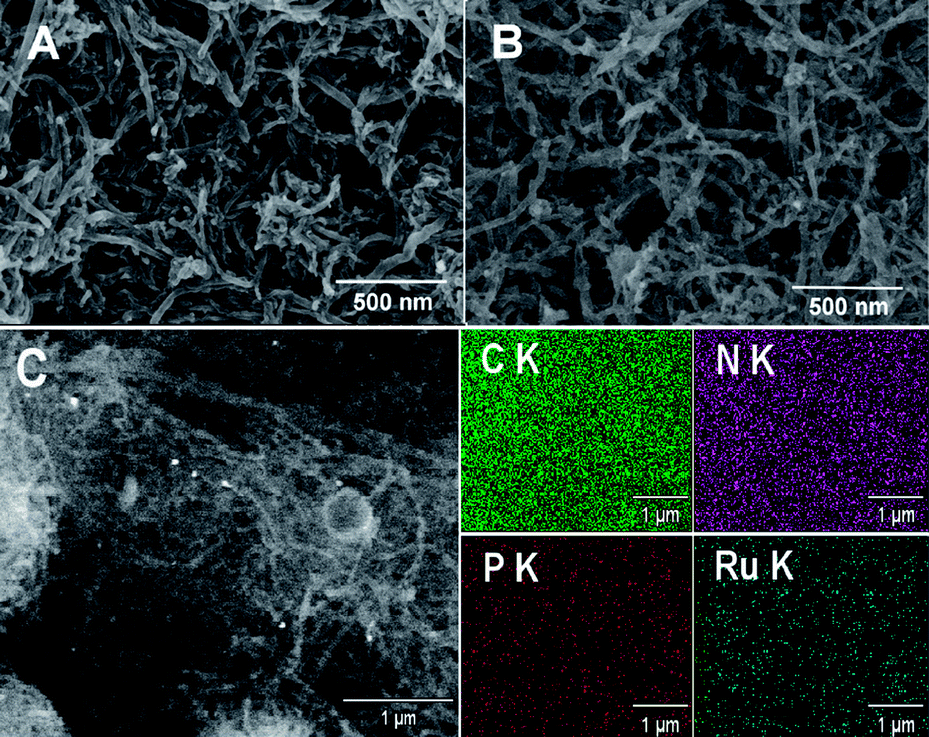
The morphology of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode was investigated by SEM. Compared with the {PAH/MWCNT-COOH}8/GC electrode (Fig. 6A), the EDTMP–RuIII complex located at the {PAH/MWCNT-COOH}8/GC electrode surface is not visible (Fig. 6B), which may originate from the small molecule size of the EDTMP–RuIII complex. However, the large-area EDS mapping patterns show that Ru and P elements are uniformly distributed on the {PAH/MWCNT-COOH}8/GC surface (Fig. 6C), confirming that the EDTMP–RuIII complex is successfully adsorbed onto the {PAH/MWCNT-COOH}8/GC electrode surface. Similar to the {PAH/MWCNT-COOH}8/GC electrode (Fig. 6A), the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode shows a three-dimensionally network-like structure (Fig. 6B). Obviously, the interconnected MWCNTs can provide continuous conducting pathways for the electron transportation. Meanwhile, the highly porous structure can essentially increase the electrode area and facilitate mass transport of analytes, and consequently increase the sensitivity of electrochemical response.60,73–78
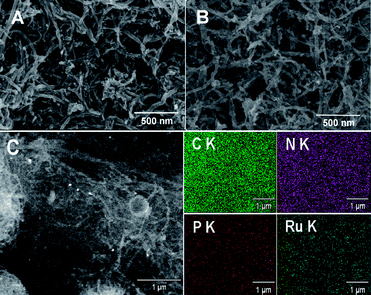 |
| | Fig. 6 Typical SEM images of the (A) {PAH/MWCNT-COOH}8/GC electrode and (B) EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode. (C) Representative large-area SEM image of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode and the corresponding EDS mapping patterns. | |
Direct electrochemistry of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode
