
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlation between the local scale structure and the electrochemical properties in lithium orthosilicate cathode materials†
Alessandro
Mancini
a,
Victoria Raissa
Barbieri
a,
Joerg C.
Neuefeind
b,
Katharine
Page
bc and
Lorenzo
Malavasi
*a
aDepartment of Chemistry and INSTM, University of Pavia, Pavia 27100, Italy. E-mail: lorenzo.malavasi@unipv.it
bOak Ridge Natl Lab, Chem & Engn Mat Div, Spallat Neutron Source, Oak Ridge, TN 37831, USA
cLos Alamos Natl Lab, Lujan Neutron Scattering Ctr, Los Alamos, NM 87545, USA
First published on 3rd September 2014
Abstract
Lithium metal orthosilicates with general formula Li2MSiO4 (M = Mn, Fe and Co) are among the most attractive new materials as potential high-specific-energy cathodes for lithium batteries. All the members of this family present a rich polymorphism with at least three clearly identified crystal structures of each Li2MSiO4 compound. Several theoretical investigations have highlighted that the energy stability of the different polymorphs is very close to each other irrespective of their average crystal structures. At the same time, the calculated and experimental electrochemical performances are again very similar among different polymorphs. By means of neutron total scattering investigation of different polymorphs (monoclinic and orthorhombic) of Li2FeSiO4 and Li2MnSiO4 orthosilicates coupled to Pair Distribution Function (PDF) analysis we showed that, at the local scale, all the polymorphs have the same structure (in particular the structure of the monoclinic polymorph) irrespective of the average structure they possess. This experimental evidence of a strong similarity at the local scale can be correlated with the observed electrochemical similarity (such as the lithium extraction voltages) among the different orthosilicate polymorphs, thus providing an approach to elucidate the relevance of local versus long-range structure.
Introduction
Lithium batteries are still among the most attractive clean and efficient energy storage systems developed in the last few decades in order to face the well-known problems related to global warming, diminishing fossil-fuel reserves, and increasing demand for portable electronic and grid storage systems.1–3 As a matter of fact, the discovery and optimization of high performance materials are critical for next-generation rechargeable batteries.4 The Li2MSiO4 (M = Fe, Mn, Co, and Ni) family is very attractive as a positive electrode for lithium batteries due to its improved safety and at least theoretical possibility to deintercalate two Li ions from the structure. In addition, these cathode materials contain abundant, non-toxic and cheap constituent elements. These materials are the object of a very deep and recent experimental and theoretical investigation aimed at improving their properties and performance in Li-batteries.Li2FeSiO4 and Li2MnSiO4 orthosilicates are characterized by a distorted hexagonal close packing of the oxide ions in which half of the tetrahedral sites are occupied by cations. In this lattice the cation site ordering can vary and the tetrahedra can be distorted thus resulting in a rich and complex polymorphism.5–18 The known polymorphs are related to the low- or high-temperature forms of Li3PO4, which are indicated as β-Li3PO4 or γ-Li3PO4, respectively. Currently, four polymorphs have been reported for Li2FeSiO4 in the literature and crystallized in the P21, P21/n, Pmnb and Pmn21 space groups.14,19 Analogously, four crystal structures have been identified for the Li2MnSiO4 orthosilicate with space groups P21/n, Pn, Pmnb and Pmn21. The orthorhombic forms (Pmnb and Pmn21) have a 2D pathway for Li-diffusion while the monoclinic structures (P21, P21/n and Pn) have framework structures with a 3D Li-pathway.
