
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic studies on lithium intercalation in a lithium-rich layered material using Li2RuO3 epitaxial film electrodes and in situ surface X-ray analysis†
Sou
Taminato
a,
Masaaki
Hirayama
*a,
Kota
Suzuki
a,
KyungSu
Kim
a,
Yueming
Zheng
a,
Kazuhisa
Tamura
b,
Jun'ichiro
Mizuki
b and
Ryoji
Kanno
a
aDepartment of Electronic Chemistry, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midoriku-ku, Yokohama, 226-8502, Japan. E-mail: hirayama@echem.titech.ac.jp; Fax: +81 45 924 5403; Tel: +81 45 924 5403
bJapan Atomic Energy Agency, Synchrotron Radiation Research Center, Kansai Research Establishment, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5148, Japan
First published on 3rd September 2014
Abstract
The surface structure of a lithium-rich layered material and its relation to intercalation properties were investigated by synchrotron X-ray surface structural analyses using Li2RuO3 epitaxial-film model electrodes with different lattice planes of (010) and (001). Electrochemical charge–discharge measurements confirmed reversible lithium intercalation activity through both planes, corresponding to three-dimensional lithium diffusion within the Li2RuO3. The (001) plane exhibited higher discharge capacities compared to the (010) plane under high rate operation (over 5 C). Direct observations of surface structural changes by in situ surface X-ray diffraction (XRD) and surface X-ray absorption near edge structure (XANES) established that an irreversible phase change occurs at the (010) surface during the first (de)intercalation process, whereas reversible structural changes take place at the (001) surface. These experimental findings suggest that the surface reconstructed phase limits lithium intercalation between the electrode and the electrolyte, leading to the poor rate capability of the (010) film. Surface structural changes at the initial cycling therefore have a pronounced effect on the power characteristics and stability of lithium-rich layered materials during battery operation.
Introduction
Lithium-rich layered oxides, xLi2MO3-(1−x)LiMO2 (M = 3d and/or 4d transition metal) have attracted significant attention as promising intercalation cathodes for use in lithium batteries, since they allow high discharge capacities over 200 mA h g−1 and high operating voltages (over 3.5 V vs. Li/Li+ on average).1–6 The discharge voltage of such cathodes, however, is gradually reduced during electrochemical cycling, resulting in the fading of energy density.1,2,7,8 Another deficit is their poor rate capability compared to that obtainable with LiCoO2.9–11 These drawbacks make it difficult to consider lithium-rich layered oxides as viable candidates for the replacement of conventional layered rock salt cathodes (Li(Co, Ni, Mn)O2). Structural observations of deteriorated electrodes have indicated that reductions in the reaction voltage are caused by a phase transition to form spinel and/or rock salt structures during the lithium (de)intercalation process.12–14 A comprehensive understanding of changes in the crystal structure during the lithium intercalation reactions is therefore an essential step in elucidating the detailed reaction mechanisms of lithium-rich layered oxide electrodes.The crystal structures of lithium-rich layered oxides are derived from rhombohedral α-NaFeO2, which is composed of alternate layers of lithium and transition metal atoms, both occupying the octahedral sites of a cubic close packing of oxide ions. One-third of the positions in the transition metal plane are replaced by lithium to form an ordered Li1/3Mn2/3 slab.7,15–17 During the intercalation process, lithium ions are believed to diffuse primarily through the infinite two-dimensional [LiO6] layers in the ab plane by occupying tetrahedral sites.18–20 Furthermore, it has been demonstrated that lithium ions diffuse through the lithium sites in the transition metal layer when lithium vacancies are formed in the transition metal layers during the deintercalation process,21,22 leading to three-dimensional lithium diffusion.23–26 The anisotropic lithium diffusion could be a factor in determining the stability and rate capability of the lithium-rich layered oxide electrodes.
Further to lithium diffusion in the bulk structure, electrochemically-driven lithium intercalation involves reaction processes at the electrode/electrolyte interface. These include adsorption of solvated lithium at the electrode surface, desolvation, surface diffusion, charge transfer and intercalation into the structure. Electrochemical and spectroscopic studies have emphasized the importance of surface reactions in terms of their effects on the power and calendar-life characteristics.27–29 Although there have been several reports for layered and spinel type electrode materials that surface structure changes at the electrochemical interface are significantly different from the bulk structure changes,30,31 no detailed information regarding the surface structures of lithium-rich layered oxide electrodes during battery operation is available.