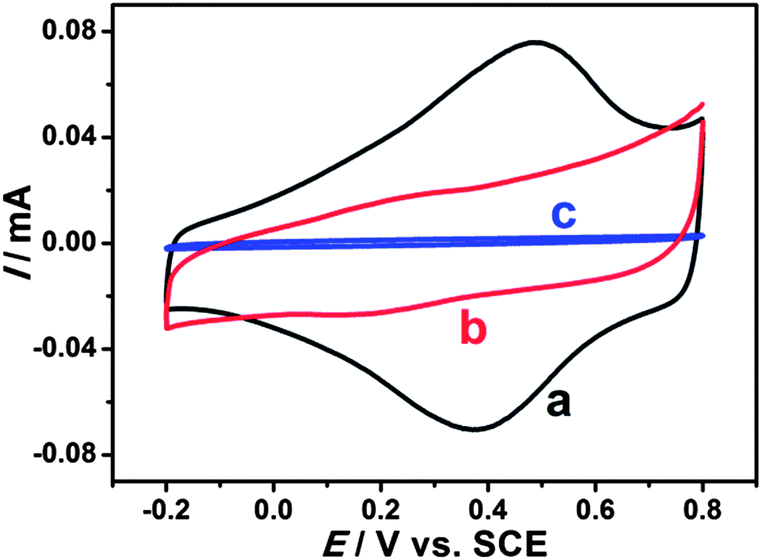
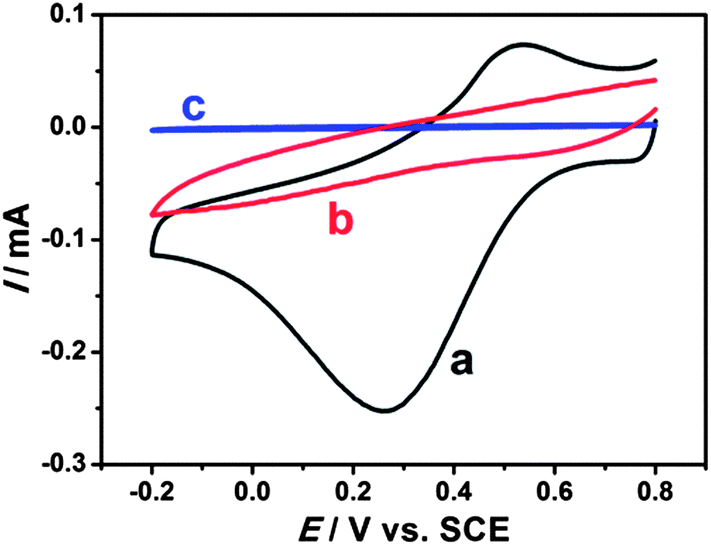
Electrochemical properties of modified electrodes were investigated by cyclic voltammetry. Well-defined and nearly reversible redox peaks with formal potentials of 0.431 V are obtained at the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode (Fig. 7a), while no peak is observed at {PAH/MWCNT-COOH}8/GC and GC electrodes (Fig. 7b and c). These results suggest that the electrochemical response of EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC originates from the direct electron transfer of the immobilized EDTMP–RuIII complex. With the increase of scan rate, the anodic and cathodic peak currents of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode increase linearly (Fig. S3, ESI†), showing a surface-controlled electrode reaction. For a surface process, according to Laviron's equation, the charge-transfer rate constant of the EDTMP–RuIII at the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode is estimated to be 0.71 s−1 at a scan rate of 0.1 V s−1. Moreover, the peak-to-peak separation values of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode increase with scan rate, indicating that the direct electrochemistry of the immobilized EDTMP–RuIII complex is a quasi-reversible reaction. In addition, the stability of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode is tested by cyclic voltammetry. After successive scanning for 100 cycles at a scan rate of 100 mV s−1, no obvious decrease of the peak current was found (Fig. S4, ESI†).