A comparison of the electrochemical properties of the P21, Pmnb and Pmn21 Li2FeSiO4 polymorphs, carried out by several groups, showed that the removal of the first Li ion occurs at about 3.1 V during the first charge of the cell, independent of the polymorph.19,20 Considering that different average structures correspond to different spatial arrangements of the polyhedra and inter-connections between them, this is a quite unexpected observation. Density Functional Theory (DFT) calculations also showed that the different crystal structures all have similar energies with differences of the order of few kJ mol−1 at room temperature.20 In addition, upon delithiation, the most stable polymorph is the one characterized by a 3D framework and during this process there is a 2D → 3D transformation.20
Similar results have been found considering the different Li2MnSiO4 polymorphs which have been the object of several experimental and computational studies as well. As for the previous case (Li2FeSiO4), the thermodynamic stability of the Pmnb, Pmn21, P21/n and Pn structures determined by DFT and atomistic simulation revealed very small energy differences with a slightly higher stability found for the orthorhombic polymorphs.14,21 Calculation of the Li-migration pathways for the different structures revealed a strong difference in the activation energies as a function of the space group, passing from 0.65 eV for the Pn to 1.13 eV for the Pmn21 structure.21 However, the experimental electrochemical behavior of all the Li2MnSiO4 polymorphs is very similar irrespective of the actual crystal structure and framework dimensionality.6
This brief overview points out a quite unusual behavior within the orthosilicate cathode materials. Irrespective of the actual average crystal structure, which may vary significantly from orthorhombic to monoclinic, the electrochemical performances and the calculated stabilities as well as Li extraction voltages are very similar. Since these similarities can be hardly correlated with the different average structures found in the Li2MSiO4 orthosilicate polymorphs, a possible origin of this behavior may be found in their microscopic structural features. As a matter of fact, the computational studies which found significant differences in the behavior of the various polymorphs (in terms, for example, of ion migration pathways and activation energies) were carried out considering the local structure characteristics. However, the computed relaxed local structures were constrained to be derived from the corresponding average structures.
What is clear from the current literature is that there is a strong discrepancy between the observed experimental results of the electrochemical properties of various orthosilicate polymorphs – i.e. all are very close to each other – and the expected differences are based on the average structures. Since such behavior may be closely correlated with the short-range structural features, which have a strong influence on the electrochemical properties (e.g., inter- and intra-polyhedra patterns), in order to shed some light on this aspect, we carried out an experimental investigation of the short-range order in different polymorphs of Li2FeSiO4 and Li2MnSiO4 orthosilicates. This study was carried out by means of neutron total scattering and PDF analysis. Our results indicate that – at the local scale – the structural arrangement of each polymorph corresponds to that of the monoclinic structure and that only at a medium distance range the structure can be properly described considering the relative average structure.
Experimental
The synthesis of the samples investigated in the present work was done as follows. Reagent powders of LiOH·H2O, MnCO3, Fe(C2O4)·2H2O and SiO2 (fumed silica, 7 nm) were purchased from Sigma Aldrich, all of them were of purity >99.9%, and used without further purifications. Stoichiometric amounts of reactants were mixed in acetone under continuous moderate stirring and N2 bubbling for 4 hours. Once acetone evaporated, mixed powders were recovered and pelletized using an isostatic oil press at 300 bar for 360 seconds (for Pmnb and P21/n-P21 polymorphs) or hand-pressed (for Pmn21 polymorphs). Pellets were positioned in a Pt crucible inside a sealed conditioning quartz tube. Argon > 99.9% was then fluxed inside the conditioning tube for 2 hours before starting the annealing process and during all the thermal treatments, in order to prevent Mn2+ and Fe2+ oxidation. Pmnb and monoclinic polymorphs were heated up to 900 °C for 12 h, while Pmn21 was annealed at 700 °C for 8 h. The annealing temperatures were always reached with a heating ramp of 2 °C min−1 while for the cooling process three different paths were followed: cooling ramp of 1 °C min−1 for Pmnb polymorphs, cooling ramp of 5 °C min−1 for Pmn21 polymorphs and fast quenching to room temperature for monoclinic polymorphs. Phase purity check was carried out by laboratory XRD (Bruker D8 Advance).Neutron powder diffraction (NPD) measurements at 300 K were carried out on a NPDF diffractometer at the Lujan Center at the Los Alamos National Laboratory and on a NOMAD diffractometer at the Oak Ridge Laboratories in a cylindrical vanadium tube. Data from an empty container were also collected to subtract the blank. For the NPDF data the corrected total scattering structure function, S(Q), was calculated with the program PDFGETN,22 and the PDF was obtained by Fourier transformation of S(Q). Qmax = 29.0 Å−1 was used. For the NOMAD data the G(r) value was extracted by using a program developed by the NOMAD group. Modeling of the experimental neutron PDF data was carried out with the aid of PDFGUI and PDFFit2 software.23
Results
Li2MnSiO4
Three different polymorphs of the Li2MnSiO4 orthosilicate have been synthesized belonging to the Pmnb, Pmn21, and P21/n space groups as confirmed by the Rietveld refinement of the neutron diffraction patterns (reported in the ESI†). Table 1 reports the structural parameters obtained from the refinement based on the ND patterns. Our synthesis approach for the sample preparation is a solid-state reaction where the different polymorphs are stabilized as a function of the calcination temperatures and/or cooling ramps. This allowed us to prepare samples with similar morphologies and crystallite dimensions while in the current literature different polymorphs are usually prepared by means of a range of synthesis routes.| Sample | Space group | a (Å) | b (Å) | c (Å) | β (°) |
|---|---|---|---|---|---|
| Li2MnSiO4 | Pmnb | 6.3049(2) | 10.7430(3) | 4.9914(2) | |
| Li2MnSiO4 | Pmn21 | 6.3060(2) | 5.3683(2) | 4.9761(3) | |
| Li2MnSiO4 | P21/n | 6.3270(1) | 10.9072(2) | 5.0691(1) | 90.995(1) |
Fig. 1a and b present the neutron PDFs for the three Li2MnSiO4 polymorphs discussed in the 1–20 Å range (Fig. 1a) and in the 1–6 Å range (Fig. 1b), respectively.