Epitaxial film electrodes with a single orientation and an atomically small roughness of less than a few nanometers can simplify the surface reaction field, and thus are ideal models for clarifying crystal structures and lithium diffusion processes at the electrode surface.30,32 Recently, epitaxial Li2RuO3(001) films having a lithium-rich layered structure have been successfully grown on Al2O3(0001) by pulsed laser deposition (PLD) followed by a post-annealing treatment.25,26 In this study, we performed mechanistic studies concerning lithium intercalation in Li2RuO3 using epitaxial thin film electrodes as a model system of lithium-rich layered material. Lattice plane dependences of intercalation properties with lattice plane and surface structure changes were investigated by electrochemical measurements and surface X-ray structural analyses using the Li2RuO3 (010) and (001) planes. Based on the results, the effects of the stability of surface structures on intercalation processes are discussed.
Experimental
Epitaxial Li2RuO3 films were synthesized on 0.5% Nb-doped SrTiO3 (111) and (110) single crystal substrates (10 × 10 × 0.5 mm, Crystal Base Co., Ltd.) using a pulsed laser deposition (PLD) method in conjunction with a post-annealing process. The PLD system consisted of 248 nm KrF excimer laser (Lambda Physik, COMPex102 and COMPex201) and vacuum chambers (PLAD131 and PLAD-312, AOV Inc.). Li2.4RuO3 was used as the target to compensate for any loss of lithium during the deposition process. The film fabrication conditions were as follows: temperature (T) = 500–550 °C, distance between substrate and target (d) = 7.0 cm, laser frequency (f) = 10 Hz, deposition time (t) = 10–30 min, laser energy (E) = 0.68–1.56 J cm−2 and oxygen pressure (pO2) = 3.3 Pa. Film deposition under these conditions was followed by annealing in air at 600 °C to improve crystallinity of the films. The orientation of each film was characterized by both out-of-plane and in-plane X-ray diffraction (XRD) measurements using a thin-film X-ray diffractometer (Rigaku ATX-G) with CuKα1 radiation. The thickness, roughness and density values of the films were analysed using X-ray reflectivity curves collected using the thin-film X-ray diffractometer (see details in ESI†). Charge–discharge characteristics were examined using 2032-type coin cells assembled in an argon-filled glove box. The counter/reference electrode and electrolyte were lithium metal and 1 mol dm−3 LiPF6 in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mixture of ethylene carbonate (EC) and diethyl carbonate (DEC). The charge–discharge capacities of the cells were calculated using the area over which the film was deposited (10 × 8 mm), the film thickness and the theoretical density of Li2RuO3 (5.15 g cm−3).
:7 mixture of ethylene carbonate (EC) and diethyl carbonate (DEC). The charge–discharge capacities of the cells were calculated using the area over which the film was deposited (10 × 8 mm), the film thickness and the theoretical density of Li2RuO3 (5.15 g cm−3).
Crystal structure changes during electrochemical reactions were investigated by in situ surface XRD analysis using a spectroelectrochemical cell.33,34 The counter electrode of the electrochemical cell was made of lithium metal and the electrolyte solution consisted of 3:7 EC/DEC containing 1 mol dm−3 LiPF6 or 1 mol dm−3 LiClO4 when working with Li2RuO3 on SrTiO3(111) or on SrTiO3(110), respectively. The XRD patterns of Li2RuO3 on SrTiO3(111) were acquired using a multi-axis diffractometer (HUBER) installed on the in-vacuum undulator beamline BL22XU at SPring-8. The X-rays were monochromated using a Si(111) double crystal system and focused via two Rh-coated bent mirrors. The details of the experimental conditions have been described elsewhere.31,32 The XRD patterns of Li2RuO3 on SrTiO3(110) were collected using a κ-type six-circle diffractometer (New Port) installed on the bending-magnet beamline BL14B1 at the synchrotron beam facilities SPring-8, Japan. An X-ray wavelength of 0.82518 Å (15 keV) was employed. The measurements were performed using reciprocal coordinate systems (H,K,L) with two components (H,K) lying parallel to the surface and a third (L) normal to the surface.35 To investigate the surface structural changes, the in-plane XRDs were collected at the incident angle to the half value of the critical angle θc. The penetration depth of X-ray (15 keV) could be calculated to be 2.6 nm at 0.5θc (0.08°). Deintercalation and intercalation were induced by the potentiostatic method using a potentiostat (Ivium, Compactstat). The XRD patterns were observed in air, after the cell construction, at 4.0 V (vs. Li/Li+) after charging, and 3.0 V after discharging. The diffraction peaks during electrochemical cycling were indexed based on a monoclinic lattice.