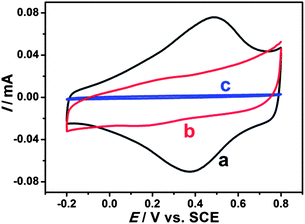 |
| | Fig. 7 Cyclic voltammograms of (a) EDTMP–Ru(III)/{PAH/MWCNT-COOH}8/GC, (b) {PAH/MWCNT-COOH}8/GC, and (c) GC electrodes in a 0.1 M pH 2.0 K2SO4–H2SO4 solution at a scan rate of 100 mV s−1. | |
Electrocatalysis of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode towards iodate reduction
In order to investigate the electrocatalytic behavior of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode towards iodate reduction, cyclic voltammetry measurements were performed in pH 2.0 K2SO4–H2SO4 solution containing 0.80 mM KIO3.46,51,52 A pronounced reduction peak occurs at 0.26 V at the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC (Fig. 8a), but no electrochemical response is observed at {PAH/MWCNT-COOH}8/GC and bare GC electrodes in the potential range from −0.2 to 0.8 V (Fig. 8b and c). These results indicate that the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode exhibits effective electrocatalytic activity for iodate reduction, and the EDTMP–RuIII complex in the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode is responsible for iodate electroreduction.
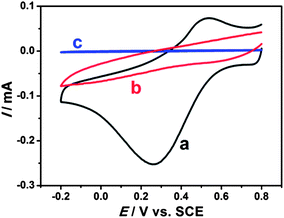 |
| | Fig. 8 Cyclic voltammograms of (a) EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC, (b) {PAH/MWCNT-COOH}8/GC and (c) GC electrodes in a 0.1 M pH 2.0 K2SO4–H2SO4 solution containing 0.80 mM KIO3 at a scan rate of 100 mV s−1. | |
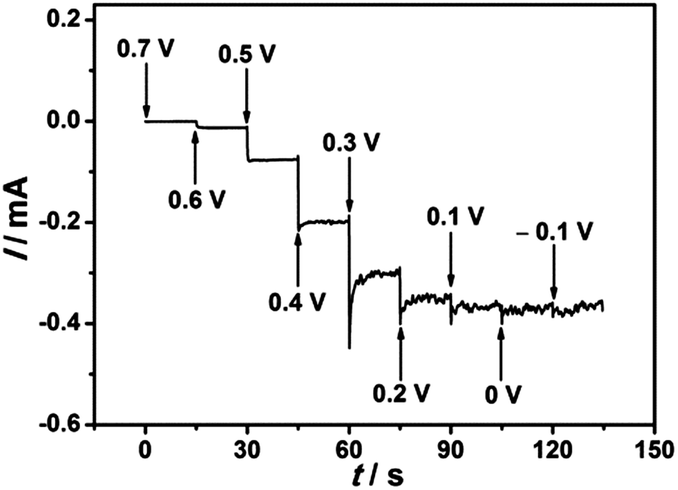
The iodate reduction at the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode was further explored for application as an amperometric iodate sensor. To investigate the effect of applied potential on the iodate reduction current, the multipotential step test was performed. As shown in Fig. 9, the detection potentials determine the sensitivity of detection. Within the potential range of 0.7–0.1 V, the steady-state current of iodate reduction increases considerably with applied potentials. Moving the applied potential to negative values further, the iodate reduction current does not increase accordingly. Thus, an applied potential of 0.1 V is selected as the optimized value for chronoamperometry.