 | ||
| Fig. 1 (a) Neutron PDFs for the three Li2MnSiO4 polymorphs in the 1–20 Å range. Data for the Pmn21 and P21/n structures have been shifted by +20 and +40, respectively. (b) Neutron PDFs for the three Li2MnSiO4 polymorphs in the 1–6 Å range. Data for the Pmn21 and P21/n structures have been shifted by +20 and +40, respectively. | ||
The simple visual inspection of the neutron PDFs reported in Fig. 1a and b shows that the two Li2MnSiO4 orthorhombic polymorphs share several similarities in terms of peak numbers and shapes while the PDF of the monoclinic structure has a greater number of pairs, which is more evident above ca. 10 Å. However, if one looks carefully at Fig. 1b, which is limited to the 1–6 Å range and which encompasses both intra-polyhedra and inter-polyhedra atom pairs, it is possible to observe a strong similarity, in terms of peak number, shape and position, between the PDFs of the three samples considered, irrespective of their average structure.
Fig. 2a–c show the fits of the 1–6 Å range for each Li2MnSiO4 polymorph carried out using as starting parameters those obtained from the Rietveld refinement of the ND patterns and keeping the correct space group for each polymorph.
 | ||
| Fig. 2 (a) Fit for the Pmnb polymorph with the Pmnb symmetry. (b) Fit for the Pmn21 polymorph with the Pmn21 symmetry. (c) Fit for the P21/n polymorph with the P21/n symmetry. (d) Fit for the Pmnb polymorph with the P21/n symmetry. (e) Fit for the Pmn21 polymorph with the P21/n symmetry. Legend for all the figures: empty blue circles: experimental data; red lines: fitted structure; green lines: residual. Residuals have been shifted by −20. | ||
As can be appreciated, the fit of the data works well for the monoclinic P21/n sample while for the other two orthorhombic samples the agreement between the experimental and calculated structures is quite poor (the agreement factors, Rwp, are 0.44 (Pmnb), 0.26 (Pmn21), and 0.11 (P21/n)). This implies that the short-range structure for the two orthorhombic polymorphs should be analyzed by means of another symmetry in order to properly describe the experimental data. This is also in agreement with the data reported in Fig. 2b that suggested a very similar local structure for the three Li2MnSiO4 samples. Accordingly, the fits of the Pmnb and Pmn21 samples were done in the P21/n space group. The lattice parameters for the three samples refined in the P21/n symmetry are reported in Table 2 together with the results obtained by fitting with the correct space group.
| Sample | Space group (average) | Space group fit 1–6 Å | a (Å) | b (Å) | c (Å) | β (°) | R wp |
|---|---|---|---|---|---|---|---|
| Li2MnSiO4 | Pmnb | P21/n | 6.4353(6) | 10.7173(9) | 4.8999(8) | 93.74(8) | 0.131 |
| Li2MnSiO4 | Pmn21 | P21/n | 6.4019(7) | 10.746(1) | 4.9177(5) | 90.78(2) | 0.145 |
| Li2MnSiO4 | P21/n | P21/n | 6.620(1) | 11.040(1) | 5.0915(3) | 92.305(7) | 0.109 |
| Li2MnSiO4 | Pmnb | Pmnb | 6.2189(7) | 11.466(1) | 4.9622(4) | 90 | 0.443 |
| Li2MnSiO4 | Pmn21 | Pmn21 | 6.1510(5) | 5.4927(3) | 5.0314(3) | 90 | 0.255 |
It is clear that the description of the neutron PDFs with the monoclinic space group for the two orthorhombic polymorphs leads to a very good description of the experimental data. This reduction of symmetry at the local scale is consistent with the similarity in the PDF features discussed above (Fig. 1b). Of course, the monoclinic structure has more refineable parameters than the two orthorhombic structures but we took care to avoid an over-description of the data (for example, we kept fixed the (x, y, z) coordinates of Si and Mn ions in the fits and used one isotropic a.d.p. for each type of atom).