In situ X-ray absorption near edge structure (XANES) measurements were performed in the fluorescence mode using a germanium single-element solid-state detector installed on beamline BL14B2 at SPring-8. The synchrotron radiation was monochromated by Si(111) double crystal system that was used as the crystal plane of the monochromator for Ru K-edge. XANES data were collected at an oblique angle of incidence (4°) and at a low glancing angle (0.1°) below the critical angle of Li2RuO3 using a θ–2θ stage mounted on the beamline. The penetration depths were calculated to be 2.80–2.85 nm in the range of X-ray energy used (21.8 to 23.4 keV). The XANES spectra collected at an incident angle of 4° correspond to the oxidation states of Ru ions throughout the films, while XANES spectra collected at a low glancing angle enhance the X-ray fluorescence of the top surface (the upper few nanometers) of the films, so as to investigate electronic structural changes in the electrode bulk and at the surface, respectively. The counter electrode of the electrochemical cell was lithium metal, and the electrolyte solution was 1 mol dm−3 LiClO4 in 3:7 EC/DEC. The XANES spectra were observed, after the cell construction, at 3.5 V and 4.0 V at the first charging, and 3.4 and 3.0 V after the first discharging. Pre-edge background and post-edge normalization to unity of the Ru K-edge spectra were performed using the ATHENA,36 and the adsorption edge E0 was defined as an energy value of 0.5 in the normalized absorption spectra. The standard deviation was evaluated from the average for at least 5 times.
Results and discussion
Film orientation
Fig. 1 shows the XRD patterns of a Li2RuO3 film on a Nb:SrTiO3(111) substrate. The pattern exhibits diffraction peaks at 18.2°, 37.2° and 78.6° along the out-of-plane [111] direction (Fig. 1a), which are attributed to the 00l diffraction peaks of Li2RuO3 with the C2/c space group.15 Along the in-plane [1−10] direction, the 020 and 060 diffraction peaks of Li2RuO3 are observed at 20.3° and 63.4°, respectively (Fig. 1b). The 020 peak is due to a superlattice structure resulting from a honeycomb-type ordering of Li/Ru in the transition metal layer.37 This result indicates the formation of Li2RuO3 with a lithium-rich layered structure. The rocking curve of the 060 reflection indicates a six-fold symmetry at intervals of 60° (Fig. 1c). The 060 reflection should show two-fold symmetry and so the observed six-fold symmetry demonstrates that the Li2RuO3(001) film is composed of 60°-rotated domains: Li2RuO3[010]//SrTiO3[1−10], Li2RuO3[010]//SrTiO3[0−11] and Li2RuO3[010]//SrTiO3[−101]. The cell parameters are a = 5.116(14) Å, b = 8.861(16) Å, c = 9.772(4) Å and β = 100.09(4)°. | ||
| Fig. 1 X-ray diffraction patterns of Li2RuO3 films deposited on the SrTiO3(111) substrate. (a) 2θ/ω scan along the [111] direction of the SrTiO3, (b) 2θχ/ϕ scan along the [1−10] direction of the SrTiO3 and (c) ϕ scan of the 060 reflection at 2θχ = 63.4°. | ||
Fig. 2 shows the XRD patterns of a Li2RuO3 film on the SrTiO3(110) substrate. The film has 0k0 and h0h orientations along the out-of-plane [110] and in-plane [001] directions of the SrTiO3 substrate, respectively (Fig. 2(a) and (b)). The rocking curve of the 202 reflection indicates two-fold symmetry at intervals of 180° (Fig. 2c), which is consistent with the lattice symmetry of Li2RuO3. The cell parameters are a = 5.07(17) Å, b = 8.82(3) Å, c = 9.77(2) Å and β = 99.7(2)°. These thin-film XRD results confirm the formation of epitaxial Li2RuO3 (010) and (001) films on the SrTiO3 (110) and (111) substrates, respectively.
 | ||
| Fig. 2 X-ray diffraction patterns of Li2RuO3 films deposited on the SrTiO3(110) substrate. (a) 2θ/ω scan along the [110] direction of the SrTiO3, (b) 2θχ/ϕ scan along the [001] direction of the SrTiO3 and (c) ϕ scan of the 202 reflection at 2θχ = 43.4°. | ||
The thickness, roughness, and density of each film were analysed using X-ray reflectivity curves (see details in ESI†). The thicknesses of the films ranged from 25 to 65 nm, depending on the duration of the deposition. The cell parameters showed no dependence of the lattice orientations on the thicknesses of the films (ESI†). We therefore succeeded in controlling the Li2RuO3 film orientation without inducing any significant differences in cell parameters and surface roughness.