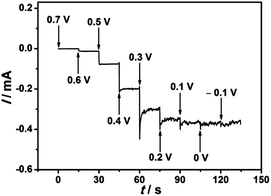 |
| | Fig. 9 Background subtracted multipotential step curve of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode in N2-saturated 0.1 M pH 2.0 K2SO4–H2SO4 solution containing 0.8 mM iodate. Step increase: 0.1 V; step time: 15 s. | |
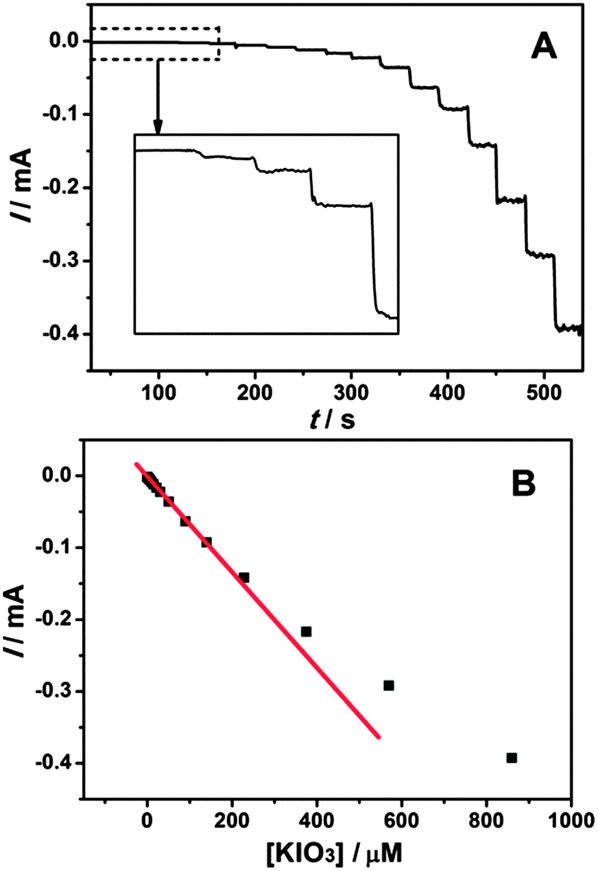
Fig. 10A presents the dynamic amperometric response of the sensor at a working potential of 0.1 V with successive injections of KIO3 into 0.1 M K2SO4–H2SO4 solution (pH 2.0) on the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode. Upon addition of KIO3, the reduction current increases rapidly and sensitively. And the response time, considering the time to reach 95% of the steady state signal, is less than 3 s. The corresponding calibration plot shows the current response and the concentration of KIO3 has a linear relationship in the concentration range from 8 × 10−8 to 2.3 × 10−4 M with a correlation coefficient of 0.9993 (Fig. 10B). The linear regression equation is expressed as ipa (mA) = −0.00182 − 0.000661c (μM). The detection limit of 3 × 10−8 M is estimated at a signal/noise (S/N) of three. The sensitivity and detection limit of the present biosensor are better than various metal nanoparticles (Pt45 and Au46), metal oxides (W,47 Mo,48 and Ru49), metal complexes (V,50 Os,51 and Fe52) and enzyme (catalase53) based iodate electrochemical sensors (Table S1, ESI†). Thus, it is concluded that the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode can be used to construct an electrochemical sensor towards the determination of iodate.
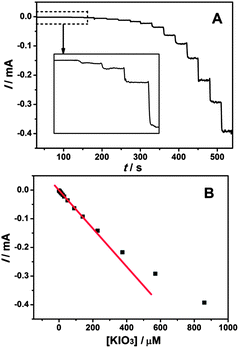 |
| | Fig. 10 (A) Amperometric response at the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode in the 0.1 M pH 2.0 K2SO4–H2SO4 solution held at 0.1 V for successive addition of iodate under continuous stirring. (B) Plot of amperometric currents vs. iodate concentrations. | |
The anti-interference ability of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode was also studied by chronoamperometry. It is observed that 100-fold Na+, K+, NO33−, SO42−, PO43−, BrO3−, NO2−, H2O2, and glucose species do not interfere with the detection of iodate, indicating that the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode possesses excellent selectivity towards iodate reduction. To characterize the reproducibility of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode, repetitive measurements were carried out in solution containing 0.1 mM iodate. Ten successive measurements showed a relative standard deviation of 3.2%, which was indicative of acceptable detection reproducibility for practical analysis. The storage stability of the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode was evaluated for a period of 30 days. When not in use, the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode was stored in air at room temperature. There was an approximately 4.3% decrease in the current response for 0.1 mM iodate after 30 days, indicating that the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode maintained its catalytic activity and could be used for an extended period of time.