In order to further extend the description of the PDF data with respect to the average structure, the data were tested against a medium r-range. Fig. 3a–c report the fits in the 11–20 Å range for the three polymorphs carried out with the monoclinic P21/n space group used for the 1–6 Å range, while Fig. 3d and e display the same data for the orthorhombic structures fitted within the Pmnb and Pmn21 symmetry, respectively, i.e. with the space group of the average structure.
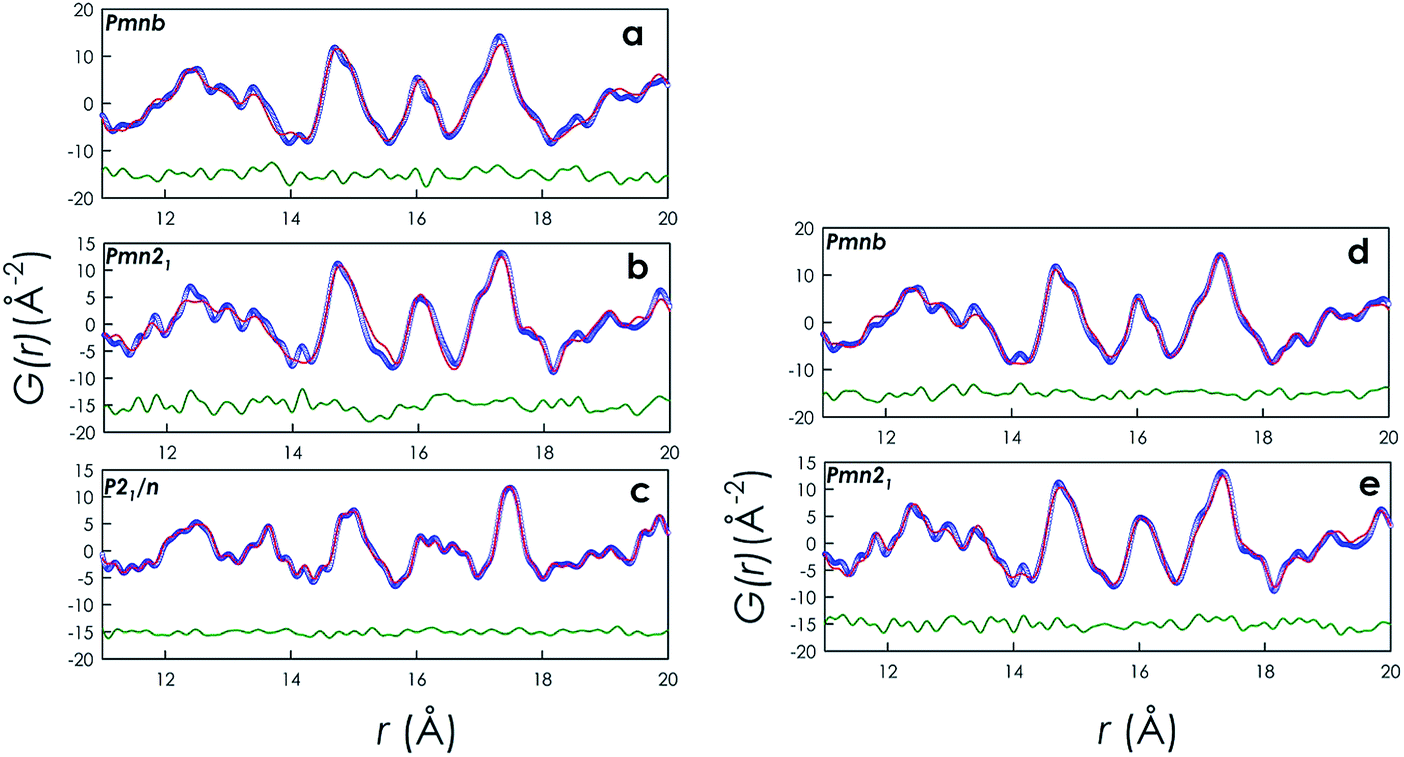 | ||
| Fig. 3 (a) Fit for the Pmnb polymorph with the P21/n symmetry. (b) Fit for the Pmn21 polymorph with the P21/n symmetry. (c) Fit for the P21/n polymorph with the P21/n symmetry. (d) Fit for the Pmnb polymorph with the Pmnb symmetry. (e) Fit for the Pmn21 polymorph with the Pmn21 symmetry. Legend for all the figures: empty blue circles: experimental data; red lines: fitted structure; green lines: residual. Residuals have been shifted by −15. | ||
The Rwp for the Pmnb phase (Fig. 3a and d) is lowered from 0.19 to 0.14 if the PDF in the 11–20 Å range is described with the “correct” orthorhombic space group instead of the monoclinic P21/n. Analogously, for the Pmn21 sample, Rwp changes from 0.24 to 0.17 passing from the P21/n to the Pmn21 space group. This result is quite interesting because it witnesses that, already in a medium range (i.e., 11–20 Å), the description of the experimental PDFs is most correctly done by using the symmetry of the average structure and that the use of a more distorted space group, such as the monoclinic P21/n (that more accurately described the short-range order), gives a worse description of the experimental data. This is also a nice proof of the reliability of the fit carried out for the 1–6 Å range.