Electrochemical properties
Fig. 3(a) and (b) show the charge–discharge and differential capacity (dQ/dV) curves of 26.4 nm thick Li2RuO3(010) film over the potential range from 3.0 to 4.0 V. The (010) film exhibited first charge and discharge capacities of 150 and 120 mA h g−1, respectively, with an irreversible capacity of 30 mA h g−1. In the dQ/dV plots, two oxidation and reduction peaks were observed at 3.38 and 3.66 V and 3.30 and 3.60 V, respectively. These peak positions are in good agreement with those of polycrystalline Li2RuO3, as reported by Kobayashi et al.15 The lithium (de)intercalation proceeds by a two-phase reaction consisting of a phase transition from monoclinic Li2RuO3 to monoclinic Li1.4RuO3 around 3.4 V and a phase transition from monoclinic Li1.4RuO3 to rhombohedral Li0.9RuO3 in the vicinity of 3.6 V. The oxidation state of the Ru ions changes from Ru4+ to Ru5+, followed by the oxygen participating in the charge compensation process, when Ru ions are fully oxidized to pentavalent.38 The discharge capacity gradually increased from the second to the fifth cycle with a concurrent decrease in the irreversible capacity, while no significant changes in the peak positions were observed in the dQ/dV plots. The discharge capacity was slightly less than that obtained from polycrystalline Li2RuO3 electrodes (160 mA h g−1), since a secondary RuO2 phase which has no electrochemical activity between 3.0 and 4.0 V (ref. 39) is present in the Li2RuO3 film electrodes. The 26.4 nm thick Li2RuO3(001) film exhibited first charge and discharge capacities of 134 and 105 mA h g−1 (Fig. 3c). In the dQ/dV plots, two oxidation and reduction peaks are observed at 3.37 and 3.61 V and 3.30 and 3.55 V, respectively (Fig. 3d). The discharge capacity gradually increased from the second to the fifth cycle with a corresponding decrease in the irreversible capacity. The fifth discharge capacity of 113 mA h g−1 is comparable to that of the (010) film (124 mA h g−1). These results agree with a three-dimensional lithium diffusion in the Li2RuO3.26 | ||
| Fig. 3 Charge–discharge curves and differential capacity plots of 26.4 nm thick (a) and (b) Li2RuO3(010) and (c) and (d) Li2RuO3(001) films from the first to the fifth cycles. A current density of 91.9 mA g−1 was applied. | ||
Fig. 4 shows the discharge capacity retentions of Li2RuO3(010) and Li2RuO3(001) films depending on the cycle number and C rate. Both the (010) and (001) films showed high capacity retention (above 90%), indicating highly stable intercalation through the (010) and (001) planes. In contrast, the rate capability tests demonstrate anisotropic behavior at C rates from 5 to 20 C. The (001) film exhibits higher power characteristics than the (010) film during the discharge process. Based on structural considerations, the 〈010〉 direction along the ab plane is better suited to lithium diffusion than the 〈001〉 direction, due to the larger bottleneck size in Li2RuO3 (ref. 15) (see details in ESI†). The anisotropic activation energies of lithium diffusion through the lithium-rich layered material Li2MnO3, which has a similar crystal structure to Li2RuO3, has been determined using first principle calculations. The results of these calculations show that lithium diffusion along the ab plane is associated with a lower activation energy (0.61 eV) than diffusion along the c axis (0.73 eV).40 These predictions based on the bulk structure, however, cannot explain the higher power characteristics of the (001) film observed in the present study. Therefore, to obtain a better understanding of the origin of the intercalation properties of Li2RuO3, surface structural changes of the Li2RuO3 electrodes were investigated by in situ X-ray diffraction and absorption techniques.