Conclusions
We have demonstrated a facile strategy for the construction of PAH functionalized MWCNT multilayer films through an LBL self-assembly technique. The noncovalent functionalization of the MWCNT-COOH with PAH created a distribution of positive charges at the tube surface, which made {PAH/MWCNT-COOH}8 films excellent substrates for immobilization of the negative charged materials. Thus, the negative EDTMP–RuIII complex could self-assemble on the {PAH/MWCNT-COOH}8 film surface owing to the strong electrostatic and/or hydrogen bonding interactions between –P–OH groups and –NH2 groups. The fabricated modified electrode displayed a fast electron transfer rate, and offered excellent electrocatalytic activity for the iodate reduction. Therefore, the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode was employed to detect iodate, and a wide linear range (8 × 10−8 to 2.3 × 10−4 M) and a low detection limit (3 × 10−8 M) were obtained. Moreover, the EDTMP–RuIII/{PAH/MWCNT-COOH}8/GC electrode showed good storage stability, reproducibility and anti-interference ability for promising practical applications.
Acknowledgements
This work was financially supported by the National “973” program of China (2012CB215500), NSFC (21005039), University Postgraduate Research and Innovation Project in Jiangsu Province (CXZZ13_0393), Outstanding Doctoral Dissertation Incubation Program of Jiangsu Province (2012BS0004), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Notes and references
- T. Y. Ma, X. J. Zhang and Z. Y. Yuan, J. Mater. Sci., 2009, 44, 6775 CrossRef CAS.
- J. Luo, M. Dong, F. Lin, M. Liu, H. Tang, H. Li, Y. Zhang and S. Yao, Analyst, 2011, 136, 4500 RSC.
- J. B. Han, X. Y. Xu, X. Y. Rao, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem., 2011, 21, 2126 RSC.
- X. J. Zhang, T. Y. Ma and Z. Y. Yuan, J. Mater. Chem., 2008, 18, 2003 RSC.
- Y. A. Li, N. H. Tai, S. K. Chen and T. Y. Tsai, ACS Nano, 2011, 5, 6500 CrossRef CAS PubMed.
- M. Zheng, P. Li, G. Fu, Y. Chen, Y. Zhou, Y. Tang and T. Lu, Appl. Catal., B, 2013, 129, 394 CrossRef CAS PubMed.
- L. Shi, R. P. Liang and J. D. Qiu, J. Mater. Chem., 2012, 22, 17196 RSC.
- H. Q. Li, L. N. Han, J. Cooper-White and I. Kim, Green Chem., 2012, 14, 586 RSC.
- S. Mantha, V. A. Pedrosa, E. V. Olsen, V. A. Davis and A. L. Simonian, Langmuir, 2010, 26, 19114 CrossRef CAS PubMed.
- C. H. Xue, R. J. Zhou, M. M. Shi, Y. Gao, G. Wu, X. B. Zhang, H. Z. Chen and M. Wang, Nanotechnology, 2008, 19, 215604 CrossRef PubMed.
- W. Yang, X. Wang, F. Yang, C. Yang and X. Yang, Adv. Mater., 2008, 20, 2579 CrossRef CAS.
- K. Balasubramanian and M. Burghard, Small, 2005, 1, 180 CrossRef CAS PubMed.
- I. Capek, Adv. Colloid Interface Sci., 2009, 150, 63 CrossRef CAS PubMed.