Li2FeSiO4
The investigation presented above for the Li2MnSiO4 orthosilicates has been carried out in an analogous way for the three polymorphs of the Li2FeSiO4 orthosilicates (P21, Pmnb and Pmn21). These have been again prepared by means of a solid state reaction by tuning the temperatures and the heating and cooling ramps in order to get the three different polymorphs. The lattice parameters, as obtained from the refinement of the ND patterns, are reported in Table 3.| Sample | Space group | a (Å) | b (Å) | c (Å) | β (°) |
|---|---|---|---|---|---|
| Li2FeSiO4 | Pmnb | 6.2660(2) | 10.6434(2) | 5.0210(1) | |
| Li2FeSiO4 | Pmn21 | 6.3049(2) | 10.7430(3) | 4.9914(2) | |
| Li2FeSiO4 | P21 | 8.2257(1) | 5.0056(3) | 8.2142(1) | 99.220(4) |
Fig. 4 reports the experimental neutron PDFs for the three samples of Li2FeSiO4 investigated. As for Li2MnSiO4, increasing the distance (r) leads to a progressive increase of the differences between the three experimental PDFs while, in the short-range, a strong similarity between the data for the three polymorphs is evident irrespective of the average structure.
 | ||
| Fig. 4 (a) Neutron PDFs for the three Li2FeSiOs4 polymorphs in the 1–20 Å range. Data for the Pmn21, and P21 structures have been shifted by +4 and +8, respectively. (b) Neutron PDFs for the three Li2FeSiO4 polymorphs in the 1–6 Å range. Data for the Pmn21, and P21 structures have been shifted by +4 and +8, respectively. | ||
Fig. 5a–c show the fit in the short range (1–6 Å) of the three experimental PDFs using, as starting parameters, those obtained from the Rietveld refinement of the ND patterns and keeping the space groups of the average structure. The structural results obtained for the fits, together with Rwp, are reported in Table 4.
 | ||
| Fig. 5 (a) Fit for the Pmnb polymorph with the Pmnb symmetry. (b) Fit for the Pmn21 polymorph with the Pmn21 symmetry. (c) Fit for the P21 polymorph with the P21 symmetry. (d) Fit for the Pmnb polymorph with the P21 symmetry. (e) Fit for the Pmn21 polymorph with the P21 symmetry. Legend for all the figures: empty blue circles: experimental data; red lines: fitted structure; green lines: residual. Residuals have been shifted by −5. | ||
| Sample | Space group (average) | Space group fit 1–6 Å | a (Å) | b (Å) | c (Å) | β (°) | R wp |
|---|---|---|---|---|---|---|---|
| Li2FeSiO4 | Pmnb | P21 | 8.294(1) | 5.0452(5) | 8.3694(4) | 102.27(1) | 0.114 |
| Li2FeSiO4 | Pmn21 | P21 | 8.2912(5) | 5.0758(5) | 8.2567(1) | 100.60 | 0.120 |
| Li2FeSiO4 | P21 | P21 | 8.3195(7) | 5.0655(3) | 8.3173(3) | 102.81(1) | 0.091 |
| Li2FeSiO4 | Pmnb | Pmnb | 6.281(1) | 10.571(2) | 4.982(1) | 90 | 0.236 |
| Li2FeSiO4 | Pmn21 | Pmn21 | 6.5052(5) | 4.8237(4) | 4.5911(5) | 90 | 0.320 |
As for the Li2MnSiO4 case, the description of the short-range order for the two orthorhombic polymorphs of Li2FeSiO4 is significantly improved if the data are described using the space group of the most distorted polymorph (i.e., the monoclinic P21 structure). Rwp lowers from 0.236 to 0.114 for the Pmnb polymorph and from 0.320 to 0.120 for the Pmn21 polymorph. Again, in order to avoid an over-description of the experimental data, the atomic positions of the Fe and Si atoms were kept fixed during the refinement and temperature factors were described with only one isotropic factor for both Li and O atoms.