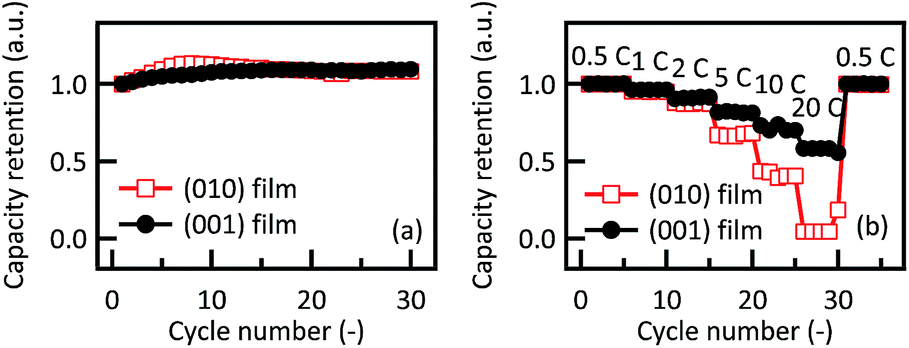 | ||
| Fig. 4 Variations in discharge capacity retention of Li2RuO3(010) and Li2RuO3(001) films with (a) cycle number and (b) charge–discharge C rates. | ||
Crystal structure changes during electrochemical cycling
 | ||
| Fig. 5 In situ XRD patterns of 29.0 nm thick Li2RuO3(010) film along the out-of-plane (a) [0,0,L] and (b) [0,K,K] and the in-plane (c) [H,0] and (d) [H,−2H] directions of the SrTiO3(110) substrate. Patterns were obtained in air (Dry), after cell construction (OCV) and during the charge–discharge process. The dark and red dashed lines in Fig. 5(c) correspond to the 202 diffraction peak of Li2RuO3 and a diffraction peak of the surface phase formed at the electrochemical process, respectively. | ||
During the first charge from the OCV state to 4.0 V, the 060 and 1−3−3 peaks shift to higher angles with decreasing peak intensities, due to lattice contraction of the Li2RuO3(010) bulk on lithium deintercalation. It has been reported that the phase transition from monoclinic layered rock salt-type Li2RuO3 to hexagonal ilmenite-type Li0.9RuO3 occurs on applying a cell voltage from 3.0 to 4.0 V, due to lithium deintercalation from the lithium layer.15 This deintercalation induces a structural change from a cubic close-packed to a hexagonal close-packed oxide-ion array, which leads to the lattice contraction. As a result, the monoclinic 060 and 13−3 peaks shifted to higher angles. The lattice change of the Li2RuO3(010) film is therefore consistent with that of polycrystalline Li2RuO3. The 060 and 1−3−3 peaks shift reversibly with a decrease in cell voltage from 4.0 to 3.0 V, corresponding to reversible intercalation through the edge plane of the two-dimensional structure of the (010) film. The XRD patterns of the 060 and 1−3−3 peaks at the fourth discharging and at the fifth charging are similar to those at the first cycle, which indicates the high stability of the Li2RuO3(010) bulk during the (de)intercalation process. The 202 and 004 surface diffraction peaks slightly shift to higher and lower angles on the charging to 4.0 V and the discharging to 3.0 V compared to the out-of-plane diffraction peaks. The small lattice change is often observed for in-plane diffraction peaks of epitaxial film electrodes, because the lattice change is strongly constrained by the substrate lattice.30–32 In the case of the 202 peak, the diffraction peak at H = 1.89 decreases in intensity on the initial charging to 4.0 V, and the intensity of the H = 1.89 peak is recovered on discharging to 3.0 V. During the subsequent cycles, the H = 1.89 peak changes reversibly, which corresponds to reversible structural changes during lithium (de)intercalation processes.
It is noteworthy that an additional peak appears at H = 1.86 on the initial charging to 4.0 V and remains stable after the first charging. The additional peak could not be detected in the in-plane XRD pattern collected using a higher incident angle than the critical angle (see details in ESI†). An irreversible transition therefore occurred at the top surface of the (010) plane during the first deintercalation process. The 004 surface diffraction peak shows a slight peak shift to a higher angle with the decrease in the peak intensity on the initial charging to 4.0 V, and the reversible peak shift and intensity changes are observed during the subsequent processes. No diffraction peak of the irreversible phase is observed along the SrTiO3[H,−2H] direction, which confirms that the new phase has a crystal structure different from that of Li2RuO3. It has been reported that the electrode surfaces are reconstructed to form a stable structure at the electrode/organic electrolyte interface.30,31 Thus, lithium intercalation proceeds between the Li2RuO3 and the electrolyte through the reconstructed surface phase.
 | ||
| Fig. 6 In situ XRD patterns of 61.6 nm thick Li2RuO3(001) film along out-of-plane (a) [0,0,L] and (b) [0,K,K] and in-plane (c) [2H,−K] directions of the SrTiO3(111) substrate. Patterns were obtained in air (Dry), after cell construction (OCV) and during the charge–discharge process. | ||
Electronic structure changes during electrochemical cycling
Fig. 7 shows the bulk and surface-enhanced XANES spectra at the Ru K-edge and K-edge adsorption energies of the Li2RuO3(010) film during the first cycle. In the bulk region, the absorption edge shifts to a higher energy during the charging process and then shifts reversibly to a lower energy during the discharge. These reversible changes in the electronic structures occur due to lithium (de)intercalation into the Li2RuO3(010) bulk. At the surface region, the absorption edge shifts to a lower energy on going from OCV to 3.5 V during the charging process, and no significant changes are observed during the following process. The electronic structure at the Li2RuO3(010) surface therefore changes irreversibly on the initial deintercalation, which is in agreement with the results of the surface XRD analyses. | ||
| Fig. 7 In situ Ru-K edge XANES spectra of Li2RuO3(010) in (a) bulk and (b) surface regions and (c) K edge absorption energies during the first charging and discharging processes. | ||
Fig. 8 presents the bulk and surface-enhanced XANES spectra and adsorption energies at the Ru K-edge of the Li2RuO3(001) film during the first cycle. In the bulk region, the absorption edge shifts to a higher energy during the charging process and then shifts reversibly to a lower energy during the discharge. A reversible shift of the absorption edge is also observed in the surface region, although the energies of the absorption edge are less than those of the bulk region. The (001) film thus exhibits reversible electronic structure changes in both the bulk and surface regions.