- A. I. Zamaleeva, I. R. Sharipova, A. V. Porfireva, G. A. Evtugyn and R. F. Fakhrullin, Langmuir, 2009, 26, 2671 CrossRef PubMed.
- H. Chen, L. Guo, A. R. Ferhan and D. H. Kim, J. Phys. Chem. C, 2011, 115, 5492 CAS.
- J. Zhu, B. S. Shim, M. Di Prima and N. A. Kotov, J. Am. Chem. Soc., 2011, 133, 7450 CrossRef CAS PubMed.
- L. Guo, N. Yin, D. Nie, F. Fu and G. Chen, Chem. Commun., 2011, 47, 10665 RSC.
- B. Qiu, Z. Lin, J. Wang, Z. Chen, J. Chen and G. Chen, Talanta, 2009, 78, 76 CrossRef CAS PubMed.
- X. Wang, J. Wang, H. Cheng, P. Yu, J. Ye and L. Mao, Langmuir, 2011, 27, 11180 CrossRef CAS PubMed.
- K. E. Tettey, J. W. C. Ho and D. Lee, J. Phys. Chem. C, 2011, 115, 6297 CAS.
- K. E. Tettey, M. Q. Yee and D. Lee, Langmuir, 2010, 26, 9974 CrossRef CAS PubMed.
- B. P. Pichon, P. Louet, O. Felix, M. Drillon, S. Begin-Colin and G. Decher, Chem. Mater., 2011, 23, 3668 CrossRef CAS.
- C. Peng, Y. S. Thio, R. A. Gerhardt, H. Ambaye and V. Lauter, Chem.
Mater., 2011, 23, 4548 CrossRef CAS.
- J. R. Siqueira, C. F. Werner, M. Backer, A. Poghossian, V. Zucolotto, O. N. Oliveira and M. J. Schoning, J. Phys. Chem. C, 2009, 113, 14765 CAS.
- A. Bouvree, J. F. Feller, M. Castro, Y. Grohens and M. Rinaudo, Sens. Actuators, B, 2009, 138, 138 CrossRef CAS PubMed.
- B. Y. Wu, S. H. Hou, F. Yin, Z. X. Zhao, Y. Y. Wang, X. S. Wang and Q. Chen, Biosens. Bioelectron., 2007, 22, 2854 CrossRef CAS PubMed.
- C. F. Ou, R. Yuan, Y. Q. Chai, M. Y. Tang, R. Chai and X. L. He, Anal. Chim. Acta, 2007, 603, 205 CrossRef CAS PubMed.
- S. Komathi, A. I. Gopalan and K. P. Lee, Biosens. Bioelectron., 2009, 24, 3131 CrossRef CAS PubMed.
- Z. A. Xu, N. Gao, H. J. Chen and S. J. Dong, Langmuir, 2005, 21, 10808 CrossRef CAS PubMed.
- Q. C. Zhao, X. D. Feng, S. L. Mei and Z. X. Jin, Nanotechnology, 2009, 20, 105101 CrossRef PubMed.
- M. N. Hyder, S. W. Lee, F. Ç. Cebeci, D. J. Schmidt, Y. Shao-Horn and P. T. Hammond, ACS Nano, 2011, 5, 8552 CrossRef CAS PubMed.
- J. Li, S. Srivastava, J. G. Ok, Y. Zhang, M. Bedewy, N. A. Kotov and A. J. Hart, Chem. Mater., 2011, 23, 1023 CrossRef CAS.
- D. Lee and T. Cui, Langmuir, 2011, 27, 3348 CrossRef CAS PubMed.
- N. Du, H. Zhang, J. Yu, P. Wu, C. Zhai, Y. Xu, J. Wang and D. Yang, Chem. Mater., 2009, 21, 5264 CrossRef CAS.
- B. P. Vinayan, R. Nagar, N. Rajalakshmi and S. Ramaprabhu, Adv. Funct. Mater., 2012, 22, 3519 CrossRef CAS.