In order to test the validity of the proposed model for a wider r-range, Fig. 6a–e report the refinement of the experimental data in the 11–20 Å range. In particular, Fig. 6a–c report the fits in the 11–20 Å range for the three polymorphs carried out with the monoclinic P21 space group, i.e. the space group that best described the 1–6 Å range, while Fig. 6d and e show the fit results of the two orthorhombic structures carried out with the two orthorhombic space groups (Pmnb and Pmn21).
 | ||
| Fig. 6 (a) Fit for the Pmnb polymorph with the P21 symmetry. (b) Fit for the Pmn21 polymorph with the P21 symmetry. (c) Fit for the P21 polymorph with the P21 symmetry. (d) Fit for the Pmnb polymorph with the Pmnb symmetry. (e) Fit for the Pmn21 polymorph with the Pmn21 symmetry. Legend for all the figures: empty blue circles: experimental data; red lines: fitted structure; green line: residual. Residuals have been shifted by −3. | ||
For Li2FeSiO4, the inspection of the fit results in this range leads to a different conclusion with respect to Li2MnSiO4. For the orthorhombic Pmnb phase, the agreement factor is 0.291 when refining the data with the P21 space group and it lowers to 0.214 if the space group of the average structure is used (i.e., Pmnb). This difference is relatively small, and the visual inspection of the fits reported in Fig. 5a and d does not indicate a huge difference in terms of fit quality between the two structural models. The situation is more clear for the other orthorhombic phase (Pmn21), where the quality of the fit is good if the data in the 11–20 Å range are again modelled with the P21 space group instead of Pmn21 of the average structure (the agreement factor is 0.189 in the first case and 0.389 in the second case). These results indicate that for Li2FeSiO4, the difference between the average and the local structure extends over a wider r-range with respect to Li2MnSiO4. In order to see whether by further extending the analysis it was possible to reconcile the average and local structure, the two orthorhombic samples were tested against the two possible models (i.e., the monoclinic and the corresponding orthorhombic space groups) up to 30 Å.
The results of the fits from 21 to 30 Å for the two orthorhombic polymorphs of Li2FeSiO4 clearly indicate a huge improvement of the results if the average structure space group is used instead of the monoclinic one. The agreement factor lowers from 0.506 to 0.189 for the Pmnb polymorphs fitting the data from 21 to 30 Å with the P21 space group or the Pmnb structure, respectively. On the same line behaves the Pmn21 polymorph, where the description of the local structure with the orthorhombic space group lowers Rwp from 0.365 (P21 space group) to 0.156 (Pmn21 space group). The corresponding plot of the refinements up to 30 Å is reported in the ESI.†
Discussion and conclusions
Lithium orthosilicates are very attractive materials in the current research as possible Li-battery cathodes even though the proposed two-Li ion exchange process is unlikely to be realized. However, the orthosilicates have also shown problems related to achieving phase purity. On the other hand, the electrochemical and theoretical investigations on lithium orthosilicates have shown strong similarities irrespective of the exact polymorph under investigation and, in addition, it seems that all the starting structures tend to stabilize the same delithiated phase.20 As we discussed in the Introduction section, this seems to be a quite unusual behavior in the field of Li-cathode materials where the electrochemical properties are strongly connected to the structural features, which in turn influence the Li-migration paths. However, it is also true that most of the electrochemical probes are closely affected by the actual local structure of the investigated materials and that the computed electrochemical properties are based on local structure optimization tools.In this work we have considered in close detail the local structure behavior of three polymorphs (two orthorhombic and one monoclinic) for the two orthosilicates Li2MnSiO4 and Li2FeSiO4. Discussing first the average structure, each polymorph can be described in a reasonable way with the corresponding space group model if neutron diffraction patterns are taken in consideration and analyzed by means of Rietveld refinement. However, the most careful investigations of the structural features of Li orthosilicates that can be found in the current literature point towards the presence of small deviations from the average structures employed to model the X-ray or neutron diffraction data.10 All these observations were carried out by means of transmission electron microscopy (TEM) which can provide important structural information at the local scale but which cannot be considered a bulk probe. Deviations from the expected long range is also visible from the presence of modulations in the background of high quality ND patterns and, more clearly, in the inspection of the S(Q) vs. Q data we collected in this experiment.