 | ||
| Fig. 8 In situ Ru-K edge XANES spectra of Li2RuO3(001) in (a) bulk and (b) surface regions and (c) K edge absorption energies during the first charging and discharging processes. | ||
Electrode reactions of Li2RuO3
Fig. 9 shows schematic representations of the proposed reactions of the Li2RuO3 electrodes, based on our electrochemical, in situ XRD and in situ XAFS measurements. Within the Li2RuO3 structure, lithium ions can diffuse along the c-axis between neighboring two-dimensional lithium layers through the lithium sites in the transition metal layer.26 | ||
| Fig. 9 Schematic representations of lithium intercalation reactions in the Li2RuO3 films. | ||
Thus, the intercalation and deintercalation of lithium ions proceed through both the (010) and (001) surfaces due to the three-dimensional lithium diffusion. No capacity fading was observed over the course of 30 cycles, which indicates highly stable (de)intercalation within the Li2RuO3 electrodes. In the bulk region, the crystal structure undergoes reversible changes with lithium (de)intercalation in the case of both the (010) and the (001) film, which is in agreement with the electrochemical results. Layered Li2MO3 electrodes based on 3d transition metals generally suffer from lowering of the reaction voltage due to a phase transition to the spinel and/or rock salt structures.12–14 In such 3d transition metal systems, it has been reported that Li/M interlayer mixing and the occupation of Li ions at the tetrahedral sites in the Li layer leads to the phase transformation.41 In contrast, 4d metal ions form strong M–O bonds due to significant orbital hybridization.42,43 These strong M–O bonds could stabilize the Li2RuO3 lattice during the lithium (de)intercalation reaction. Recently, Sathiya et al. reported that the substitution of Ru for Mn ions in Li2MnO3 improves the structural stability and facilitates electrochemical reactions of substituted compound.38
In the surface region, the irreversible phase change occurs at the top surface of the (010) plane during the first deintercalation process, whereas reversible structural changes are observed at the (001) surface. It has been demonstrated that surface structure changes during the initial cycle may be attributed to surface reconstruction phenomena that are typically induced by instability of the termination structure.31–33 Surface reconstruction accompanied by rearrangement of cations and anions could form a stable surface structure in response to changes in interfacial conditions during the (de)intercalation processes. No published results are available for the Li2RuO3 surface, but the surface energies of the (010) and (001) planes of Li(Li0.17Ni0.25Mn0.58)O2, which have a lithium-rich layered rock salt structure, have been investigated based on density functional theory (DFT).44 The (010) plane of Li(Li0.17Ni0.25Mn0.58)O2 has a higher surface energy than the (001) plane, which suggests lower stability of the (010) surface. Assuming that the relative stabilities of the Li2RuO3 surfaces are similar to those of Li(Li0.17Ni0.25Mn0.58)O2, it is reasonable to suggest that significant structural change occurs at the less stable (010) surface during the reconstruction process. No substantial effects of the surface reconstructed phase on the discharge capacity were observed under low current density operation. In contrast, the discharge capacities of the (010) film decreased to a greater extent than those of the (001) film under high current density operation from 5 C to 20 C. First principle simulations indicate that bulk lithium diffusion in the 〈010〉 direction is more favourable to high rate operation than diffusion which proceeds along the 〈001〉 direction.40 Hence, we conclude that the surface reconstructed phase limits the intercalation reaction through the (010) surface, likely as a result of high resistance to lithium ion diffusion and/or charge transfer reactions. Anisotropic properties such as those confirmed by our experimental results could be a common phenomenon in polycrystalline Li2MO3 electrodes containing 3d transition metals and intended for practical applications. The stabilization of the (010) surface is therefore a key issue with regard to improving the power characteristics of lithium-rich layered materials.