-
A. G. Gilman, L. S. Goodman, T. W. Rad and F. Murad, The Pharmacological Basis of Therapeutics, Macmillan, New York, 7th edn, 1985 Search PubMed.
- Z. Wu, Y. Lv, Y. Xia, P. A. Webley and D. Zhao, J. Am. Chem. Soc., 2011, 134, 2236 CrossRef PubMed.
- Z. S. Li, X. M. Huang, X. F. Zhang, L. Zhang and S. Lin, J. Mater. Chem., 2012, 22, 23602 RSC.
- C. G. Hu, H. H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. M. Dai and L. T. Qu, Adv. Mater., 2012, 24, 5493 CrossRef CAS PubMed.
- A. Ponrouch, S. Garbarino, E. Bertin, C. Andrei, G. A. Botton and D. Guay, Adv. Funct. Mater., 2012, 22, 4172 CrossRef CAS.
- H. J. Wang, M. Imura, Y. Nemoto, L. Wang, H. Y. Jeong, T. Yokoshima, O. Terasaki and Y. Yamauchi, Chem.–Eur. J., 2012, 18, 13142 CrossRef CAS PubMed.
- W. Zhu, A. X. Yin, Y. W. Zhang and C. H. Yan, Chem.–Eur. J., 2012, 18, 12222 CrossRef CAS PubMed.
- J. C. Claussen, A. Kumar, D. B. Jaroch, M. H. Khawaja, A. B. Hibbard, D. M. Porterfield and T. S. Fisher, Adv. Funct.
Mater., 2012, 22, 3399 CrossRef CAS.
- M. N. Cao, D. S. Wu, S. Y. Gao and R. Cao, Chem.–Eur. J., 2012, 18, 12978 CrossRef CAS PubMed.
- Y. Li, Y. Zhou, H. Xian, L. Wang and J. Huo, Anal. Sci., 2011, 27, 1223 CrossRef CAS.
- X. Huang, Y. Li, Y. Chen and L. Wang, Sens. Actuators, B, 2008, 134, 780 CrossRef CAS PubMed.
- B. X. Zou, X. X. Liu, D. Diamond and K. T. Lau, Electrochim. Acta, 2010, 55, 3915 CrossRef CAS PubMed.
- L. Tian, L. Liu, L. Chen, N. Lu and H. Xu, Sens. Actuators, B., 2005, 105, 484 CrossRef CAS PubMed.
- F. Chatraei and H. R. Zare, Mater. Sci. Eng., C, 2013, 33, 721 CrossRef CAS PubMed.
- A. Salimi, H. Mamkhezri and S. Mohebbi, Electrochem. Commun., 2006, 8, 688 CrossRef CAS PubMed.
- A. Salimi, B. Kavosi, A. Babaei and R. Hallaj, Anal. Chim. Acta, 2008, 618, 43 CrossRef CAS PubMed.
- A. Salimi, H. MamKhezri, R. Hallaj and S. Zandi, Electrochim. Acta, 2007, 52, 6097 CrossRef CAS PubMed.
- A. Salimi, A. Noorbakhsh and M. Ghadermarzi, Sens. Actuators, B., 2007, 123, 530 CrossRef CAS PubMed.
- G. Fu, W. Han, L. Yang, J. Lin, S. Wei, Y. Chen, Y. Zhou, Y. Tang, T. Lu and X. Xia, J. Mater. Chem., 2012, 22, 17604 RSC.
- G. Fu, K. Wu, X. Jiang, L. Tao, Y. Chen, J. Lin, Y. Zhou, S. Wei, Y. Tang, T. Lu and X. Xia, Phys. Chem. Chem. Phys., 2013, 15, 3793 RSC.
- G. Fu, L. Tao, M. Zhang, Y. Chen, Y. Tang, J. Lin and T. Lu, Nanoscale, 2013, 5, 8007 RSC.