The analysis of the neutron PDFs for the three different polymorphs of each compound has led to an important observation. Considering the local structure in the range up to 6 Å all the polymorphs of Li2MnSiO4 and Li2FeSiO4 are properly described by making use of a single monoclinic structure irrespective of the average structure. This range roughly corresponds to one unit cell of the silicates. Extending the PDF analysis up to 20 or 30 Å, the different polymorphs are properly described making use of the average structure space groups reported in the literature and used to analyze the ND patterns. From a structural point of view, the use of a monoclinic local structure, also for the orthorhombic samples indicates that, at the local scale, the lithium orthosilicates are more distorted than expected from the average structure. Fig. 7 reports an illustrative graphical representation of the changes occurring at the structure passing from an orthorhombic (Pmnb) to a monoclinic symmetry (P21/n).
 | ||
| Fig. 7 Arrangement within in the unit cell for Li2MnSiO4 in the Pmnb s.g. (a) and in the P21/n s.g. (b). Structural data taken from the PDF refinements. | ||
The use of a monoclinic local structure leads to a broader bond-length distribution with respect to the orthorhombic symmetry. For example, the Mn(Fe)–O and Si–O polyhedra have three distinct bond lengths (with two equal distances between Si (and Mn(Fe)) and the O-atoms running along the a-axis) ranging from ca. 1.65 Å to more than 2 Å in the orthorhombic symmetry. The use of monoclinic local symmetry results in four bond lengths within the same distance range (bond lengths for the samples investigated are reported in the ESI†). In addition, the PDF results indicated that, at the local scale, all the polymorphs present two distinct Li-sites with respect to the single Li site in the orthorhombic structures. Finally, the slight deviation of the β angle from 90° induces some rotation of the Si–O and Mn(Fe)–O polyhedra.
From our neutron PDF results it seems quite clear that the increased disorder of the orthorhombic polymorphs in the orthosilicates is mostly confined at the local scale. The common structural features among all the polymorphs at the local scale have important consequences on the electrochemical performance. It is well known that the local arrangement of Li-ions has the strongest impact on the Li-diffusion and, in turn, on the electrochemical behavior. A recent modelling study has shown that orthorhombic polymorphs of lithium silicates have simpler Li migration paths with respect to the monoclinic structures but that the activation energies for the higher symmetric structures have higher energies with respect to the monoclinic arrangements.21 For example, the first jump in the Pmn21 and Pmnb polymorphs of Li2MnSiO4 has been calculated to be more than 1 eV with distances above 3 Å while in the Pn symmetry Ea is around 0.65 eV with a jump distance of below 3 Å. Second jump energies and distances among the different polymorphs are very similar. The results of this modelling work indirectly corroborate the actual PDF results reported in this work. Most of the electrochemical properties are strongly linked to the Li extraction and in particular to the local arrangement of Li ions within the orthosilicate structure. The extremely similar experimental extraction voltages suggest that in all the polymorphs the Li-environment is very similar leading to similar electrochemical results, which should not be the case if very different activation energies and local migration paths are present. For example, the modelling results predict that the electrochemical properties of the Pmn21 polymorph of Li2MnSiO4 should be significantly poorer with respect to the other polymorphs by considering the activation energies and migration paths at the sort range as calculated by using an energy relaxed local structure based on the Pmn21 symmetry. However, this is not the case, and this polymorph behaves in the same way as the other Li2MnSiO4 polymorphs. The results of the present work indicate as well that the delithiation performance does not depend on the long range crystal structure of the selected polymorph and that the efforts in optimizing the lithium orthosilicate properties should not be directed towards the optimization of highly phase-pure materials since even mixtures of different polymorphs should work well. This is further true in view of the fact that after delithiation all the polymorphs tend to transform to a single stable structure.