Conclusions
Epitaxial Li2RuO3 films with different lattice planes were successfully synthesized on SrTiO3 single-crystal substrates by pulsed laser deposition. The Li2RuO3 films exhibited (010) and (001) orientations on Nb-doped SrTiO3 (110) and (111) substrates, respectively. These films had similar cell parameters and surface roughness and provided a restricted reaction field suitable for mechanistic studies of lithium diffusion and interfacial reactions. The (010) and (001) films exhibited a first discharge capacity of 120 and 105 mA h g−1 at 0.5 C rate, respectively. The (001) film, however, showed better rate capability than the (010) film, even though the structural features expect a superior Li diffusion along the 〈010〉 direction. In situ surface XRD and surface XANES analyses showed that an irreversible phase transition occurs at the (010) surface during the first deintercalation process, while the (001) surface undergoes reversible structural changes. The observed phase formation on the (010) surface might originate from the difference in surface energies between the (010) and (001) planes, resulting in anisotropic intercalation properties during high rate operation. The surface structures could therefore determine the power density and stability of lithium-rich layered material electrodes.Acknowledgements
The author thanks Dr T. Honma and Dr H. Ofuchi of the Japan Synchrotron Radiation Research Institute (JASRI) for assistance with synchrotron XAFS experiments performed using the BL14B2 line at the SPring-8 facility at JASRI. This work was conducted in collaboration with the Genesis Research Institute. The research was also partially supported by a Grant-in-Aid for Scientific Research (A), a Grant-in-Aid for Young Scientists (B), and a Grant-in-Aid for Scientific Research on Innovative Areas “Exploration of nanostructure–property relationships for materials innovation” from the Japan Society for the Promotion of Science and a Grant-in-Aid from the Advanced Low Carbon Technology Research and Development Program (ALCA) of the JST. The synchrotron experiments were performed under the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposals no. 2010A1742, 2010A3672 and 2011B3671).Notes and references
- Z. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191–A194 CrossRef CAS PubMed.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef CAS PubMed.
- A. Ito, D. Li, Y. Ohsawa and Y. Sato, J. Power Sources, 2008, 183, 344–346 CrossRef CAS PubMed.
- D. Y. W. Yu, K. Yanagida, Y. Kato and H. Nakamura, J. Electrochem. Soc., 2009, 156, A417–A424 CrossRef CAS PubMed.
- T. Ohzuku, M. Nagayama, K. Tsuji and K. Ariyoshi, J. Mater. Chem., 2011, 21, 10179–10188 RSC.
- K. Kubota, T. Kaneko, M. Hirayama, M. Yonemura, Y. Imanari, K. Nakane and R. Kanno, J. Power Sources, 2012, 216, 249–255 CrossRef CAS PubMed.
- H. Kobayashi, R. Kanno, M. Tabuchi, H. Kageyama, O. Nakamura and M. Takano, J. Power Sources, 1997, 68, 686–691 CrossRef CAS.
- B. Ammundsen, J. Paulsen, I. Davidson, R.-S. Liu, C.-H. Shen, J.-M. Chen, L.-Y. Jang and J.-F. Lee, J. Electrochem. Soc., 2002, 149, A431–A436 CrossRef CAS PubMed.
- J. Choi and A. Manthiram, Solid State Ionics, 2005, 176, 2251–2256 CrossRef CAS PubMed.
- J. Jiang, K. W. Eberman, L. J. Krause and J. R. Dahn, J. Electrochem. Soc., 2005, 152, A1879–A1889 CrossRef CAS PubMed.
- Z. Li, N. A. Chernova, J. Feng, S. Upreti, F. Omenya and M. S. Whittingham, J. Electrochem. Soc., 2012, 159, A116–A120 CrossRef CAS PubMed.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef CAS.
- B. Song, Z. Liu, M. O. Lai and L. Lu, Phys. Chem. Chem. Phys., 2012, 14, 12875–12883 RSC.
- A. Boulineau, L. Simonin, J.-F. Colin, E. Canévet, L. Daniel and S. Patoux, Chem. Mater., 2012, 24, 3558–3566 CrossRef CAS.
- H. Kobayashi, R. Kanno, Y. Kawamoto, M. Tabuchi, O. Nakamura and M. Takano, Solid State Ionics, 1995, 82, 25–31 CrossRef CAS.
- H. Kobayashi, R. Kanno, Y. Kawamoto, M. Tabuchi and O. Nakamura, Solid State Ionics, 1996, 86–88, 859–863 CrossRef CAS.
- K. Asakura, S. Okada, H. Arai, S.-i. Tobishima and Y. Sakurai, J. Power Sources, 1999, 81–82, 388–392 CrossRef CAS.
- G. Nuspl, M. Nagaoka, K. Yoshizawa, F. Mohri and T. Yamabe, Bull. Chem. Soc. Jpn., 1998, 71, 2259–2265 CrossRef CAS.