- Y. Chen, J. Xu, X. Liu, Y. Tang and T. Lu, Appl. Catal., B, 2013, 140–141, 552 CrossRef CAS PubMed.
- J. H. Rouse, P. T. Lillehei, J. Sanderson and E. J. Siochi, Chem. Mater., 2004, 16, 3904 CrossRef CAS.
- H. Paloniemi, M. Lukkarinen, T. Aaritalo, S. Areva, J. Leiro, M. Heinonen, K. Haapakka and J. Lukkari, Langmuir, 2005, 22, 74 CrossRef PubMed.
- H. Liu, Y. Cui, P. Li, Y. Zhou, X. Zhu, Y. Tang, Y. Chen and T. Lu, Analyst, 2013, 138, 2647 RSC.
- H. Paloniemi, T. ritalo, T. Laiho, H. Liuke, N. Kocharova, K. Haapakka, F. Terzi, R. Seeber and J. Lukkari, J. Phys. Chem. B, 2005, 109, 8634 CrossRef CAS PubMed.
- M. Fagnoni, A. Profumo, D. Merli, D. Dondi, P. Mustarelli and E. Quartarone, Adv. Mater., 2009, 21, 1761 CrossRef CAS.
- V. Di Marco, M. Kilyen, T. Jakusch, P. Forgó, G. Dombi and T. Kiss, Eur. J. Inorg. Chem., 2004, 2004, 2524 CrossRef.
- B. Nowack and A. T. Stone, Environ. Chem. Lett., 2003, 1, 24 CrossRef CAS PubMed.
- Y. Liang, Y. Zhou, J. Ma, J. Zhao, Y. Chen, Y. Tang and T. Lu, Appl. Catal., B, 2011, 103, 388 CrossRef CAS PubMed.
- J. Galezowska, R. Janicki, A. Mondry, R. Burgada, T. Bailly, M. Lecouvey and H. Kozlowski, Dalton Trans., 2006, 4384 RSC.
- R. Janicki and A. Mondry, Polyhedron, 2008, 27, 1942 CrossRef CAS PubMed.
- J. Wu, H. Hou, H. Han and Y. Fan, Inorg. Chem., 2007, 46, 7960 CrossRef CAS PubMed.
- P. Fiurasek and L. Reven, Langmuir, 2007, 23, 2857 CrossRef CAS PubMed.
- A. Ojida, M. Inoue, Y. Mito-oka, H. Tsutsumi, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 2052 CrossRef CAS PubMed.
- L. N. Johnson and R. J. Lewis, Chem. Rev., 2001, 101, 2209 CrossRef CAS PubMed.
- S. Anderson, U. Neidlein, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed., 1995, 34, 1596 CrossRef CAS.
- P. Li, Y. Ding, A. Wang, L. Zhou, S. Wei, Y. Zhou, Y. Tang, Y. Chen, C. X. Cai and T. Lu, ACS Appl. Mater. Interfaces, 2013, 5, 2255 CAS.
- P. Li, H. Liu, Y. Ding, Y. Wang, Y. Chen, Y. Zhou, Y. Tang, H. Wei, C. Cai and T. Lu, J. Mater. Chem., 2012, 22, 15370 RSC.
- H. Liu, Y. Cui, P. Li, Y. Zhou, Y. Chen, Y. Tang and T. Lu, Anal. Chim. Acta, 2013, 776, 24 CrossRef CAS PubMed.
- F. Li, Z. Wang, W. Chen and S. S. Zhang, Biosens. Bioelectron., 2009, 24, 3030 CrossRef CAS PubMed.
- G. Zhang, L. Zhang, L. Shen, Y. Chen, Y. Zhou, Y. Tang and T. Lu, ChemPlusChem, 2012, 77, 936 CrossRef CAS.
- J. Xu, X. Liu, Y. Chen, Y. Zhou, T. Lu and Y. Tang, J. Mater. Chem., 2012, 22, 23659 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table. See DOI: 10.1039/c3tb21433h |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.