Finally, the results reported here also support the evidence that the Li-cathode material performance is mostly dependent upon the local structure of Li-ions within a peculiar structure and that the actual longer range diffusion (being 2D or 3D) has a less important impact on the electrochemical properties. In view of this comprehensive structural investigation of the local scale structure of prototype Li-battery cathode materials (i.e., orthosilicates), it can be argued that the design of improved and optimized new compositions should pay strong attention to the local structural features more than the symmetry on the long range.
Acknowledgements
Funding from the INTSM-RL research project “ATLANTE” is acknowledged. Research conducted at the ORNL's Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, and US Department of Energy. This work has benefited from the use of the Lujan Center at the Los Alamos Neutron Science Center, funded by the Department of Energy Office of Basic Energy Sciences.References
- M. S. Whittingham, MRS Bull., 2008, 33, 411 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- J. Liu, J.-G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. Li, Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929 CrossRef CAS.
- M. S. Islam and C. A. Fisher, Chem. Soc. Rev., 2014, 43, 185 RSC.
- V. V. Politaev, A. A. Petrenko, V. B. Nalbandyan, B. S. Medvedev and E. S. Shvetsova, J. Solid State Chem., 2007, 180, 1045 CrossRef CAS PubMed.
- R. J. Gummow and Y. He, J. Power Sources, 2014, 253, 315 CrossRef CAS PubMed.
- A. R. Armstrong, C. Sirisopanaporn, P. Adamson, J. Billaud, R. Dominko, C. Masquelier and P. G. Bruce, Z. Anorg. Allg. Chem., 2014, 640, 1043 CAS.
- C. Masquelier and L. Croguennec, Chem. Soc. Rev., 2013, 113, 6552 CrossRef CAS PubMed.
- G. Mail, C. Sirisopanaporn, C. Masquelier, D. Hanzel and R. Dominko, Chem. Mater., 2011, 23, 2735 CrossRef.
- A. Boulineau, C. Sirisopanaporn, R. Domink, A. R. Armstrong, P. G. Bruce and C. Masquelier, Dalton Trans., 2010, 6310 RSC.
- R. Dominko, M. Bele, M. Gaberscek, A. Meden, M. Remskar and J. Jamnik, Electrochem. Commun., 2006, 8, 217 CrossRef CAS PubMed.
- M. S. Islam, R. Dominko, C. Masquelier, C. Sirisopanaporn, A. R. Armstrong and P. G. Bruce, J. Mater. Chem., 2011, 21, 9811 RSC.
- M. E. Arroyo-de Dompablo, M. Armand, J. M. Tarascon and U. Amador, Electrochem. Commun., 2006, 8, 1292 CrossRef CAS PubMed.
- M. E. Arroyo-de Dompablo, R. Dominko, J. M. Gallardo-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik and E. Morán, Chem. Mater., 2008, 20, 574 Search PubMed.
- S. Nishimura, S. Hayase, R. Kanno, M. Yashima, N. Nakayama and A. Yamada, J. Am. Chem. Soc., 2008, 130, 13212 CrossRef CAS PubMed.
- C. Sirisopanaporn, R. Dominko, C. Masquelier, A. R. Armstrong, G. Maliad and P. G. Bruce, J. Mater. Chem., 2011, 21, 17823 RSC.
- R. J. Gummow, N. Sharma, V. K. Peterson and Y. He, J. Solid State Chem., 2012, 188, 32 CrossRef CAS PubMed.
- H. Duncan, A. Kondamreddy, P. H. J. Mercier, Y. Le Page, Y. Abu-Lebdeh, M. Couillard, P. S. Whifield and I. J. Davidson, Chem. Mater., 2011, 23, 5446 CrossRef CAS.
- C. Eames, A. R. Armstrong, P. G. Bruce and M. S. Islam, Chem. Mater., 2012, 24, 2155 CrossRef CAS.
- A. Saracibar, A. Van der Ven and M. E. Arroyo-de Dompablo, Chem. Mater., 2012, 24, 495 CrossRef CAS.
- C. A. J. Fisher, N. Kuganathan and M. S. Islam, J. Mater. Chem. A, 2013, 1, 4207 CAS.
- P. F. Peterson, M. Gutmann, T. Proffen and S. J. L. Billinge, J. Appl. Crystallogr., 2000, 33, 1192 CrossRef CAS.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Bozin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta04063e |
| This journal is © The Royal Society of Chemistry 2014 |