- A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2000, 3, 301–304 CrossRef CAS PubMed.
- A. Hirano, K. Kanie, T. Ichikawa, N. Imanishi, Y. Takeda, R. Kanno, T. Kamiyama and F. Izumi, Solid State Ionics, 2002, 152–153, 207–216 CrossRef CAS.
- A. Van der Ven and G. Ceder, Electrochem. Commun., 2004, 6, 1045–1050 Search PubMed.
- C. P. Grey, W.-S. Yoon, J. Reed and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A290–A293 CrossRef CAS PubMed.
- Y. Koyama, I. Tanaka, M. Nagao and R. Kanno, J. Power Sources, 2009, 189, 798–801 CrossRef CAS PubMed.
- M. Jiang, B. Key, Y. S. Meng and C. P. Grey, Chem. Mater., 2009, 21, 2733–2745 CrossRef CAS.
- Y. Zheng, S. Taminato, Y. Xu, K. Suzuki, K. Kim, M. Hirayama and R. Kanno, J. Power Sources, 2012, 208, 447–451 CrossRef CAS PubMed.
- Y. Zheng, S. Taminato, K. Suzuki, M. Hirayama and R. Kanno, Thin Solid Films, 2012, 520, 4889–4893 CrossRef CAS PubMed.
- J. Cho, G. B. Kim, H. S. Lim, C.-S. Kim and S.-I. Yoo, Electrochem. Solid-State Lett., 1999, 2, 607–609 CrossRef CAS PubMed.
- G. Amatucci, A. Du Pasquier, A. Blyr, T. Zheng and J. M. Tarascon, Electrochim. Acta, 1999, 45, 255–271 CrossRef CAS.
- S. K. Martha, J. Nanda, G. M. Veith and N. J. Dudney, J. Power Sources, 2012, 216, 179–186 CrossRef CAS PubMed.
- K. Sakamoto, M. Hirayama, N. Sonoyama, D. Mori, A. Yamada, K. Tamura, J. Mizuki and R. Kanno, Chem. Mater., 2009, 21, 2632–2640 CrossRef CAS.
- M. Hirayama, H. Ido, K. Kim, W. Cho, K. Tamura, J. Mizuki and R. Kanno, J. Am. Chem. Soc., 2010, 132, 15268–15276 CrossRef CAS PubMed.
- K. Sakamoto, M. Hirayama, H. Konishi, N. Sonoyama, N. Dupré, D. Guyomard, K. Tamura, J. Mizuki and R. Kanno, Phys. Chem. Chem. Phys., 2010, 12, 3815–3823 RSC.
- M. Hirayama, N. Sonoyama, M. Ito, M. Minoura, D. Mori, A. Yamada, K. Tamura, J. Mizuki and R. Kanno, J. Electrochem. Soc., 2007, 154, A1065–A1072 CrossRef CAS PubMed.
- M. Hirayama, N. Sonoyama, T. Abe, M. Minoura, M. Ito, D. Mori, A. Yamada, R. Kanno, T. Terashima, M. Takano, K. Tamura and J. Mizuki, J. Power Sources, 2007, 168, 493–500 CrossRef CAS PubMed.
- B. M. Ocko, J. Wang, A. Davenport and H. Isaacs, Phys. Rev. Lett., 1990, 65, 1466–1469 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Breger, M. Jiang, N. Dupré, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575–2585 CrossRef CAS PubMed.
- M. Sathiya, K. Ramesha, G. Rousse, D. Foix, D. Gonbeau, A. S. Prakash, M. L. Doublet, K. Hemalatha and J. M. Tarascon, Chem. Mater., 2013, 25, 1121–1131 CrossRef CAS.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621–625 CrossRef CAS.
- R. Xiao, H. Li and L. Chen, Chem. Mater., 2012, 24, 4242–4251 CrossRef CAS.
- B. Xu, C. R. Fell, M. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223–2233 CAS.
- J.-H. Park, J. Lim, J. Yoon, K.-S. Park, J. Gim, J. Song, H. Park, D. Im, M. Park, D. Ahn, Y. Paik and J. Kim, Dalton Trans., 2012, 41, 3053–3059 RSC.
- M. D. Johannes, A. M. Stux and K. E. Swider-Lyons, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075124 CrossRef.
- G.-Z. Wei, X. Lu, F.-S. Ke, L. Huang, J.-T. Li, Z.-X. Wang, Z.-Y. Zhou and S.-G. Sun, Adv. Mater., 2010, 22, 4364–4367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta02795g |
| This journal is © The Royal Society of Chemistry 2014 |